
Cross-Domain Text Mining of Pathophysiological Processes Associated with Diabetic Kidney Disease

 , and
, and
Abstract
:1. Introduction
- The developed workflow with SemNet 2.0 and additional post hoc analysis with Gene Ontology (GO) networks and Cytoscape support the integrative prioritization of literature relationships that advance the study of DKD.
- The top-ranked source nodes identified by SemNet 2.0 enable the more comprehensive construction of protein–protein interactions, the efficient modeling of biochemical pathways, and the expedited testing of literature-based hypotheses through experiments.
- The application of artificial intelligence-enabled LBD techniques to DKD research enables faster integration and processing of new biomedical literature towards secondary analyses that improve drug discovery and target treatment design.
2. Results
2.1. Source Nodes Revealed by Cross-Domain Analysis
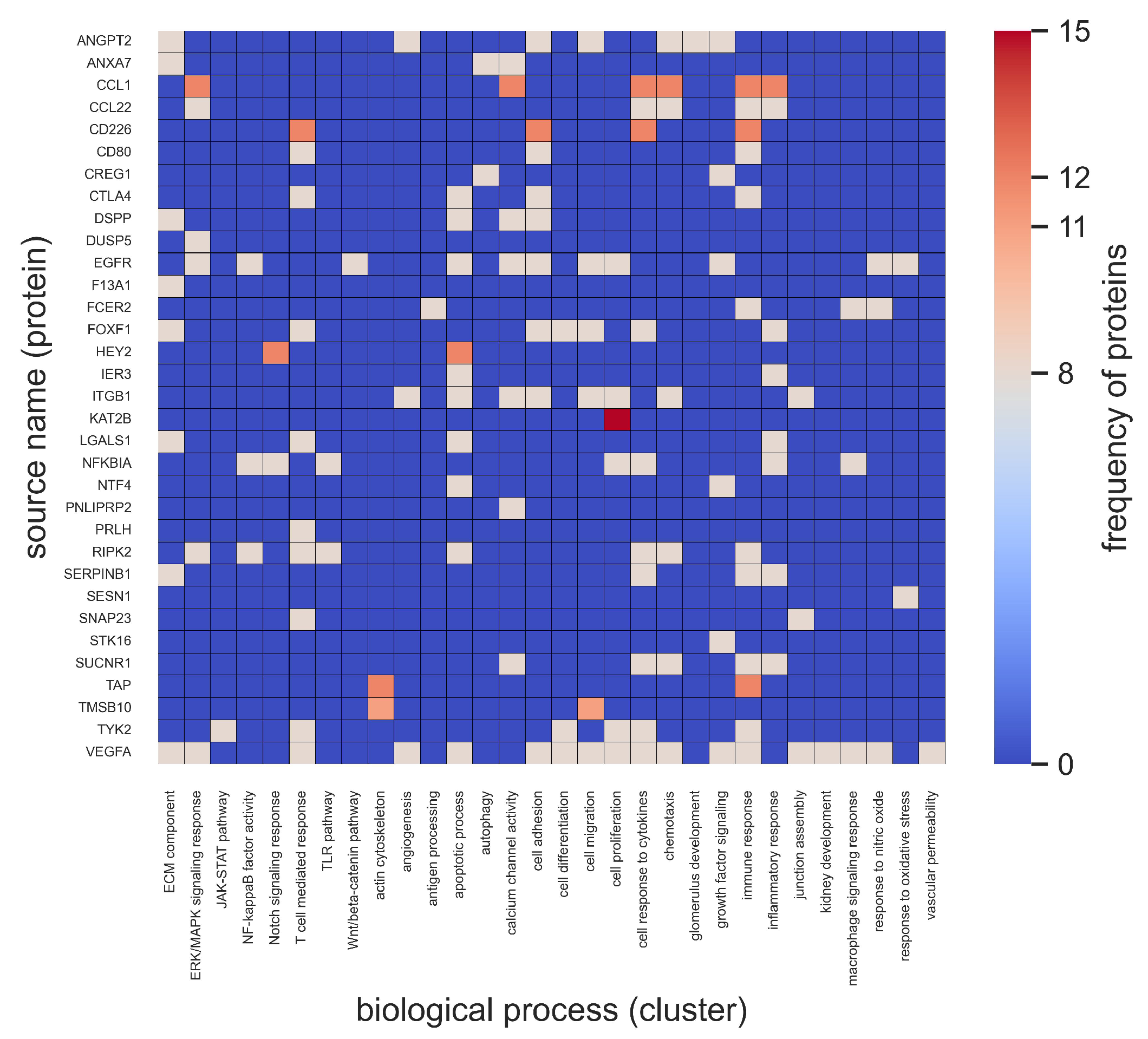
2.2. Biological Themes Associated with Top 10% of Source Nodes
2.3. Interaction Network in Cytoscape
3. Discussion
3.1. Top-Ranked Intersecting GEC-IR-DKD Nodes
3.1.1. Top-Ranked Intersecting Cellular Functions and Signaling Processes
- Role of T-cells. This finding is consistent with other studies that report T-cells as being the most studied immune cells that infiltrate kidney tissues and trigger inflammatory responses in DKD [48,49,50]. Given the prevalent role of T-cells and the T-cell-mediated cytokine response in DKD [28,51,52], their highly ranked importance by the unsupervised ranking algorithm in SemNet 2.0 was expected.
- Role of VEGF. Growth factor signaling via VEGF, fibroblast growth factors, transforming growth factor-, and insulin-like growth factors in diabetes and diabetic kidney disease has been studied in detail [56,57]. VEGF is a potent angiogenic and vascular permeability factor and is responsible for endothelial cell proliferation and differentiation and increased permeability [9,58]. VEGF also maintains endothelial cell homeostasis, and a disturbance in basal VEGF levels is implicated in diabetes-related complications, including kidney disease [59]. Specifically, VEGF-A is associated with macrophage or monocyte differentiation, which suggests its role in the macrophage response in pathological conditions [9,42,57,58].
- Role of TGF. An increase or decrease in the production of transforming growth factor -1 (TGF-1) has been associated with diabetic nephropathy and retinopathy [56]. The expression of TGF-1 is increased in endothelial cells, which, in turn, triggers the activation of TGF receptors, namely TGFBR2 and TGFBR3, on B lymphocytes, podocytes, glomerular endothelial cells, and mesangial cells, leading to epithelial–mesenchymal transition and fibrosis in the development of diabetic nephropathy [17,57,59]. Insulin-like growth factor-I is a naturally occurring single-chain polypeptide that has been widely used in the treatment of diabetic glomerular and renal tubular injuries [56,57,60].
3.1.2. Top-Ranked Intersecting Genes
- Role of succinate receptor 1 (SUCNR1). SUCNR1 is involved in both glucose homeostasis and macrophage activation. SUCNR1 is an extracellular receptor activated by succinate, and SUCNR1 accumulation in macrophages is known to activate the pro-inflammatory response [43]. Moreover, the role of SUCNR1 has been suggested in the development of fibrosis in diabetes mellitus and other diabetes-related complications such as diabetic retinopathy and metabolic syndrome [61]. SUCNR1 can serve as a potential link in understanding glucose-mediated macrophage cell polarization.
- Role of hexokinase 1 (HK1). The HK1 gene encodes a ubiquitous form of hexokinase, which localizes to the outside membrane of mitochondria. Mutations in HK1 have been associated with hemolytic anemia due to hexokinase deficiency. However, its role in DKD is more likely linked to dysregulated glucose metabolism. HK1 is also associated with the cytokine response, the inflammatory response, and growth factor signaling [62].
- Role of ephrin. Ephrin receptors make up the largest subgroup of the receptor tyrosine kinase family, which have a key role in vascular regulation. SemNet 2.0 has previously highlighted the role of tyrosine kinase pathways in resistant hypertension [36]. The protein encoded by this gene binds to ephrin-B2 and plays an essential role in vascular development. Ephrin receptor EPHB4 is associated with angiogenesis in DKD, the immune response, and GEC. EPHB4 receptor interactions between endothelial cells and monocytes/macrophages are relevant for vascular development [57,59]. The inhibition of proteins in the ephrin B family prevents endothelial cell sprouting and initiates disorders in endothelial cell assembly [59].
- Role of serpin family B member 1 (SERPINB1). SERPINB1 is among the serpin protein families that are found in GEC. SERPINB1 acts primarily to protect the cells from proteases released into the cytoplasm during stress [63]. The results presented suggest SERPINB1’s involvement in the inflammatory cytokine response. Previous research has suggested serpin proteins to be associated with macrophage motility as well [59].
- Role of integrins. Integrins regulate many biological processes, such as cell growth, migration, and signaling and cytokine activation, thereby contributing to inflammation and angiogenesis [64]. In the present study, integrin ITGB1 was associated with several cellular functions: angiogenesis, apoptosis, cell–cell junction integrity, and cell adhesion (Figure 5). Studies have also suggested combined treatment strategies through the inhibition of both ITGB and ITGA integrins to reduce macrophage filtration into the glomeruli [59,65]. Such compelling evidence suggests that ITGB1 may have the potential to be a clinical marker for the prognosis of glomerular diseases, immune cell infiltration, and glomerular endothelial viability [59].
3.1.3. Top-Ranked Intersecting Proteins
- Role of tyrosine kinsase 2 (TYK2). Previous studies found an association of TYK2 candidate with type 1 diabetes mellitus and a role of TYK2 in regulating apoptotic and pro-inflammatory pathways in pancreatic -cells through modulation of the type I interferon signaling pathway [66,67]. Likewise, tyrosine kinase inhibitor drugs were previously predicted by SemNet 2.0 to be associated with hyperglycemia in patients who were not initially diabetic [29].
- Role of NFKBIA. NFKBIA regulates the activity of NFB, which plays a role in processes such as the accumulation of advanced glycation end products and activation of the renin–angiotensin system pathways, protein kinase C, and oxidative stress in diabetic nephropathy [71].
- Role of synaptosome-associated protein 23 (SNAP23). Our analyses also identified SNAP23 associated with adherens junction assembly in correlation to GEC, the immune response, and DKD [72]. SPI1 and SNAP23 were genes highly associated with the immune response, cell differentiation, cell migration, the response to cytokines, and apoptotic processes (Figure 6 and Figure A1). Some studies have previously identified SPI1 gene involvement in regulatory mechanisms in DKD, but this may need more experimental verification [73]. The SNAP23 gene is relatively abundant in the kidney and primarily involved in exocytosis [74]. SNAP23 has been shown to reduce proteinuria, reduce podocyte foot process fusion, and reduce endothelial cell damage upon the inhibition of SNAP23-mediated exocytosis [75].
3.2. Visualization of Literature-Based Discovery Network Predictions
3.3. Comparing LBD Networks to Traditional Bioinformatics Networks
3.4. Limitations and Future Directions
4. Materials and Methods

4.1. SemNet 2.0
4.2. SemNet 2.0 Simulations for DKD
4.3. Analysis of Source Nodes Revealed by SemNet 2.0
4.3.1. Functional Ontology Mapping
4.3.2. Cytoscape
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Topic | Data Type | Model Type | Main Contribution | References |
|---|---|---|---|---|
| DKD kidney and immune crosstalk | In vitro | Logic-based ODE model | Identified critical interactions and chemical species associated with the pathophysiology of glucose and immune-mediated glomerular endothelial dysfunction using a network-based model. | [9] |
| Glomerular fibrosis in DKD | Mice, in vitro | Kinetic ODE model | Clarified mechanisms behind diabetic glomerular fibrosis and the reasons behind the failure of certain drugs to guide the creation of better diabetes-related kidney damage therapies. | [15] |
| Hyperglycemia-induced podocyte injury in DKD | Human | Kinetic ODE model | Integrated mathematical modeling, optimization, and sensitivity analyses to simulate the effects of glucose on the local RAS in podocytes in diabetic nephropathy to achieve physiologically plausible model outputs. | [18] |
| Podocytes in kidney diseases | In vitro | Multi-compartment ODE model | Analyzed feedforward motifs, spatial dynamics, and the roles of the PKA and MAPK pathways and provided insights into factors that could drive proliferation or differentiation in cultured podocytes. | [19] |
| Tubulointerstitial inflammation and fibrosis in lupus nephritis | Human | PDE model | Modeled disease progression and effects of dosing levels for novel therapeutics by simulating various levels of inhibition necessary to attenuate the inflammation to the fibrosis pathway. | [20] |
| Kidney and cardiac disorders | Human | Kinetic ODE model | Used QSP approach to understand the complex interactions between cardiac and renal functions in HF-rEF and the effects of pharmacological interventions such as ACEi and SGLT2i therapies. | [22] |
| Kidney and bone metabolism | Human | QSP model | Network topology enabled more effective targeting of key components and better prediction of perturbation effects. | [24] |
| CKD and bone | Mixed | QSP model | Conjugated individual existing cardiac and renal models to study pathophysiological interplay and disease relatedness. This model can be used in personalizing therapy and research settings and generating new hypotheses. | [25] |
| Multiple myeloma and kidney disorders | Human | Kinetic ODE model | Captured the qualitative behavior of the cell and protein populations and their interaction with the proximal tubule of the kidney, free light chains, renal fibroblasts, and myeloma cells. This model may support better patient prognosis in patients with multiple myeloma and renal impairment. | [23] |




References
- Hoogeveen, E.K. The Epidemiology of Diabetic Kidney Disease. Kidney Dial. 2022, 2, 433–442. [Google Scholar] [CrossRef]
- Yang, J.; Liu, Z. Mechanistic Pathogenesis of Endothelial Dysfunction in Diabetic Nephropathy and Retinopathy. Front. Endocrinol. 2022, 13, 816400. [Google Scholar] [CrossRef] [PubMed]
- Ghaderian, S.B.; Hayati, F.; Shayanpour, S.; Beladi Mousavi, S.S. Diabetes and End-Stage Renal Disease; A Review Article on New Concepts. J. Renal. Inj. Prev. 2015, 4, 28–33. [Google Scholar] [CrossRef]
- Gembillo, G.; Ingrasciotta, Y.; Crisafulli, S.; Luxi, N.; Siligato, R.; Santoro, D.; Trifirò, G. Kidney Disease in Diabetic Patients: From Pathophysiology to Pharmacological Aspects with a Focus on Therapeutic Inertia. Int. J. Mol. Sci. 2021, 22, 4824. [Google Scholar] [CrossRef]
- Wan, K.S.; Hairi, N.N.; Mustapha, F.; Mohd Yusoff, M.F.; Mat Rifin, H.; Ismail, M.; Moy, F.M.; Ahmad, N.A. Prevalence of Diabetic Kidney Disease and the Associated Factors among Patients with Type 2 Diabetes in a Multi-Ethnic Asian Country. Sci. Rep. 2024, 14, 7074. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Agarwal, R.; Alpers, C.E.; Bakris, G.L.; Brosius, F.C.; Kolkhof, P.; Uribarri, J. Molecular Mechanisms and Therapeutic Targets for Diabetic Kidney Disease. Kidney Int. 2022, 102, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Mazzieri, A.; Porcellati, F.; Timio, F.; Reboldi, G. Molecular Targets of Novel Therapeutics for Diabetic Kidney Disease: A New Era of Nephroprotection. Int. J. Mol. Sci. 2024, 25, 3969. [Google Scholar] [CrossRef]
- Patidar, K.; Ford Versypt, A.N. Logic-Based Modeling of Inflammatory Macrophage Crosstalk with Glomerular Endothelial Cells in Diabetic Kidney Disease. bioRxiv 2023. [Google Scholar] [CrossRef]
- Navarro-González, J.F.; Mora-Fernández, C.; Muros de Fuentes, M.; García-Pérez, J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat. Rev. Nephrol. 2011, 7, 327–340. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, C. Recent Advances in the Management of Diabetic Kidney Disease: Slowing Progression. Int. J. Mol. Sci. 2024, 25, 3086. [Google Scholar] [CrossRef] [PubMed]
- Ayala, T.S.; Tessaro, F.H.G.; Jannuzzi, G.P.; Bella, L.M.; Ferreira, K.S.; Martins, J.O. High glucose environments interfere with bone marrow-derived macrophage inflammatory mediator release, the TLR4 pathway and glucose metabolism. Sci. Rep. 2019, 9, 11447. [Google Scholar] [CrossRef] [PubMed]
- Tessaro, F.H.G.; Ayala, T.S.; Bella, L.M.; Martins, J.O. Macrophages from a type 1 diabetes mouse model present dysregulated PI3K/AKT, ERK 1/2 and SAPK/JNK levels. Immunobiology 2019, 225, 151879. [Google Scholar]
- Thomas, H.Y.; Ford Versypt, A.N. Pathophysiology of Mesangial Expansion in Diabetic Nephropathy: Mesangial Structure, Glomerular Biomechanics, and Biochemical Signaling and Regulation. J. Biol. Eng. 2022, 16, 19. [Google Scholar] [CrossRef] [PubMed]
- Thomas, H.Y.; Ford Versypt, A.N. A Mathematical Model of Glomerular Fibrosis in Diabetic Kidney Disease to Predict Therapeutic Efficacy. bioRxiv 2023. [Google Scholar] [CrossRef]
- Lassén, E.; Daehn, I.S. Molecular Mechanisms in Early Diabetic Kidney Disease: Glomerular Endothelial Cell Dysfunction. Int. J. Mol. Sci. 2020, 21, 9456. [Google Scholar] [CrossRef]
- Hu, S.; Hang, X.; Wei, Y.; Wang, H.; Zhang, L.; Zhao, L. Crosstalk Among Podocytes, Glomerular Endothelial Cells and Mesangial Cells in Diabetic Kidney Disease: An Updated Review. Cell Commun. Signal. 2024, 22, 136. [Google Scholar] [CrossRef] [PubMed]
- Pilvankar, M.R.; Higgins, M.A.; Ford Versypt, A.N. Mathematical Model for Glucose Dependence of the Local Renin–Angiotensin System in Podocytes. Bull. Math. Biol. 2018, 80, 880–905. [Google Scholar] [CrossRef]
- Azeloglu, E.U.; Hardy, S.V.; Eungdamrong, N.J.; Chen, Y.; Jayaraman, G.; Chuang, P.Y.; Fang, W.; Xiong, H.; Neves, S.R.; Jain, M.R.; et al. Interconnected Network Motifs Control Podocyte Morphology and Kidney Function. Sci. Signal. 2014, 7, ra12. [Google Scholar] [CrossRef]
- Hao, W.; Rovin, B.H.; Friedman, A. Mathematical Model of Renal Interstitial Fibrosis. Proc. Natl. Acad. Sci. USA 2014, 111, 14193–14198. [Google Scholar] [CrossRef]
- Ma, T.K.; Kam, K.K.; Yan, B.P.; Lam, Y.Y. Renin-Angiotensin-Aldosterone System Blockade for Cardiovascular Diseases: Current Status. Br. J. Pharmacol. 2010, 160, 1273–1292. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Basu, S.; Hallow, K.M. Cardiac and Renal Function Interactions in Heart Failure with Reduced Ejection Fraction: A Mathematical Modeling Analysis. PLoS Comput. Biol. 2020, 16, e1008074. [Google Scholar] [CrossRef] [PubMed]
- Walk, J.C.; Ayati, B.P.; Holstein, S.A. Modeling the Effects of Multiple Myeloma on Kidney Function. Sci. Rep. 2019, 9, 1726. [Google Scholar] [CrossRef] [PubMed]
- Peterson, M.C.; Riggs, M.M. A Physiologically Based Mathematical Model of Integrated Calcium Homeostasis and Bone Remodeling. Bone 2010, 46, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Gaweda, A.E.; McBride, D.E.; Lederer, E.D.; Brier, M.E. Development of a Quantitative Systems Pharmacology Model of Chronic Kidney Disease: Metabolic Bone Disorder. Am. J. Physiol. Ren. Physiol. 2020, 320, F203–F211. [Google Scholar] [CrossRef]
- Zhou, A.X.; Jeansson, M.; He, L.; Wigge, L.; Tonelius, P.; Tati, R.; Cederblad, L.; Muhl, L.; Uhrbom, M.; Liu, J.; et al. Renal Endothelial Single-Cell Transcriptomics Reveals Spatiotemporal Regulation and Divergent Roles of Differential Gene Transcription and Alternative Splicing in Murine Diabetic Nephropathy. Int. J. Mol. Sci. 2024, 25, 4320. [Google Scholar] [CrossRef]
- Shao, X.; Shi, Y.; Wang, Y.; Zhang, L.; Bai, P.; Wang, J.; Aniwan, A.; Lin, Y.; Zhou, S.; Yu, P. Single-Cell Sequencing Reveals the Expression of Immune-Related Genes in Macrophages of Diabetic Kidney Disease. Inflammation 2024, 47, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.; Zhu, E.; Li, N.; Gong, L.; Xu, H.; Zhong, Y.; Gong, K.; Jiang, S.; Wang, X.; Fei, L.; et al. Identification of Diagnostic Markers Related to Oxidative Stress and Inflammatory Response in Diabetic Kidney Disease by Machine Learning Algorithms: Evidence from Human Transcriptomic Data and Mouse Experiments. Front. Endocrinol. 2023, 14, 1134325. [Google Scholar] [CrossRef]
- Mehra, N.; Varmeziar, A.; Chen, X.; Kronick, O.; Fisher, R.; Kota, V.; Mitchell, C.S. Cross-Domain Text Mining to Predict Adverse Events from Tyrosine Kinase Inhibitors for Chronic Myeloid Leukemia. Cancers 2022, 14, 4686. [Google Scholar] [CrossRef]
- Swanson, D.R. Fish Oil, Raynaud’s Syndrome, and Undiscovered Public Knowledge. Perspect. Biol. Med. 1986, 30, 7–18. [Google Scholar] [CrossRef]
- Kirkpatrick, A.; Onyeze, C.; Kartchner, D.; Allegri, S.; Nakajima An, D.; McCoy, K.; Davalbhakta, E.; Mitchell, C.S. Optimizations for Computing Relatedness in Biomedical Heterogeneous Information Networks: SemNet 2.0. Big Data Cogn. Comput. 2022, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, A.; Onyeze, C.; Kartchner, D.; Allegri, S.; Nakajima An, D.; McCoy, K.; Davalbhakta, E.; Mitchell, C.S. SemNet 2.0. Available online: https://github.com/pathology-dynamics/semnet-2 (accessed on 9 January 2024).
- Kilicoglu, H.; Shin, D.; Fiszman, M.; Rosemblat, G.; Rindflesch, T.C. SemMedDB: A PubMed-Scale Repository of Biomedical Semantic Predications. Bioinformatics 2012, 28, 3158–3160. [Google Scholar] [CrossRef]
- McCoy, K.; Gudapati, S.; He, L.; Horlander, E.; Kartchner, D.; Kulkarni, S.; Mehra, N.; Prakash, J.; Thenot, H.; Vanga, S.V.; et al. Biomedical Text Link Prediction for Drug Discovery: A Case Study with COVID-19. Pharmaceutics 2021, 13, 794. [Google Scholar] [CrossRef] [PubMed]
- Tandra, G.; Yoone, A.; Mathew, R.; Wang, M.; Hales, C.M.; Mitchell, C.S. Literature-based Discovery Predicts Antihistamines are a Promising Repurposed Adjuvant Therapy for Parkinson’s disease. Int. J. Mol. Sci. 2023, 24, 12339. [Google Scholar] [CrossRef] [PubMed]
- Kartchner, D.; McCoy, K.; Dubey, J.; Zhang, D.; Zheng, K.; Umrani, R.; Kim, J.J.; Mitchell, C.S. Literature-Based Discovery to Elucidate the Biological Links between Resistant Hypertension and COVID-19. Biology 2023, 12, 1269. [Google Scholar] [CrossRef]
- Al-Hussaini, I.; White, B.; Varmeziar, A.; Mehra, N.; Sanchez, M.; Lee, J.; DeGroote, N.P.; Miller, T.P.; Mitchell, C.S. An Interpretable Machine Learning Framework for Rare Disease: A Case Study to Stratify Infection Risk in Pediatric Leukemia. J. Clin. Med. 2024, 13, 1788. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Cytoscape Contributors. Cytoscape User Manual. Available online: https://manual.cytoscape.org (accessed on 9 January 2024).
- Baldarelli, R.; Smith, C.; Finger, J.; Hayamizu, T.; McCright, I.; Xu, J.; Shaw, D.; Beal, J.; Blodgett, O.; Campbell, J.; et al. The Mouse Gene Expression Database (GXD): 2021 Update. Nucleic Acids Res. 2020, 49, D924–D931. [Google Scholar] [CrossRef]
- Blake, J.; Baldarelli, R.; Kadin, J.; Richardson, J.; Smith, C.; Bult, C.; Anagnostopoulos, A.; Beal, J.; Bello, S.; Blodgett, O.; et al. Mouse Genome Database (MGD): Knowledgebase for Mouse–Human Comparative Biology. Nucleic Acids Res. 2020, 49, D981–D987. [Google Scholar] [CrossRef]
- Wu, Q.; Finley, S. Mathematical Model Predicts Effective Strategies to Inhibit VEGF-eNOS Signaling. J. Clin. Med. 2020, 9, 1255. [Google Scholar] [CrossRef]
- Keiran, N.; Ceperuelo-Mallafré, V.; Calvo, E.; Hernández-Alvarez, M.I.; Ejarque, M.; Núñez-Roa, C.; Horrillo, D.; Maymó-Masip, E.; Rodríguez, M.M.; Fradera, R.; et al. SUCNR1 Controls an Anti-Inflammatory Program in Macrophages to Regulate the Metabolic Response to Obesity. Nat. Immunol. 2019, 20, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Stan, R.V.; Tse, D.; Deharvengt, S.J.; Smits, N.C.; Xu, Y.; Luciano, M.R.; McGarry, C.l.; Buitendijk, M.; Nemani, K.V.; Elgueta, R.; et al. The Diaphragms of Fenestrated Endothelia: Gatekeepers of Vascular Permeability and Blood Composition. Dev. Cell 2012, 23, 1203–1218. [Google Scholar] [CrossRef]
- Finch, N.C.; Neal, C.R.; Welsh, G.I.; Foster, R.R.; Satchell, S.C. The Unique Structural and Functional Characteristics of Glomerular Endothelial Cell Fenestrations and Their Potential as a Therapeutic Target in Kidney Disease. Am. J. Physiol. Ren. Physiol. 2023, 325, F465–F478. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Wandel, E.; Wobus, M.; Schneider, R.; Amasheh, S.; Sittig, D.; Kerner, C.; Naumann, R.; Hamann, J.; Aust, G. Overexpression of CD97 in Intestinal Epithelial Cells of Transgenic Mice Attenuates Colitis by Strengthening Adherens Junctions. PLoS ONE 2010, 5, e8507. [Google Scholar] [CrossRef]
- Satchell, S.; Tooke, J. What is the Mechanism of Microalbuminuria in Diabetes: A Role for the Glomerular Endothelium? Diabetologia 2008, 51, 714–725. [Google Scholar] [CrossRef]
- Cai, F.; Zhou, X.; Jia, Y.; Yao, W.; Lv, J.; Liu, G.; Yang, L. Identification of Key Genes of Human Advanced Diabetic Nephropathy Independent of Proteinuria by Transcriptome Analysis. Biomed. Res. Int. 2020, 2020, 7283581. [Google Scholar] [CrossRef]
- Lim, A.K.H.; Tesch, G.H. Inflammation in Diabetic Nephropathy. Mediat. Inflamm. 2012, 2012, 146154. [Google Scholar] [CrossRef] [PubMed]
- Klessens, C.Q.F.; Zandbergen, M.; Wolterbeek, R.; Bruijn, J.A.; Rabelink, T.J.; Bajema, I.M.; IJpelaar, D.H.T. Macrophages in Diabetic Nephropathy in Patients with Type 2 Diabetes. Nephrol. Dial. Transplant. 2017, 32, 1322–1329. [Google Scholar] [CrossRef]
- Liu, Y.; Lv, Y.; Zhang, T.; Huang, T.; Lang, Y.; Sheng, Q.; Liu, Y.; Kong, Z.; Gao, Y.; Lu, S.; et al. T cells and their Products in Diabetic Kidney Disease. Front. Immunol. 2023, 14, 1084448. [Google Scholar] [CrossRef]
- Roep, B.O. The Role of T-cells in the Pathogenesis of Type 1 Diabetes: From Cause to Cure. Diabetologia 2003, 46, 305–321. [Google Scholar] [CrossRef]
- Chan, L.; Schrier, R.W. Effects of Calcium Channel Blockers on Renal Function. Annu. Rev. Med. 1990, 41, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Wang, Z.; Frimpong, F.; Chen, X. Calcium-Permeable Channels and Endothelial Dysfunction in Acute Lung Injury. Curr. Issues Mol. Biol. 2022, 44, 2217–2229. [Google Scholar] [CrossRef]
- Dalal, P.J.; Muller, W.A.; Sullivan, D.P. Endothelial Cell Calcium Signaling During Barrier Function and Inflammation. Am. J. Pathol. 2020, 190, 535–542. [Google Scholar] [CrossRef]
- van Nieuwenhoven, F.A.; Jensen, L.J.N.; Flyvbjerg, A.; Goldschmeding, R. Imbalance of Growth Factor Signalling in Diabetic Kidney Disease: Is Connective Tissue Growth Factor (CTGF, CCN2) the Perfect Intervention Point? Nephrol. Dial. Transplant. 2005, 20, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liu, Y.; Guo, F.; Zhao, L.; Qin, G. Single-Cell RNA Sequencing Reveals Rac1 Involvement in Macrophages Efferocytosis in Diabetic Kidney Disease. Inflammation 2023. [Google Scholar] [CrossRef]
- Majumder, S.; Advani, A. VEGF and the Diabetic Kidney: More than too much of a good thing. J. Diabetes Complicat. 2017, 31, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Shen, K.; Li, J.; Leung, S.W.S.; Zhu, T.; Shi, Y. Glomerular Endothelial Cells are the Coordinator in the Development of Diabetic Nephropathy. Front. Med. 2021, 8, 655639. [Google Scholar] [CrossRef]
- Mohamed-Ali, V.; Pinkney, J. Therapeutic Potential of Insulin-Like Growth Factor-1 in Patients with Diabetes Mellitus. Treat. Endocrinol. 2002, 1, 399–410. [Google Scholar] [CrossRef]
- Ariza, A.C.; Deen, P.M.; Robben, J.H. The Succinate Receptor as a Novel Therapeutic Target for Oxidative and Metabolic Stress-Related Conditions. Front. Endocrinol. 2012, 3, 22. [Google Scholar] [CrossRef] [PubMed]
- Codocedo, J.F.; Landreth, G.E. The Intersection of Metabolism and Inflammation Is Governed by the Intracellular Topology of Hexokinases and the Metabolic Fate of Glucose. Immunometabolism 2022, 4, e00011. [Google Scholar] [CrossRef]
- Torriglia, A.; Martin, E.; Jaadane, I. The Hidden Side of SERPINB1/Leukocyte Elastase Inhibitor. Semin. Cell Dev. Biol. 2017, 62, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Mezu-Ndubuisi, O.J.; Maheshwari, A. The Role of Integrins in Inflammation and Angiogenesis. Pediatr. Res. 2021, 89, 1619–1626. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Tiwari, A.K.; Issekutz, T.B.; Lefkowith, J.B. Differing Roles of CD18 and VLA-4 in Leukocyte Migration/Activation during Anti-GBM Nephritis. Kidney Int. 1996, 50, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Pellenz, F.; Dieter, C.; Duarte, G.; Canani, L.; Souza, B.; Crispim, D. The rs2304256 Polymorphism in TYK2 Gene Is Associated with Protection for Type 1 Diabetes Mellitus. Diabetes Metab. J. 2021, 45, 899. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Takahashi, H.; Mine, K.; Higashimoto, K.; Inoue, K.; Kojima, M.; Kuroki, S.; Eguchi, T.; Ono, Y.; Inuzuka, S.; et al. TYK2 Promoter Variant Is Associated with Impaired Insulin Secretion and Lower Insulin Resistance in Japanese Type 2 Diabetes Patients. Genes 2021, 12, 400. [Google Scholar] [CrossRef]
- Yan, C.; Fang, P.; Zhang, H.; Tao, J.; Tian, X.; Li, Y.; Zhang, J.; Sun, M.; Li, S.; Wang, H.; et al. CREG1 Promotes Angiogenesis and Neovascularization. Front. Biosci. 2014, 19, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Goto, A.; Endo, Y.; Yamashita, H. CREG1 Stimulates AMPK Phosphorylation and Glucose Uptake in Skeletal Muscle Cells. Biochem. Biophys. Res. Commun. 2022, 641, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, G.; Araujo, L.; Jinwala, F.; Li, S.; Lee, L. The Structure and Biological Function of CREG. Front. Cell Dev. Biol. 2018, 6, 136. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhang, C. The Role of Innate Immunity in Diabetic Nephropathy and Their Therapeutic Consequences. J. Pharm. Anal. 2024, 14, 39–51. [Google Scholar] [CrossRef]
- Guo, Z.; Neilson, L.J.; Zhong, H.; Murray, P.S.; Zanivan, S.; Zaidel-Bar, R. E-Cadherin Interactome Complexity and Robustness Resolved by Quantitative Proteomics. Sci. Signal. 2014, 7, rs7. [Google Scholar] [CrossRef]
- Li, B.; Zhao, X.; Xie, W.; Hong, Z.; Zhang, Y. Integrative Analyses of Biomarkers and Pathways for Diabetic Nephropathy. Front. Genet. 2023, 14, 1128136. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Nielsen, S.; Mandon, B.; Terris, J.; Kishore, B.K.; Knepper, M.A. SNAP-23 in Rat Kidney: Colocalization with Aquaporin-2 in Collecting Duct Vesicles. Am. J. Physiol. Ren. Physiol. 1998, 275, F752–F760. [Google Scholar] [CrossRef] [PubMed]
- Caster, D.J.; Korte, E.A.; Tan, M.; Barati, M.T.; Tandon, S.; Creed, T.M.; Salant, D.J.; Hata, J.L.; Epstein, P.N.; Huang, H.; et al. Neutrophil Exocytosis Induces Podocyte Cytoskeletal Reorganization and Proteinuria in Experimental Glomerulonephritis. Am. J. Physiol. Ren. Physiol. 2018, 315, F595–F606. [Google Scholar] [CrossRef] [PubMed]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. In Proceedings of the International AAAI Conference on Web and Social Media, Oxford, UK, 19 March 2009; Volume 3, pp. 361–362. [Google Scholar] [CrossRef]
- Batt, S.; Grealis, T.; Harmon, O.; Tomolonis, P. Learning Tableau: A Data Visualization Tool. J. Econ. Ed. 2020, 51, 317–328. [Google Scholar] [CrossRef]
- Smith, M.; Ceni, A.; Milic-Frayling, N.; Shneiderman, B.; Mendes Rodrigues, E.; Leskovec, J.; Dunne, C. NodeXL: A Free and Open Network Overview, Discovery and Exploration Add-in for Excel 2007/2010/2013/2016. Available online: https://www.smrfoundation.org (accessed on 9 January 2024).
- Neo4j, Inc. Neo4j-The World’s Leading Graph Database. Available online: http://neo4j.org/ (accessed on 9 January 2024).
- Allegri, S.A.; McCoy, K.; Mitchell, C.S. CompositeView: A Network-Based Visualization Tool. Big Data Cogn. Comput. 2022, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Hojjati, F.; Roointan, A.; Gholaminejad, A.; Eshraghi, Y.; Gheisari, Y. Identification of Key Genes and Biological Regulatory Mechanisms in Diabetic Nephropathy: Meta-Analysis of Gene Expression Datasets. Nefrologia 2023, 43, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Patidar, K.; Deng, J.H.; Mitchell, C.S.; Ford Versypt, A.N. DKD_CaseStudy_SemNet2. Available online: https://github.com/ashleefv/DKD_CaseStudy_SemNet2 (accessed on 13 March 2024). [CrossRef]
- Sedler, A.; Mitchell, C.S. SemNet: Using Local Features to Navigate the Biomedical Concept Graph. Front. Bioeng. Biotechnol. 2019, 7, 156. [Google Scholar] [CrossRef]
- Ali, M.; Berrendorf, M.; Hoyt, C.T.; Vermue, L.; Sharifzadeh, S.; Tresp, V.; Lehmann, J. PyKEEN 1.0: A Python Library for Training and Evaluating Knowledge Graph Embeddings. J. Mach. Learn. Res 2021, 22, 1–6. [Google Scholar]
- Stewart, A.P.; Clatworthy, M.R. Single-Cell Genomics Sheds Light on Kidney Tissue Immunity. Nat. Rev. Nephrol. 2024, 20, 71–72. [Google Scholar] [CrossRef]
- Lake, B.B.; Menon, R.; Winfree, S.; Hu, Q.; Melo Ferreira, R.; Kalhor, K.; Barwinska, D.; Otto, E.A.; Ferkowicz, M.; Diep, D.; et al. An Atlas of Healthy and Injured Cell States and Niches in the Human Kidney. Nature 2023, 619, 585–594. [Google Scholar] [CrossRef]
- Hansen, J.; Sealfon, R.; Menon, R.; Eadon, M.T.; Lake, B.B.; Steck, B.; Anjani, K.; Parikh, S.; Sigdel, T.K.; Zhang, G.; et al. A Reference Tissue Atlas for the Human Kidney. Sci. Adv. 2022, 8, eabn4965. [Google Scholar] [CrossRef]
- He, Y.O.; Barisoni, L.; Rosenberg, A.Z.; Robinson, P.; Diehl, A.D.; Chen, Y.; Phuong, J.P.; Hansen, J.; Herr II, B.W.; Börner, K.; et al. Ontology-Based Modeling, Integration, and Analysis of Heterogeneous Clinical, Pathological, and Molecular Kidney Data for Precision Medicine. bioRxiv 2024. [Google Scholar] [CrossRef]
- Bhatia, D.; Srivastava, S.P. Editorial: Diabetic Kidney Disease: Routes to Drug Development, Pharmacology and Underlying Molecular Mechanisms. Front. Pharmacol. 2023, 14, 1252315. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K. Treatment of Diabetic Kidney Disease: Current and Future Targets. Korean J. Intern. Med. 2017, 32, 622–630. [Google Scholar] [CrossRef]
- Sebastian, Y.; Smalheiser, N.R. Editorial: Emerging Areas in Literature-Based Discovery. Front. Res. Metr. Anal. 2023, 8, 1122547. [Google Scholar] [CrossRef]
- National Library of Medicine (US). UMLS Reference Manual. Available online: https://www.ncbi.nlm.nih.gov/books/NBK9676/ (accessed on 9 January 2024).
- Shi, C.; Kong, X.; Huang, Y.; Yu, P.S.; Wu, B. HeteSim: A General Framework for Relevance Measure in Heterogeneous Networks. IEEE Trans. Knowl. Data Eng. 2014, 26, 2479–2492. [Google Scholar] [CrossRef]
- Pires, C. A Systematic Review on the Contribution of Artificial Intelligence in the Development of Medicines for COVID-2019. J. Pers. Med. 2021, 11, 926. [Google Scholar] [CrossRef]
- Ernst, P.; Siu, A.; Weikum, G. KnowLife: A Versatile Approach for Constructing a Large Knowledge Graph for Biomedical Sciences. BMC Bioinform. 2015, 16, 157. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Kim, S.; Song, M.; Jeong, M.; Kim, D.; Kang, J.; Rousseau, J.F.; Li, X.; Xu, W.; Torvik, V.I.; et al. Building a PubMed Knowledge Graph. Sci. Data 2020, 7, 205. [Google Scholar] [CrossRef]
- Chandak, P.; Huang, K.; Zitnik, M. Building a Knowledge Graph to Enable Precision Medicine. Sci. Data 2023, 10, 67. [Google Scholar] [CrossRef]
- Bang, D.; Lim, S.; Lee, S.; Kim, S. Biomedical Knowledge Graph Learning for Drug Repurposing by Extending Guilt-by-Association to Multiple Layers. Nat. Commun. 2023, 14, 3570. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhou, W.; Ren, J.; Dong, L.; Wang, Y.; Jin, S.; Cheng, L. Annotating the Function of the Human Genome with Gene Ontology and Disease Ontology. Biomed. Res. Int. 2016, 2016, 4130861. [Google Scholar] [CrossRef] [PubMed]
- Asif, M.; Martiniano, H.F.M.C.M.; Vicente, A.M.; Couto, F.M. Identifying Disease Genes Using Machine Learning and Gene Functional Similarities, Assessed through Gene Ontology. PLoS ONE 2018, 13, e0208626. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium; Ashburner, M.; Ball, C.; Blake, J.; Botstein, D.; Butler, H.; Cherry, J.; Davis, A.P.; Dolinski, K.; Dwight, S.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Gene Ontology Consortium. The Gene Ontology Resource: Enriching a GOld Mine. Nucleic Acids Res. 2020, 49, D325–D334. [Google Scholar] [CrossRef]
- Cox, B.J. Systems Biology Analyses of the Placenta. In The Guide to Investigation of Mouse Pregnancy; Academic Press: Cambridge, MA, USA, 2014; pp. 259–274. [Google Scholar]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patidar, K.; Deng, J.H.; Mitchell, C.S.; Ford Versypt, A.N. Cross-Domain Text Mining of Pathophysiological Processes Associated with Diabetic Kidney Disease. Int. J. Mol. Sci. 2024, 25, 4503. https://doi.org/10.3390/ijms25084503
Patidar K, Deng JH, Mitchell CS, Ford Versypt AN. Cross-Domain Text Mining of Pathophysiological Processes Associated with Diabetic Kidney Disease. International Journal of Molecular Sciences. 2024; 25(8):4503. https://doi.org/10.3390/ijms25084503
Chicago/Turabian StylePatidar, Krutika, Jennifer H. Deng, Cassie S. Mitchell, and Ashlee N. Ford Versypt. 2024. "Cross-Domain Text Mining of Pathophysiological Processes Associated with Diabetic Kidney Disease" International Journal of Molecular Sciences 25, no. 8: 4503. https://doi.org/10.3390/ijms25084503