
A Proximity Complementation Assay to Identify Small Molecules That Enhance the Traffic of ABCA4 Misfolding Variants
 , , , , , and
, , , , , and
Abstract
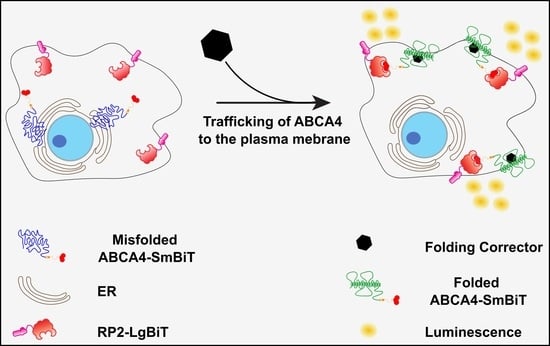
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
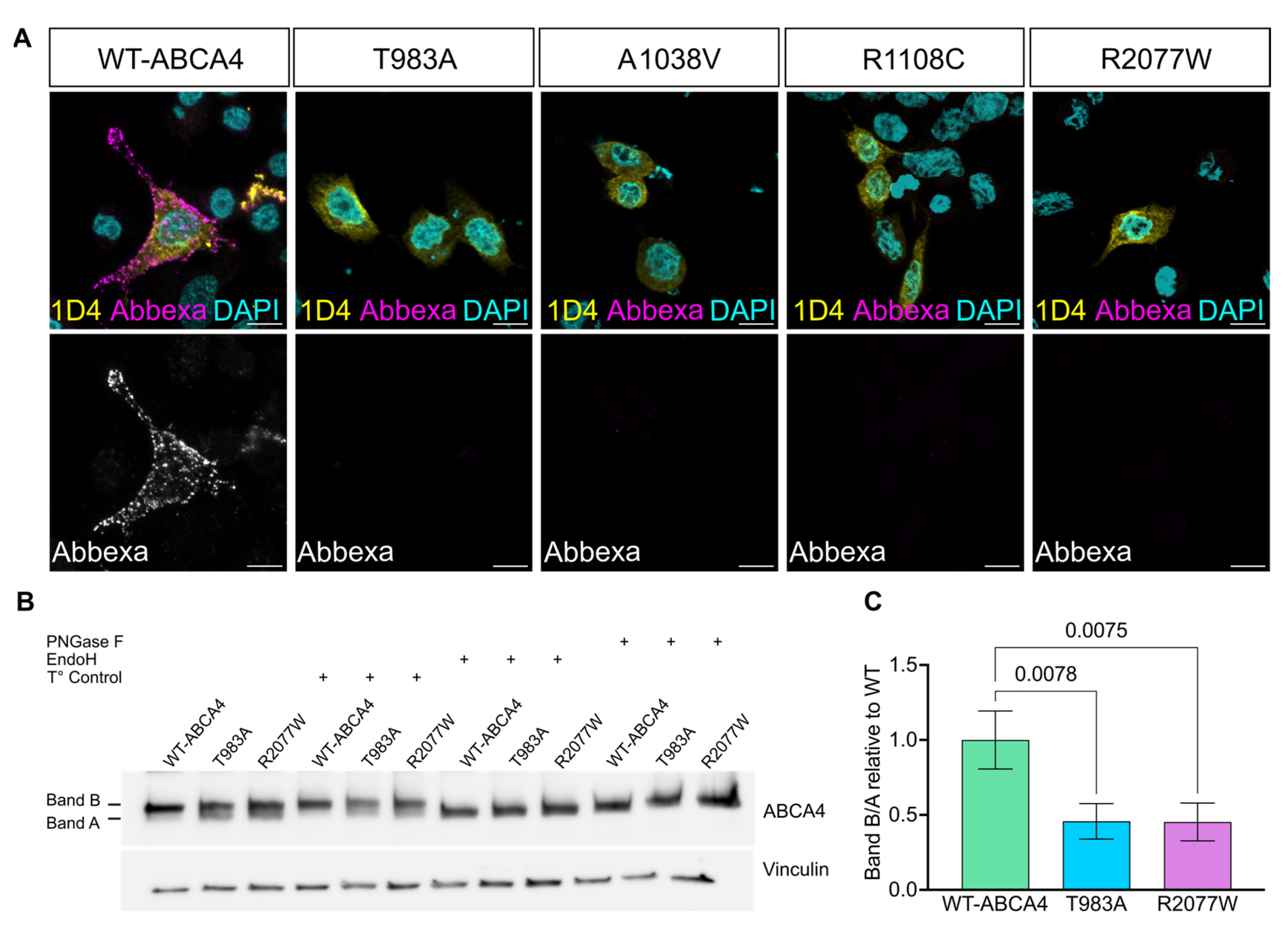
2.1. ABCA4 Missense Variants Are Retained in the ER
2.2. ABCA4 Missense Variants Do Not Traffic to the Plasma Membrane in Cultured Cells
2.3. Reduced Temperature Increases ABCA4 Protein Level and Promotes Traffic to the Cell Surface
2.4. Split NanoBit Proximity Complementation Assay: A New HTS-Friendly Platform to Study Protein Localisation to the Plasma Membrane
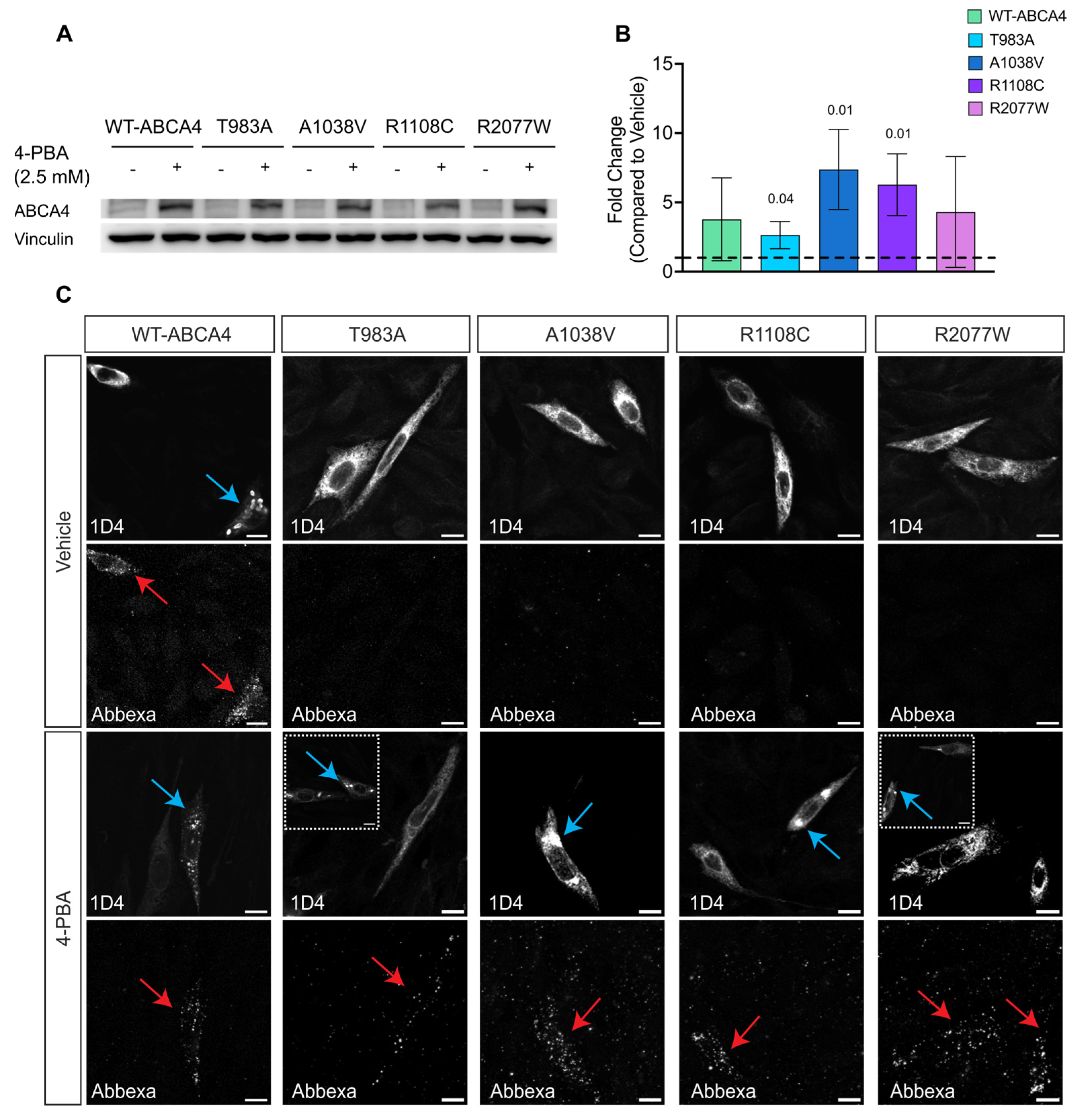
2.5. The Proximity Complementation System Can Detect Pharmacological Rescue
2.6. 4-PBA and AICAR Can Promote ABCA4 Traffic to the Plasma Membrane
3. Discussion
4. Materials and Methods
4.1. Immunofluorescence of Cells
4.2. Immunofluorescence of Live Cells
4.3. Cell Protein Extraction Using RIPA Buffer
4.4. Western Blot
4.5. Endo H and PNGase-F Digestion
4.6. Protein Translation Inhibition
4.7. Luminescence Protocol and Detection
4.8. Plasmid Production
4.9. Cell Culture and Transient Transfection of DNA Plasmids
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanany, M.; Rivolta, C.; Sharon, D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 2710–2716. [Google Scholar] [CrossRef] [PubMed]
- Cremers, F.P.M.; Lee, W.; Collin, R.W.J.; Allikmets, R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog. Retin. Eye Res. 2020, 79, 100861. [Google Scholar] [CrossRef]
- Illing, M.; Molday, L.L.; Molday, R.S. The 220-kDa rim protein of retinal rod outer segments is a member of the ABC transporter superfamily. J. Biol. Chem. 1997, 272, 10303–10310. [Google Scholar] [CrossRef] [PubMed]
- Papermaster, D.S.; Converse, C.A.; Zorn, M. Biosynthetic and immunochemical characterization of large protein in frog and cattle rod outer segment membranes. Exp. Eye Res. 1976, 23, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Papermaster, D.S.; Reilly, P.; Schneider, B.G. Cone lamellae and red and green rod outer segment disks contain a large intrinsic membrane protein on their margins: An ultrastructural immunocytochemical study of frog retinas. Vis. Vision. Res. 1982, 22, 1417–1428. [Google Scholar] [CrossRef] [PubMed]
- Molday, L.L.; Rabin, A.R.; Molday, R.S. ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nat. Genet. 2000, 25, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Allikmets, R.; Singh, N.; Sun, H.; Shroyer, N.F.; Hutchinson, A.; Chidambaram, A.; Gerrard, B.; Baird, L.; Stauffer, D.; Peiffer, A.; et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 1997, 15, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Mata, N.L.; Weng, J.; Travis, G.H. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc. Natl. Acad. Sci. USA 2000, 97, 7154–7159. [Google Scholar] [CrossRef] [PubMed]
- Quazi, F.; Lenevich, S.; Molday, R.S. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat. Commun. 2012, 3, 925. [Google Scholar] [CrossRef]
- Weng, J.; Mata, N.L.; Azarian, S.M.; Tzekov, R.T.; Birch, D.G.; Travis, G.H. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell 1999, 98, 13–23. [Google Scholar] [CrossRef]
- Molday, R.S. Insights into the Molecular Properties of ABCA4 and Its Role in the Visual Cycle and Stargardt Disease. Prog. Mol. Biol. Transl. Sci. 2015, 134, 415–431. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, S.S.; Runhart, E.H.; Bauwens, M.; Corradi, Z.; De Baere, E.; Roosing, S.; Haer-Wigman, L.; Dhaenens, C.M.; Vulto-van Silfhout, A.T.; Cremers, F.P.M. Personalized genetic counseling for Stargardt disease: Offspring risk estimates based on variant severity. Am. J. Hum. Genet. 2022, 109, 498–507. [Google Scholar] [CrossRef]
- Garces, F.; Jiang, K.; Molday, L.L.; Stohr, H.; Weber, B.H.; Lyons, C.J.; Maberley, D.; Molday, R.S. Correlating the Expression and Functional Activity of ABCA4 Disease Variants with the Phenotype of Patients with Stargardt Disease. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2305–2315. [Google Scholar] [CrossRef]
- Garces, F.A.; Scortecci, J.F.; Molday, R.S. Functional Characterization of ABCA4 Missense Variants Linked to Stargardt Macular Degeneration. Int. J. Mol. Sci. 2021, 22, 185. [Google Scholar] [CrossRef]
- Sun, H.; Smallwood, P.M.; Nathans, J. Biochemical defects in ABCR protein variants associated with human retinopathies. Nat. Genet. 2000, 26, 242–246. [Google Scholar] [CrossRef]
- Zhong, M.; Molday, L.L.; Molday, R.S. Role of the C terminus of the photoreceptor ABCA4 transporter in protein folding, function, and retinal degenerative diseases. J. Biol. Chem. 2009, 284, 3640–3649. [Google Scholar] [CrossRef]
- Zhang, N.; Tsybovsky, Y.; Kolesnikov, A.V.; Rozanowska, M.; Swider, M.; Schwartz, S.B.; Stone, E.M.; Palczewska, G.; Maeda, A.; Kefalov, V.J.; et al. Protein misfolding and the pathogenesis of ABCA4-associated retinal degenerations. Hum. Human. Mol. Genet. 2015, 24, 3220–3237. [Google Scholar] [CrossRef] [PubMed]
- Molday, R.S.; Garces, F.A.; Scortecci, J.F.; Molday, L.L. Structure and function of ABCA4 and its role in the visual cycle and Stargardt macular degeneration. Prog. Retin. Eye Res. 2022, 89, 101036. [Google Scholar] [CrossRef] [PubMed]
- Papp, E.; Csermely, P. Chemical chaperones: Mechanisms of action and potential use. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2006; pp. 405–416. [Google Scholar] [CrossRef]
- Jeon, J.H.; Im, S.; Kim, H.S.; Lee, D.; Jeong, K.; Ku, J.M.; Nam, T.G. Chemical Chaperones to Inhibit Endoplasmic Reticulum Stress: Implications in Diseases. Drug Des. Devel Ther. 2022, 16, 4385–4397. [Google Scholar] [CrossRef]
- Cortez, L.; Sim, V. The therapeutic potential of chemical chaperones in protein folding diseases. Prion 2014, 8, 197–202. [Google Scholar] [CrossRef]
- Morello, J.P.; Petäjä-Repo, U.E.; Bichet, D.G.; Bouvier, M. Pharmacological chaperones: A new twist on receptor folding. Trends Pharmacol. Sci. 2000, 21, 466–469. [Google Scholar] [CrossRef]
- Grasso, D.; Galderisi, S.; Santucci, A.; Bernini, A. Pharmacological Chaperones and Protein Conformational Diseases: Approaches of Computational Structural Biology. Int. J. Mol. Sci. 2023, 24, 5819. [Google Scholar] [CrossRef]
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Buchner, J. Molecular chaperones and protein quality control: An introduction to the JBC Reviews thematic series. J. Biol. Chem. 2019, 294, 2074–2075. [Google Scholar] [CrossRef]
- Aleksandrov, A.A.; Aleksandrov, L.A.; Riordan, J.R. CFTR (ABCC7) is a hydrolyzable-ligand-gated channel. Pflug. Arch. Eur. J. Physiol. 2007, 453, 693–702. [Google Scholar] [CrossRef]
- Cutting, G.R. Modifier genetics: Cystic fibrosis. Annu. Rev. Genom. Hum. Genet. 2005, 6, 237–260. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Straley, K.S.; Cao, D.; González, J.; Hadida, S.; Hazlewood, A.; Joubran, J.; Knapp, T.; Makings, L.R.; Miller, M.; et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L1117–L1130. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445–Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef] [PubMed]
- Goetz, D.M.; Savant, A.P. Review of CFTR modulators 2020. Pediatr. Pulmonol. 2021, 56, 3595–3606. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.C.; Pack, T.F.; Rochelle, L.K.; Chakraborty, S.K.; Zhang, M.; Eaton, A.W.; Bai, Y.; Ernst, L.A.; Barak, L.S.; Waggoner, A.S.; et al. A rapid and affordable screening platform for membrane protein trafficking. BMC Biol. 2015, 13, 107. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tang, H. High-throughput screening assays to identify small molecules preventing photoreceptor degeneration caused by the rhodopsin P23H mutation. Methods Mol. Biol. 2015, 1271, 369–390. [Google Scholar] [CrossRef] [PubMed]
- Ansbro, M.R.; Shukla, S.; Ambudkar, S.V.; Yuspa, S.H.; Li, L. Screening compounds with a novel high-throughput ABCB1-mediated efflux assay identifies drugs with known therapeutic targets at risk for multidrug resistance interference. PLoS ONE 2013, 8, e60334. [Google Scholar] [CrossRef] [PubMed]
- Ivnitski-Steele, I.; Larson, R.S.; Lovato, D.M.; Khawaja, H.M.; Winter, S.S.; Oprea, T.I.; Sklar, L.A.; Edwards, B.S. High-throughput flow cytometry to detect selective inhibitors of ABCB1, ABCC1, and ABCG2 transporters. Assay. Drug Dev. Technol. 2008, 6, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Sorrenson, B.; Suetani, R.J.; Williams, M.J.; Bickley, V.M.; George, P.M.; Jones, G.T.; McCormick, S.P. Functional rescue of mutant ABCA1 proteins by sodium 4-phenylbutyrate. J. Lipid Res. 2013, 54, 55–62. [Google Scholar] [CrossRef]
- Kinting, S.; Höppner, S.; Schindlbeck, U.; Forstner, M.E.; Harfst, J.; Wittmann, T.; Griese, M. Functional rescue of misfolding ABCA3 mutations by small molecular correctors. Hum. Human. Mol. Genet. 2018, 27, 943–953. [Google Scholar] [CrossRef]
- Liu, Q.; Sabirzhanova, I.; Bergbower, E.A.S.; Yanda, M.; Guggino, W.G.; Cebotaru, L. The CFTR Corrector, VX-809 (Lumacaftor), Rescues ABCA4 Trafficking Mutants: A Potential Treatment for Stargardt Disease. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2019, 53, 400–412. [Google Scholar] [CrossRef]
- Sabirzhanova, I.; Lopes Pacheco, M.; Rapino, D.; Grover, R.; Handa, J.T.; Guggino, W.B.; Cebotaru, L. Rescuing Trafficking Mutants of the ATP-binding Cassette Protein, ABCA4, with Small Molecule Correctors as a Treatment for Stargardt Eye Disease. J. Biol. Chem. 2015, 290, 19743–19755. [Google Scholar] [CrossRef]
- Scortecci, J.F.; Molday, L.L.; Curtis, S.B.; Garces, F.A.; Panwar, P.; Van Petegem, F.; Molday, R.S. Cryo-EM structures of the ABCA4 importer reveal mechanisms underlying substrate binding and Stargardt disease. Nat. Commun. 2021, 12, 5902. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, D.; Arendt, A.; Hargrave, P.; McDowell, J.H.; Molday, R.S. Localization of binding sites for carboxyl terminal specific anti-rhodopsin monoclonal antibodies using synthetic peptides. Biochemistry 1984, 23, 6544–6549. [Google Scholar] [CrossRef] [PubMed]
- Molday, L.L.; Wahl, D.; Sarunic, M.V.; Molday, R.S. Localization and functional characterization of the p.Asn965Ser (N965S) ABCA4 variant in mice reveal pathogenic mechanisms underlying Stargardt macular degeneration. Hum. Human. Mol. Genet. 2018, 27, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Athanasiou, D.; Aguila, M.; Opefi, C.A.; South, K.; Bellingham, J.; Bevilacqua, D.; Munro, P.M.; Kanuga, N.; Mackenzie, F.E.; Dubis, A.M.; et al. Rescue of mutant rhodopsin traffic by metformin-induced AMPK activation accelerates photoreceptor degeneration. Hum. Human. Mol. Genet. 2017, 26, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.M.; Song, H.S.; Kim, J.Y.; Kwon, E.Y.; Ha, S.Y.; Kim, M.; Choi, J.H. Functional characterization of ABCA4 genetic variants related to Stargardt disease. Sci. Rep. 2022, 12, 22282. [Google Scholar] [CrossRef] [PubMed]
- Ng, E.S.Y.; Kady, N.; Hu, J.; Dave, A.; Jiang, Z.; Pei, J.; Gorin, M.B.; Matynia, A.; Radu, R.A. Membrane Attack Complex Mediates Retinal Pigment Epithelium Cell Death in Stargardt Macular Degeneration. Cells 2022, 11, 3462. [Google Scholar] [CrossRef] [PubMed]
- Farnoodian, M.; Bose, D.; Khristov, V.; Susaimanickam, P.J.; Maddileti, S.; Mariappan, I.; Abu-Asab, M.; Campos, M.; Villasmil, R.; Wan, Q.; et al. Cell-autonomous lipid-handling defects in Stargardt iPSC-derived retinal pigment epithelium cells. Stem Cell Rep. 2022, 17, 2438–2450. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Leahy, D. Enzymatic deglycosylation of glycoproteins. Methods Enzym. Enzymol. 2013, 533, 259–263. [Google Scholar] [CrossRef]
- Pearring, J.N.; Martínez-Márquez, J.; Willer, J.R.; Lieu, E.C.; Salinas, R.Y.; Arshavsky, V.Y. The GARP Domain of the Rod CNG Channel’s β1-Subunit Contains Distinct Sites for Outer Segment Targeting and Connecting to the Photoreceptor Disk Rim. J. Neurosci. Off. J. Soc. Neurosci. 2021, 41, 3094–3104. [Google Scholar] [CrossRef]
- Wang, X.; Koulov, A.V.; Kellner, W.A.; Riordan, J.R.; Balch, W.E. Chemical and biological folding contribute to temperature-sensitive DeltaF508 CFTR trafficking. Traffic 2008, 9, 1878–1893. [Google Scholar] [CrossRef]
- Denning, G.M.; Anderson, M.P.; Amara, J.F.; Marshall, J.; Smith, A.E.; Welsh, M.J. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 1992, 358, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Matteson, J.; An, Y.; Moyer, B.; Yoo, J.S.; Bannykh, S.; Wilson, I.A.; Riordan, J.R.; Balch, W.E. COPII-dependent export of cystic fibrosis transmembrane conductance regulator from the ER uses a di-acidic exit code. J. Cell Biol. 2004, 167, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Dixon, A.S.; Schwinn, M.K.; Hall, M.P.; Zimmerman, K.; Otto, P.; Lubben, T.H.; Butler, B.L.; Binkowski, B.F.; Machleidt, T.; Kirkland, T.A.; et al. NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chem. Biol. 2016, 11, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Chapple, J.P.; Hardcastle, A.J.; Grayson, C.; Willison, K.R.; Cheetham, M.E. Delineation of the plasma membrane targeting domain of the X-linked retinitis pigmentosa protein RP2. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2015–2020. [Google Scholar]
- Saliba, R.S.; Munro, P.M.G.; Luthert, P.J.; Cheetham, M.E. The cellular fate of mutant rhodopsin: Quality control, degradation and aggresome formation. J. Cell Sci. 2002, 115, 2907–2918. [Google Scholar] [CrossRef] [PubMed]
- Wiszniewski, W.; Zaremba, C.M.; Yatsenko, A.N.; Jamrich, M.; Wensel, T.G.; Lewis, R.A.; Lupski, J.R. ABCA4 mutations causing mislocalization are found frequently in patients with severe retinal dystrophies. Hum. Human. Mol. Genet. 2005, 14, 2769–2778. [Google Scholar] [CrossRef] [PubMed]
- Noorwez, S.M.; Kuksa, V.; Imanishi, Y.; Zhu, L.; Filipek, S.; Palczewski, K.; Kaushal, S. Pharmacological chaperone-mediated in vivo folding and stabilization of the P23H-opsin mutant associated with autosomal dominant retinitis pigmentosa. J. Biol. Chem. 2003, 278, 14442–14450. [Google Scholar] [CrossRef] [PubMed]
- Mendes, H.F.; Cheetham, M.E. Pharmacological manipulation of gain-of-function and dominant-negative mechanisms in rhodopsin retinitis pigmentosa. Hum. Human. Mol. Genet. 2008, 17, 3043–3054. [Google Scholar] [CrossRef] [PubMed]
- Laselva, O.; Guerra, L.; Castellani, S.; Favia, M.; Di Gioia, S.; Conese, M. Small-molecule drugs for cystic fibrosis: Where are we now? Pulm. Pharmacol. Ther. 2022, 72, 102098. [Google Scholar] [CrossRef]
- Swanton, E.; Bulleid, N.J. Protein folding and translocation across the endoplasmic reticulum membrane. Mol. Membr. Biol. 2003, 20, 99–104. [Google Scholar] [CrossRef]
- Howell, G.J.; Holloway, Z.G.; Cobbold, C.; Monaco, A.P.; Ponnambalam, S. Cell biology of membrane trafficking in human disease. Int. Rev. Cytol. 2006, 252, 1–69. [Google Scholar] [CrossRef] [PubMed]
- Tsybovsky, Y.; Palczewski, K. Expression, purification and structural properties of ABC transporter ABCA4 and its individual domains. Protein Expr. Purif. 2014, 97, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.H.; Gregory, R.J.; Marshall, J.; Paul, S.; Souza, D.W.; White, G.A.; O’Riordan, C.R.; Smith, A.E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990, 63, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Tsybovsky, Y.; Wang, B.; Quazi, F.; Molday, R.S.; Palczewski, K. Posttranslational modifications of the photoreceptor-specific ABC transporter ABCA4. Biochemistry 2011, 50, 6855–6866. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Cao, X.; Yang, F.; Shi, D.-J.; Tang, Y.-Q.; Zheng, J.; Wang, K. A TRPV4 channel C-terminal folding recognition domain critical for trafficking and function. J. Biol. Chem. 2013, 288, 10427–10439. [Google Scholar] [CrossRef] [PubMed]
- Romei, M.G.; Boxer, S.G. Split Green Fluorescent Proteins: Scope, Limitations, and Outlook. Annu. Rev. Biophys. 2019, 48, 19–44. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Charlton, C.A.; Blau, H.M. Monitoring protein-protein interactions in intact eukaryotic cells by beta-galactosidase complementation. Proc. Natl. Acad. Sci. USA 1997, 94, 8405–8410. [Google Scholar] [CrossRef] [PubMed]
- Wehrman, T.S.; Casipit, C.L.; Gewertz, N.M.; Blau, H.M. Enzymatic detection of protein translocation. Nat. Methods 2005, 2, 521–527. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Chen, J. Mechanism of CFTR correction by type I folding correctors. Cell 2022, 185, 158–168.e111. [Google Scholar] [CrossRef]
- Lee, W.; Zernant, J.; Nagasaki, T.; Molday, L.L.; Su, P.Y.; Fishman, G.A.; Tsang, S.H.; Molday, R.S.; Allikmets, R. Cis-acting modifiers in the ABCA4 locus contribute to the penetrance of the major disease-causing variant in Stargardt disease. Hum. Human. Mol. Genet. 2021, 30, 1293–1304. [Google Scholar] [CrossRef]
- Iannitti, T.; Palmieri, B. Clinical and experimental applications of sodium phenylbutyrate. Drugs R. D 2011, 11, 227–249. [Google Scholar] [CrossRef] [PubMed]
- Brusilow, S.W.; Maestri, N.E. Urea cycle disorders: Diagnosis, pathophysiology, and therapy. Adv. Pediatr. 1996, 43, 127–170. [Google Scholar] [CrossRef] [PubMed]
- Zeitlin, P.L.; Diener-West, M.; Rubenstein, R.C.; Boyle, M.P.; Lee, C.K.; Brass-Ernst, L. Evidence of CFTR function in cystic fibrosis after systemic administration of 4-phenylbutyrate. Mol. Ther. J. Am. Soc. Gene Ther. 2002, 6, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, R.C.; Egan, M.E.; Zeitlin, P.L. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J. Clin. Investig. 1997, 100, 2457–2465. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Goldberg, E.; Goldberg, J. ER retention is imposed by COPII protein sorting and attenuated by 4-phenylbutyrate. eLife 2017, 6, e26624. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.Y.; Qian, S.B. Less is more: Improving proteostasis by translation slow down. Trends Biochem. Sci. 2013, 38, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Meriin, A.B.; Mense, M.; Colbert, J.D.; Liang, F.; Bihler, H.; Zaarur, N.; Rock, K.L.; Sherman, M.Y. A novel approach to recovery of function of mutant proteins by slowing down translation. J. Biol. Chem. 2012, 287, 34264–34272. [Google Scholar] [CrossRef]
- Meriin, A.B.; Zaarur, N.; Sherman, M.Y. Association of translation factor eEF1A with defective ribosomal products generates a signal for aggresome formation. J. Cell Sci. 2012, 125, 2665–2674. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piccolo, D.; Zarouchlioti, C.; Bellingham, J.; Guarascio, R.; Ziaka, K.; Molday, R.S.; Cheetham, M.E. A Proximity Complementation Assay to Identify Small Molecules That Enhance the Traffic of ABCA4 Misfolding Variants. Int. J. Mol. Sci. 2024, 25, 4521. https://doi.org/10.3390/ijms25084521
Piccolo D, Zarouchlioti C, Bellingham J, Guarascio R, Ziaka K, Molday RS, Cheetham ME. A Proximity Complementation Assay to Identify Small Molecules That Enhance the Traffic of ABCA4 Misfolding Variants. International Journal of Molecular Sciences. 2024; 25(8):4521. https://doi.org/10.3390/ijms25084521
Chicago/Turabian StylePiccolo, Davide, Christina Zarouchlioti, James Bellingham, Rosellina Guarascio, Kalliopi Ziaka, Robert S. Molday, and Michael E. Cheetham. 2024. "A Proximity Complementation Assay to Identify Small Molecules That Enhance the Traffic of ABCA4 Misfolding Variants" International Journal of Molecular Sciences 25, no. 8: 4521. https://doi.org/10.3390/ijms25084521