

Exploring the Anticancer Potential of Semisynthetic Derivatives of 7α-Acetoxy-6β-hydroxyroyleanone from Plectranthus sp.: An In Silico Approach
 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Results
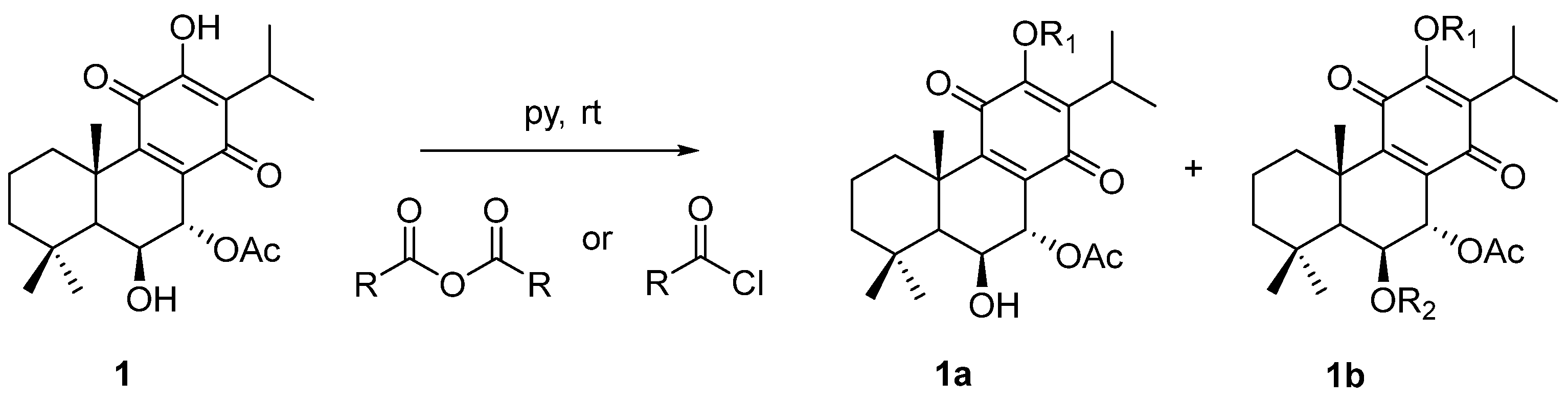
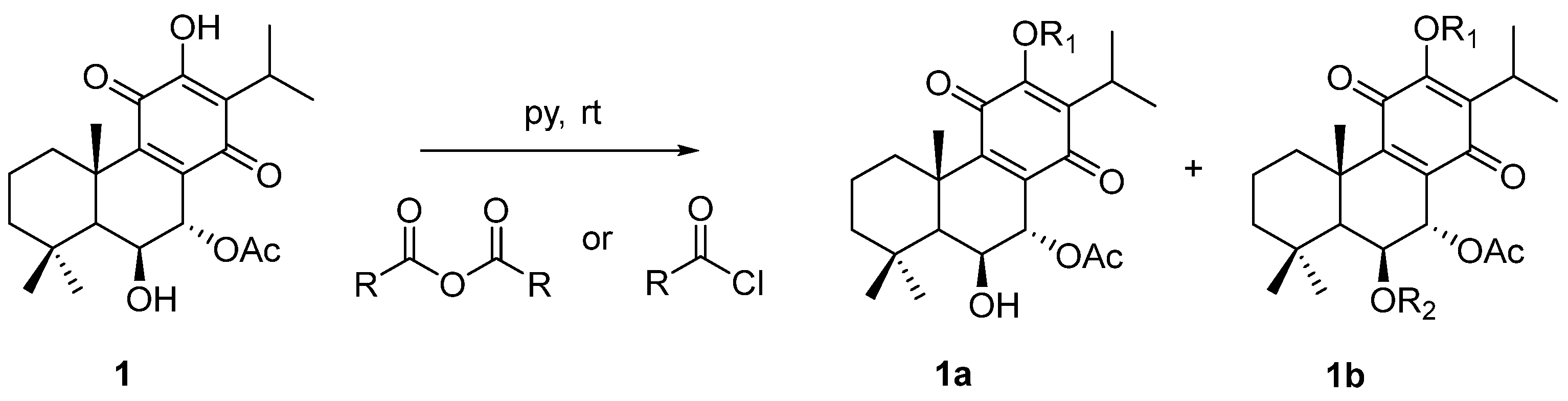
2.1. Characterization of the Synthesized Compounds
2.2. ADMET and Drug-Likeness Analysis Results
2.3. Toxicity Prediction and Molecular Property Results
2.4. Antineoplastic and Anticarcinogenic Activity Results
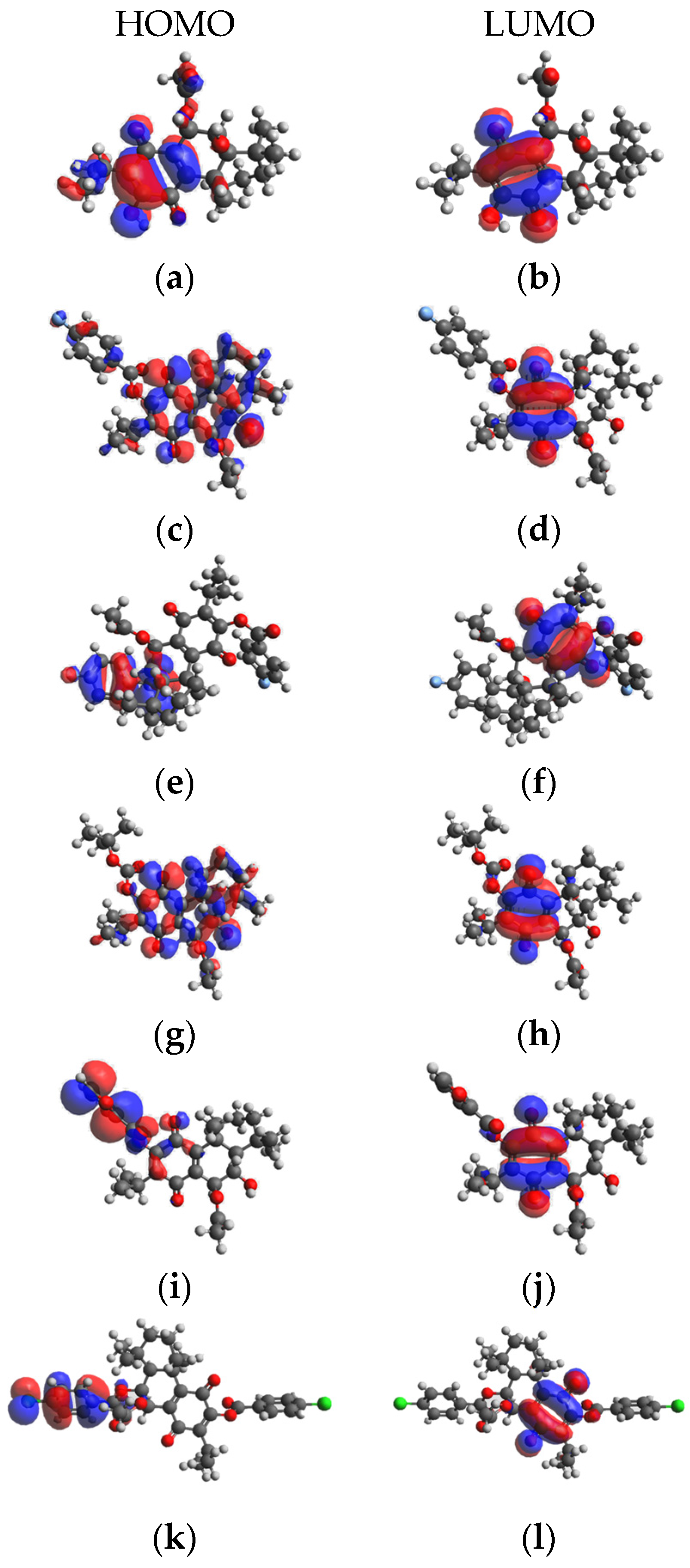
2.5. Density functional theory Calculation Results
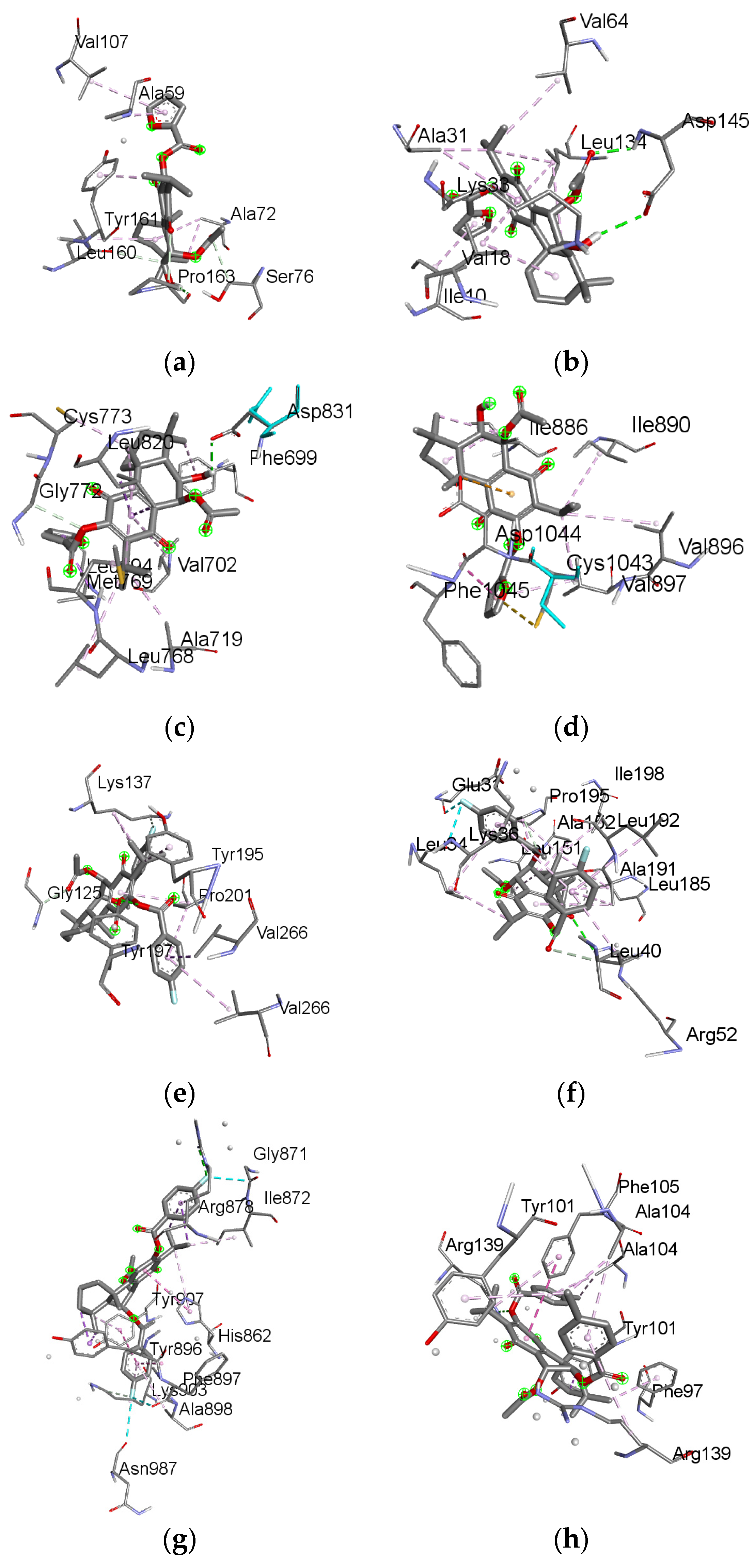
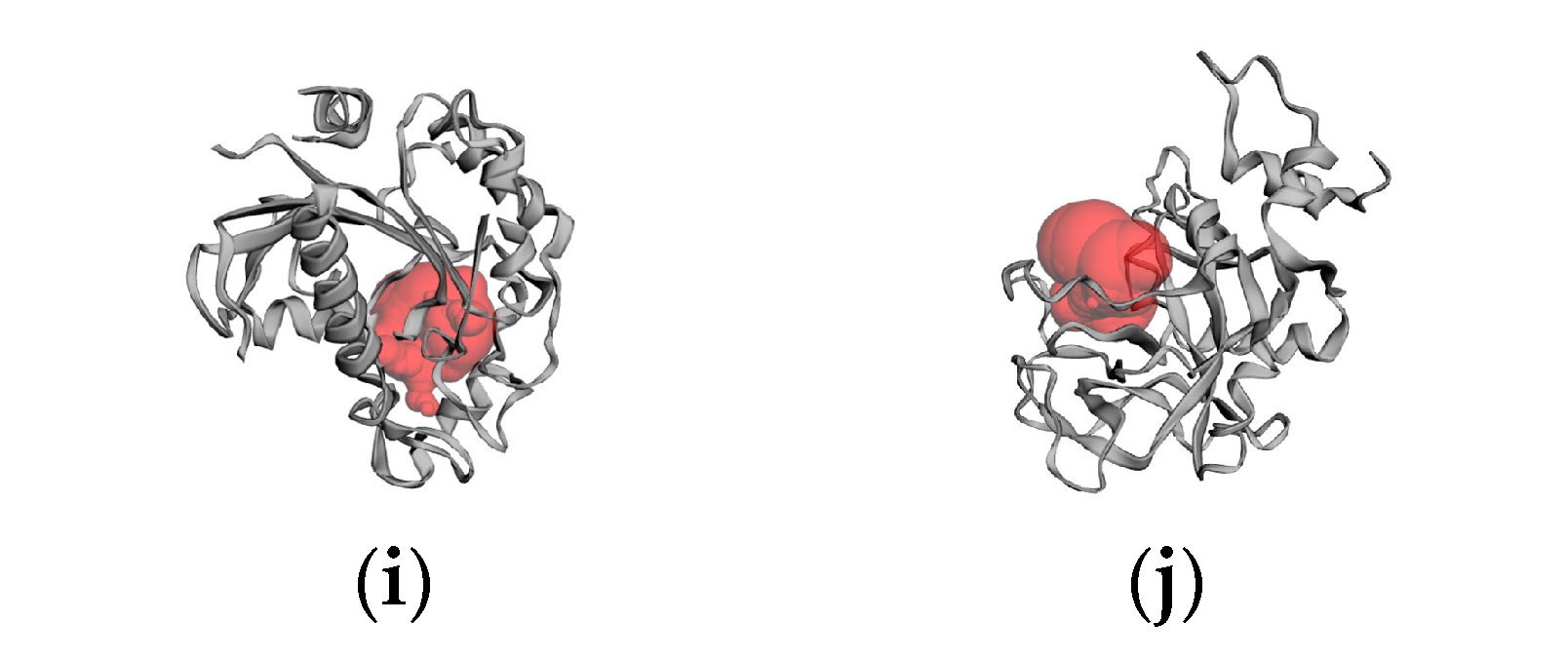
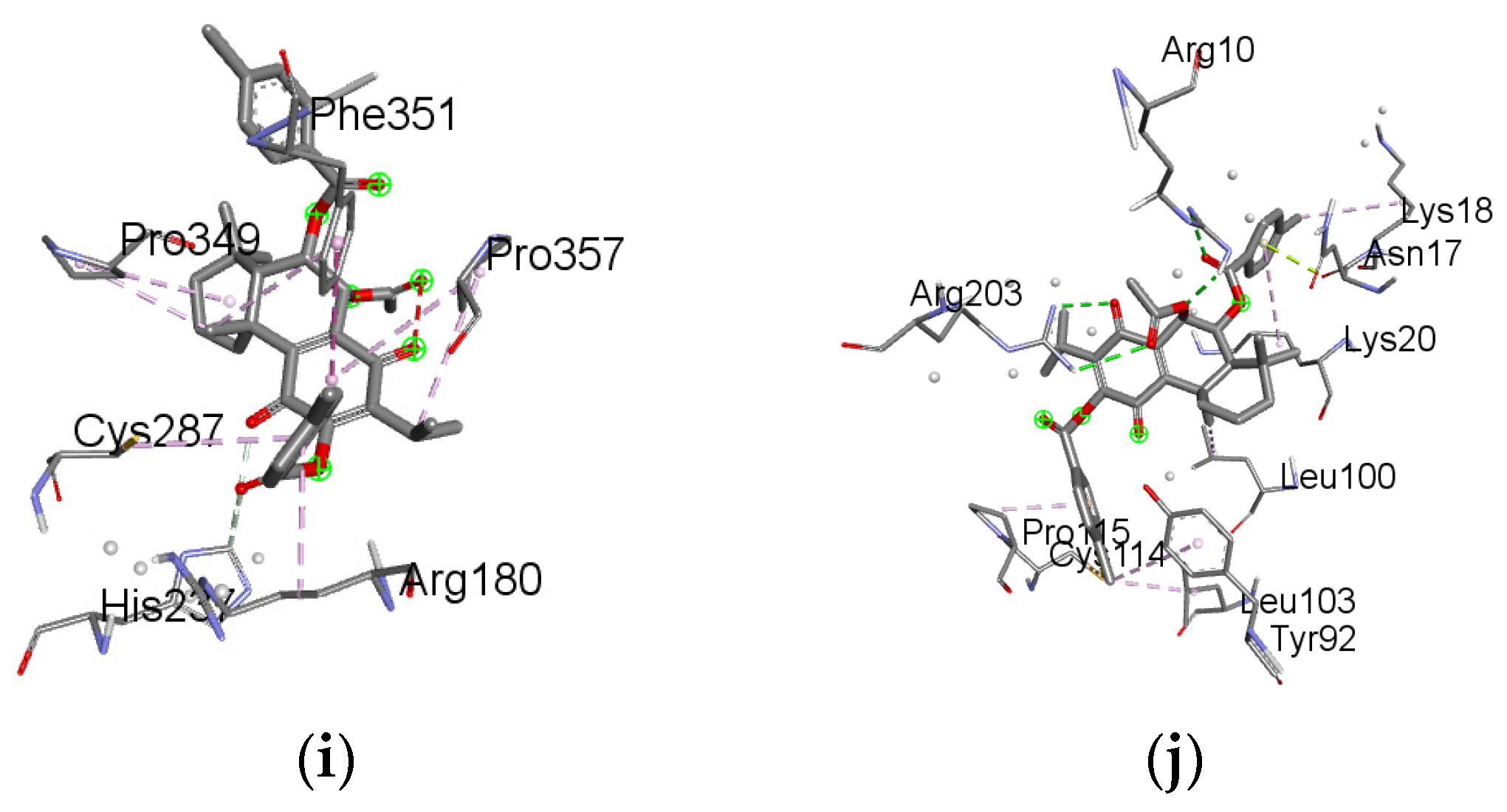
2.6. Molecular Docking Results
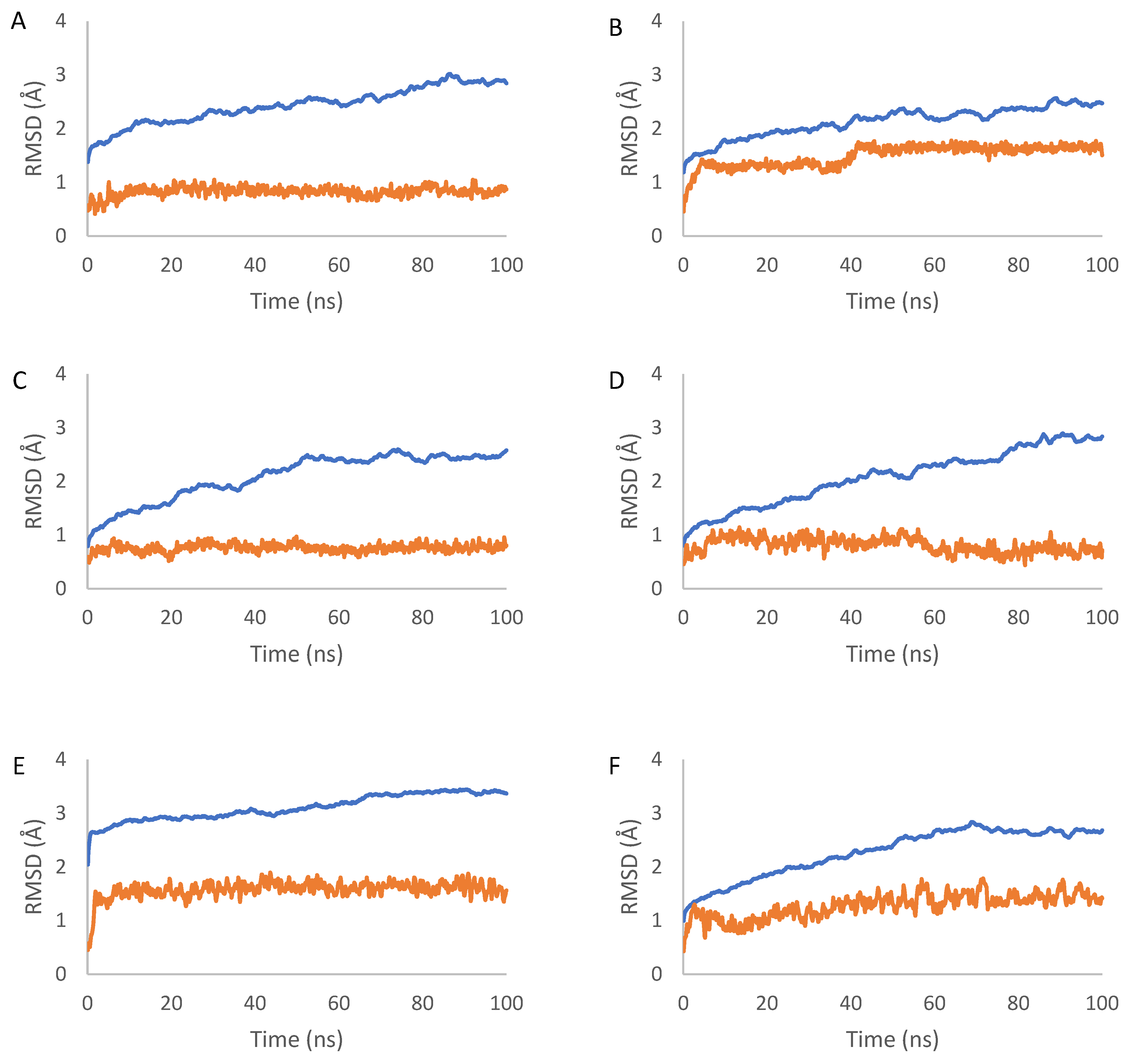
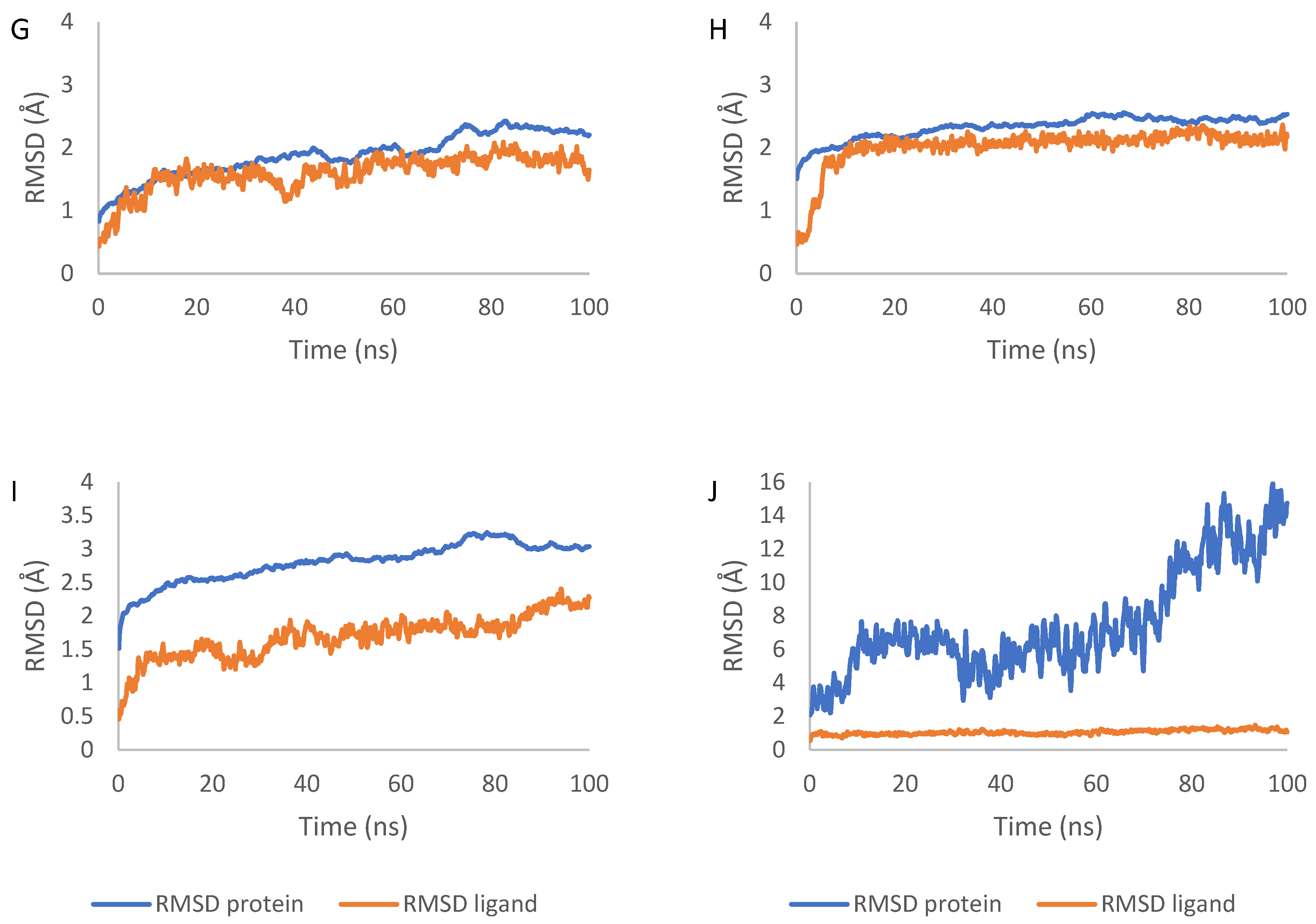
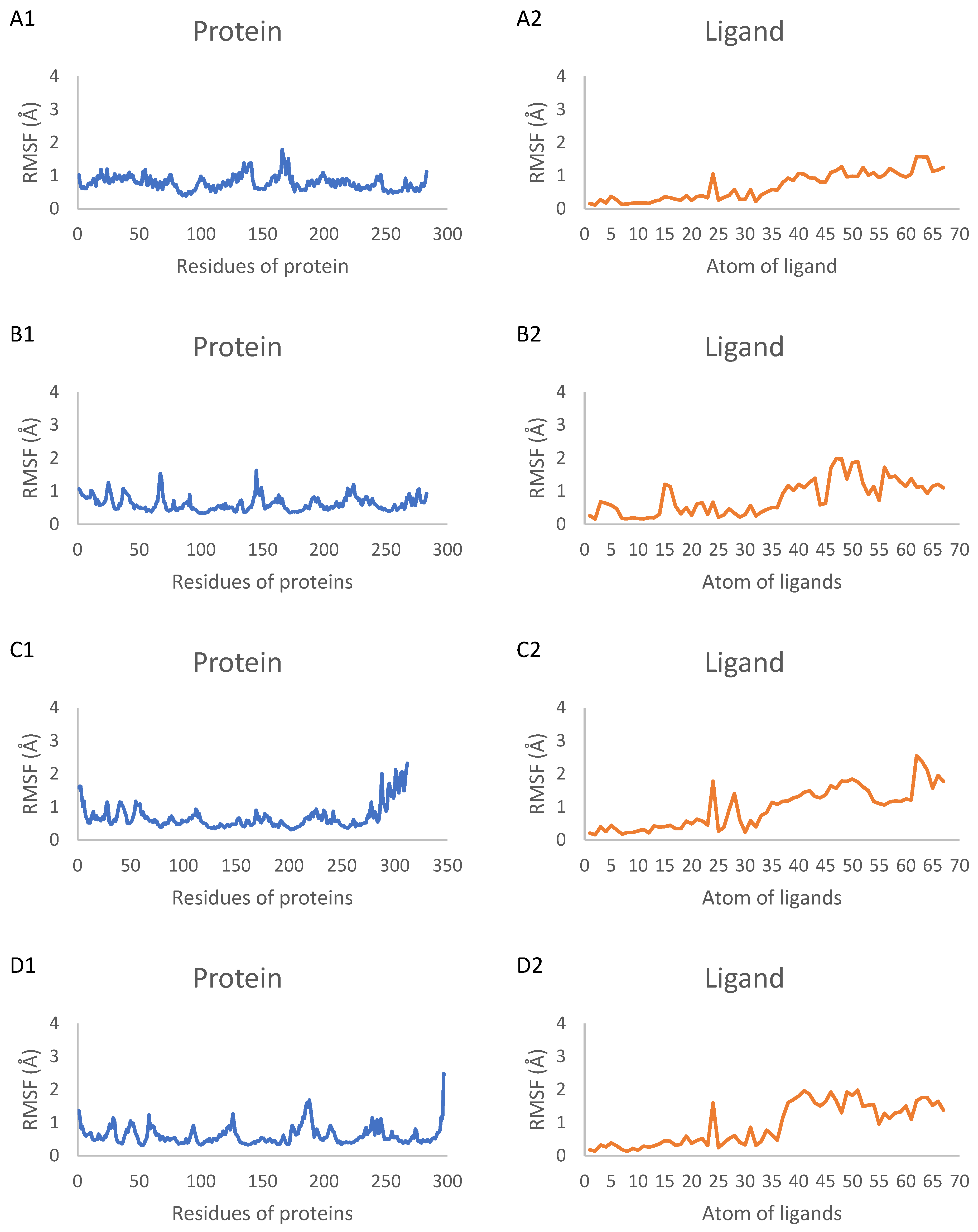
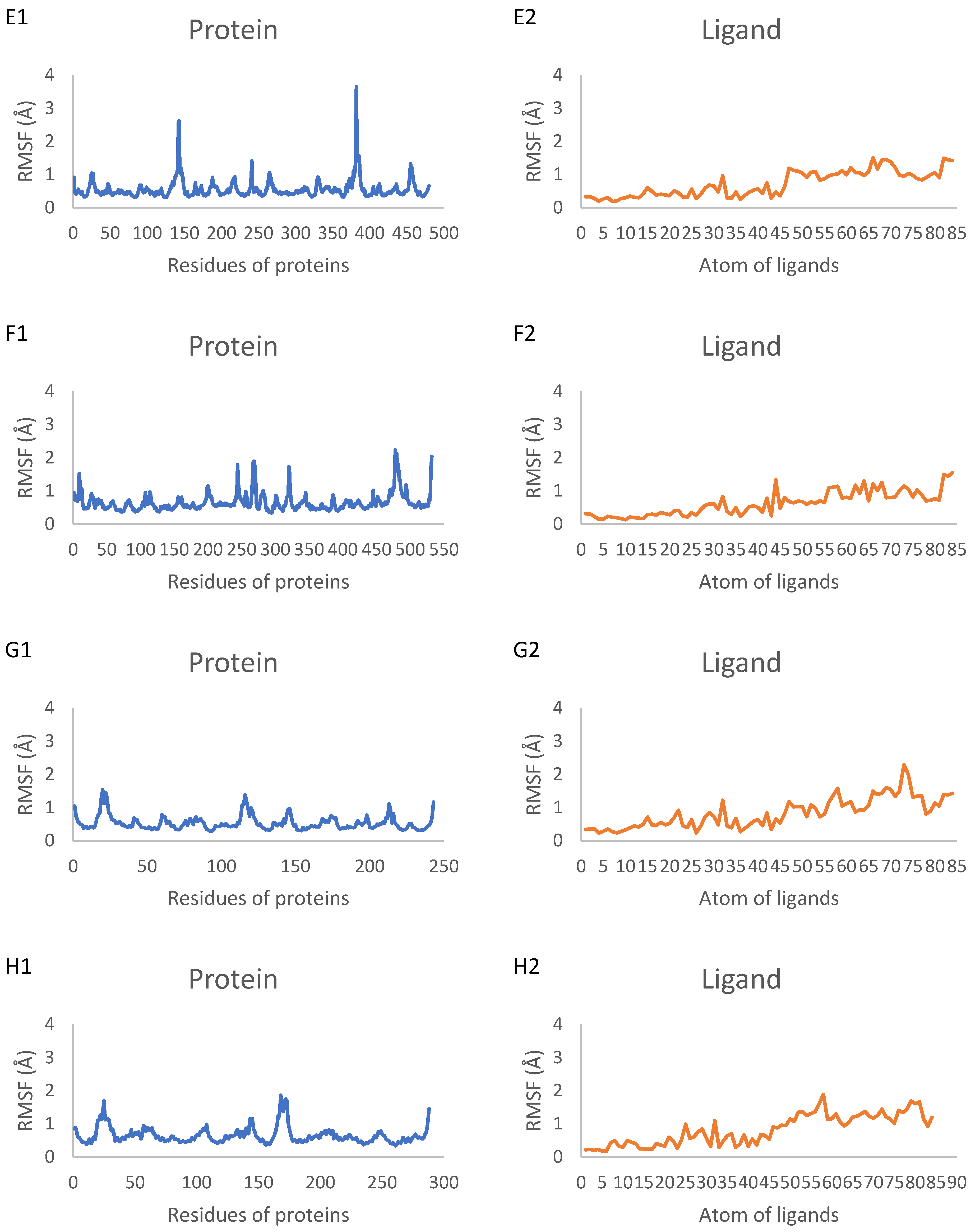
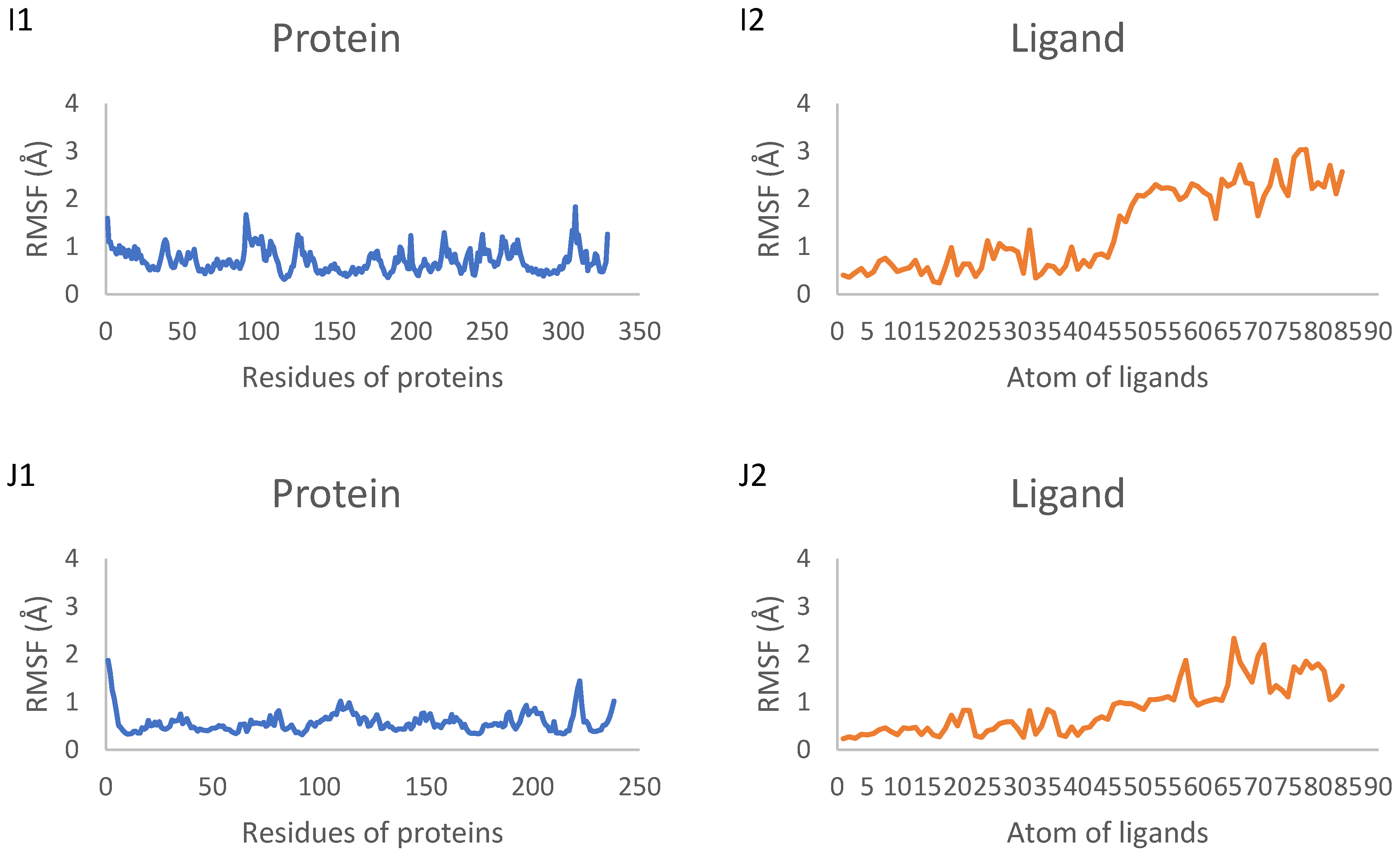
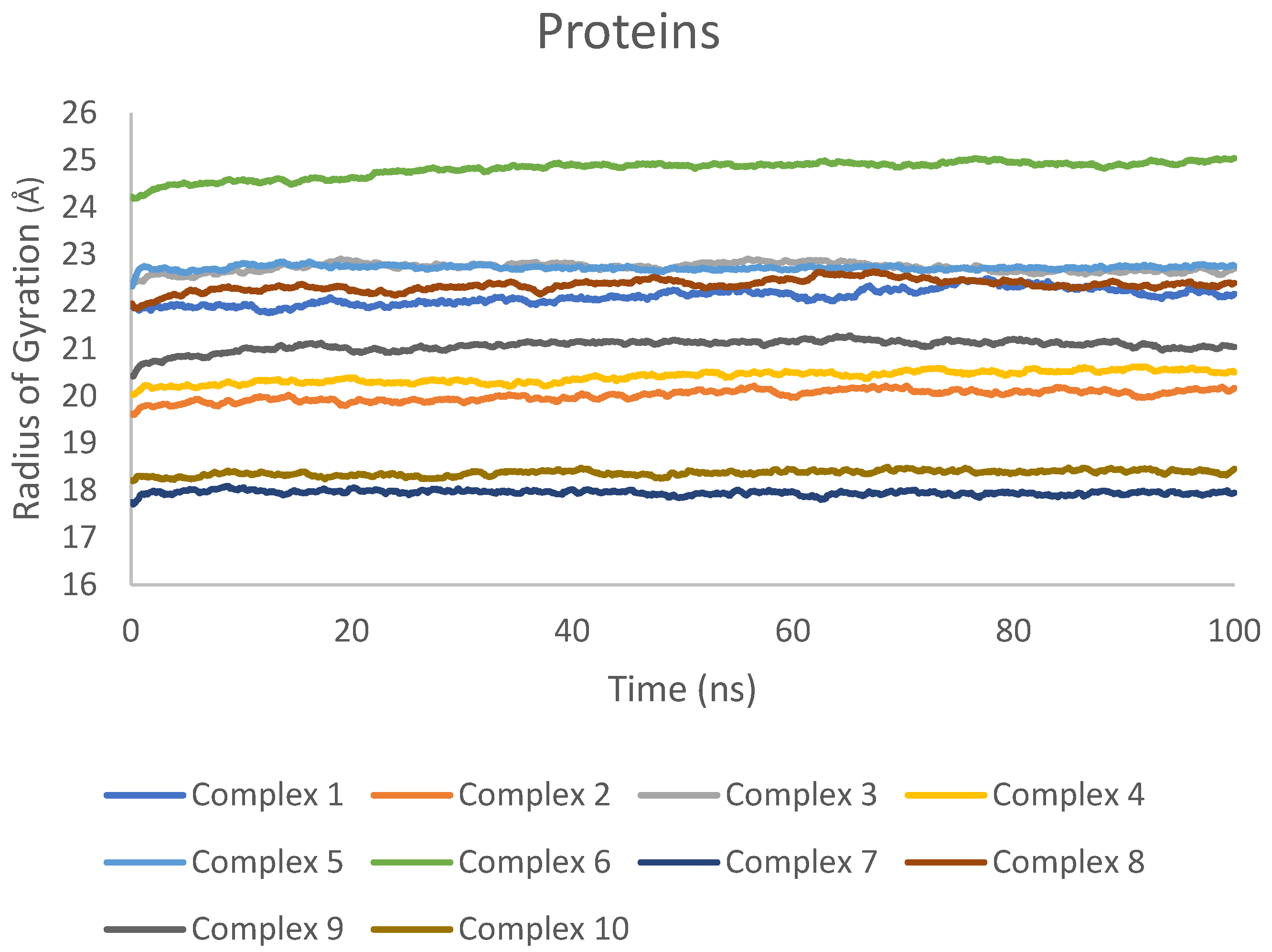
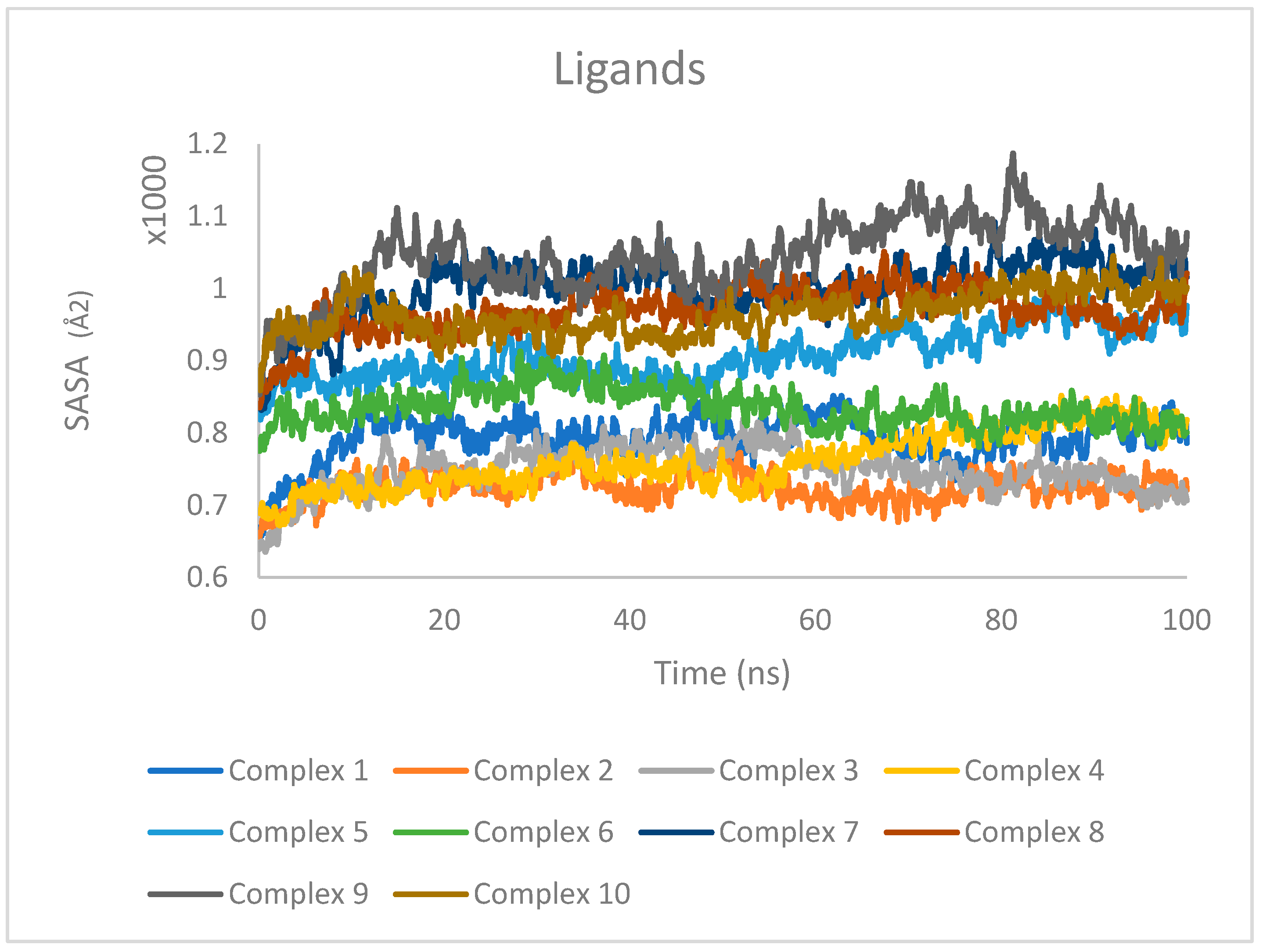
2.7. Molecular Dynamics Simulation Results
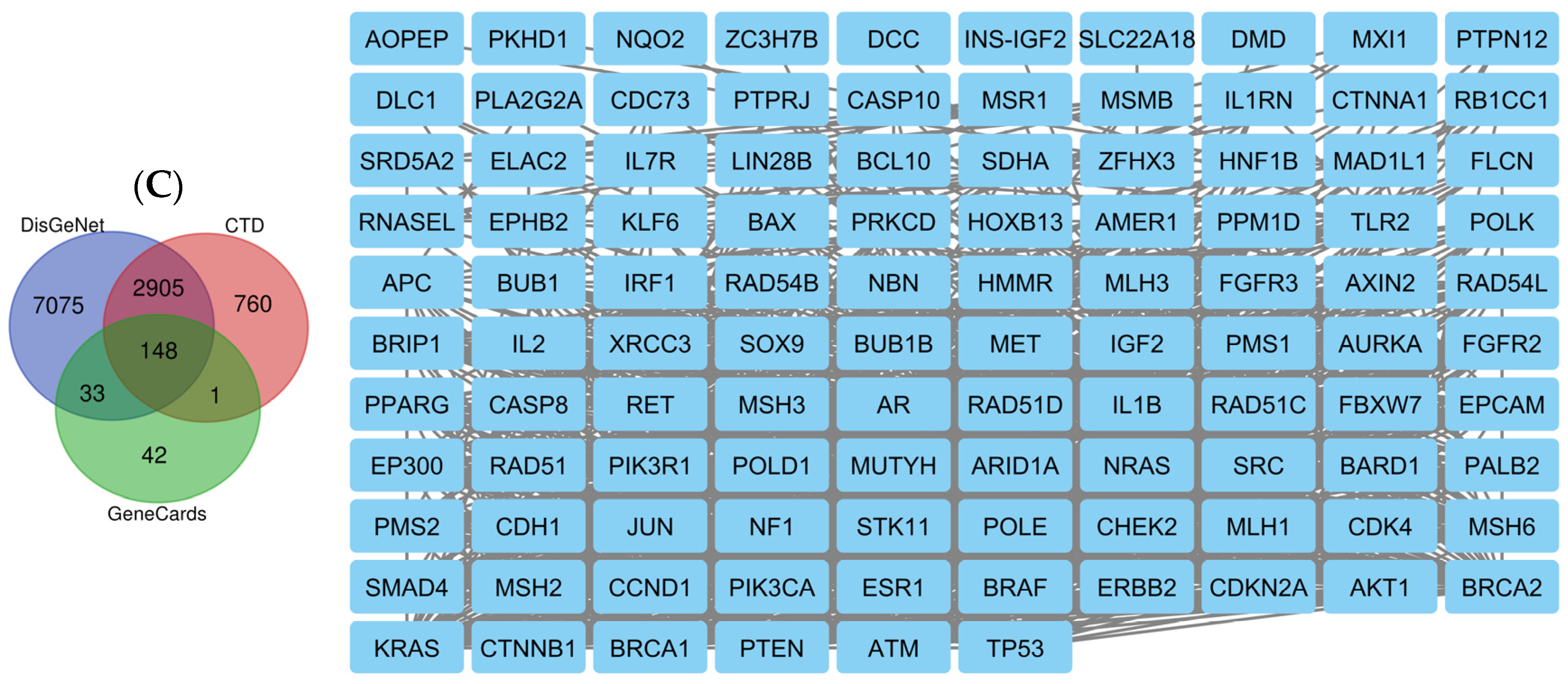
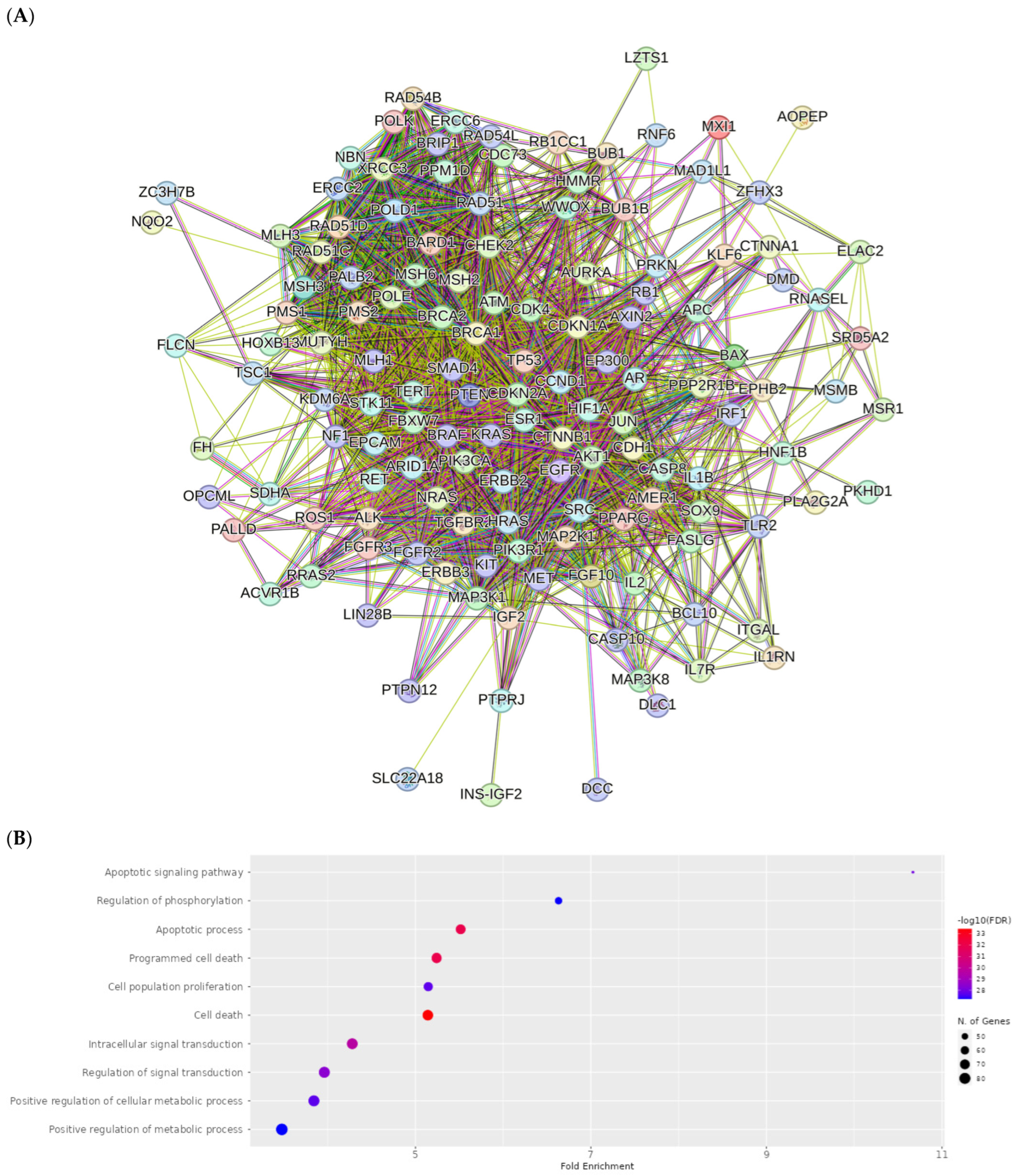
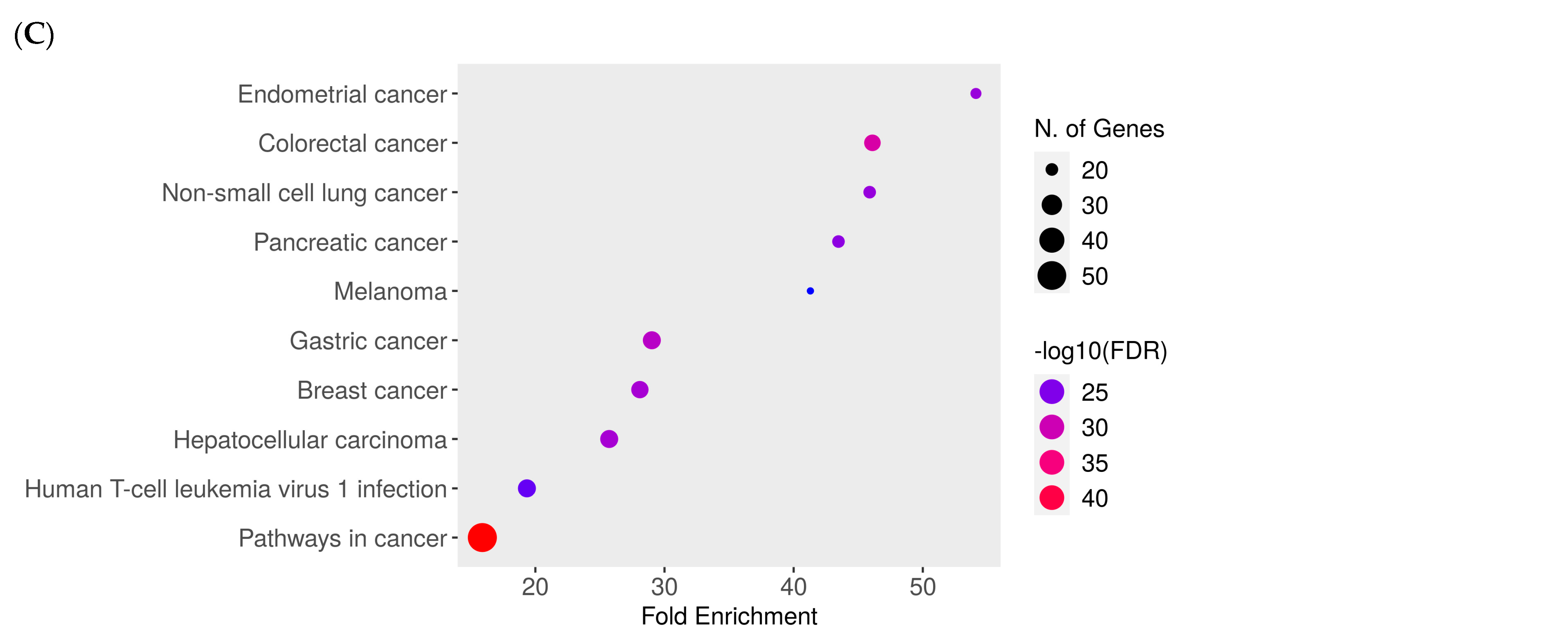
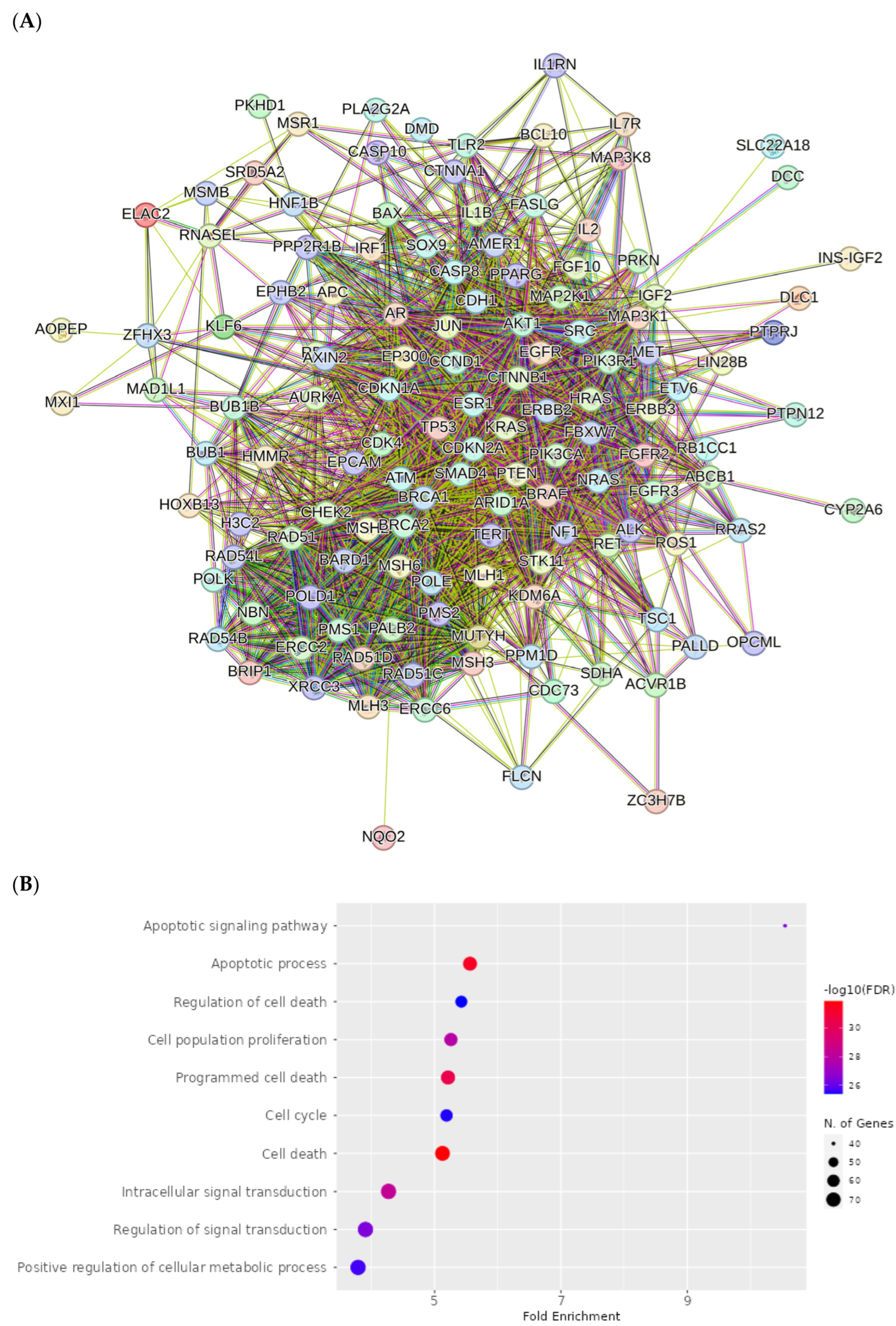
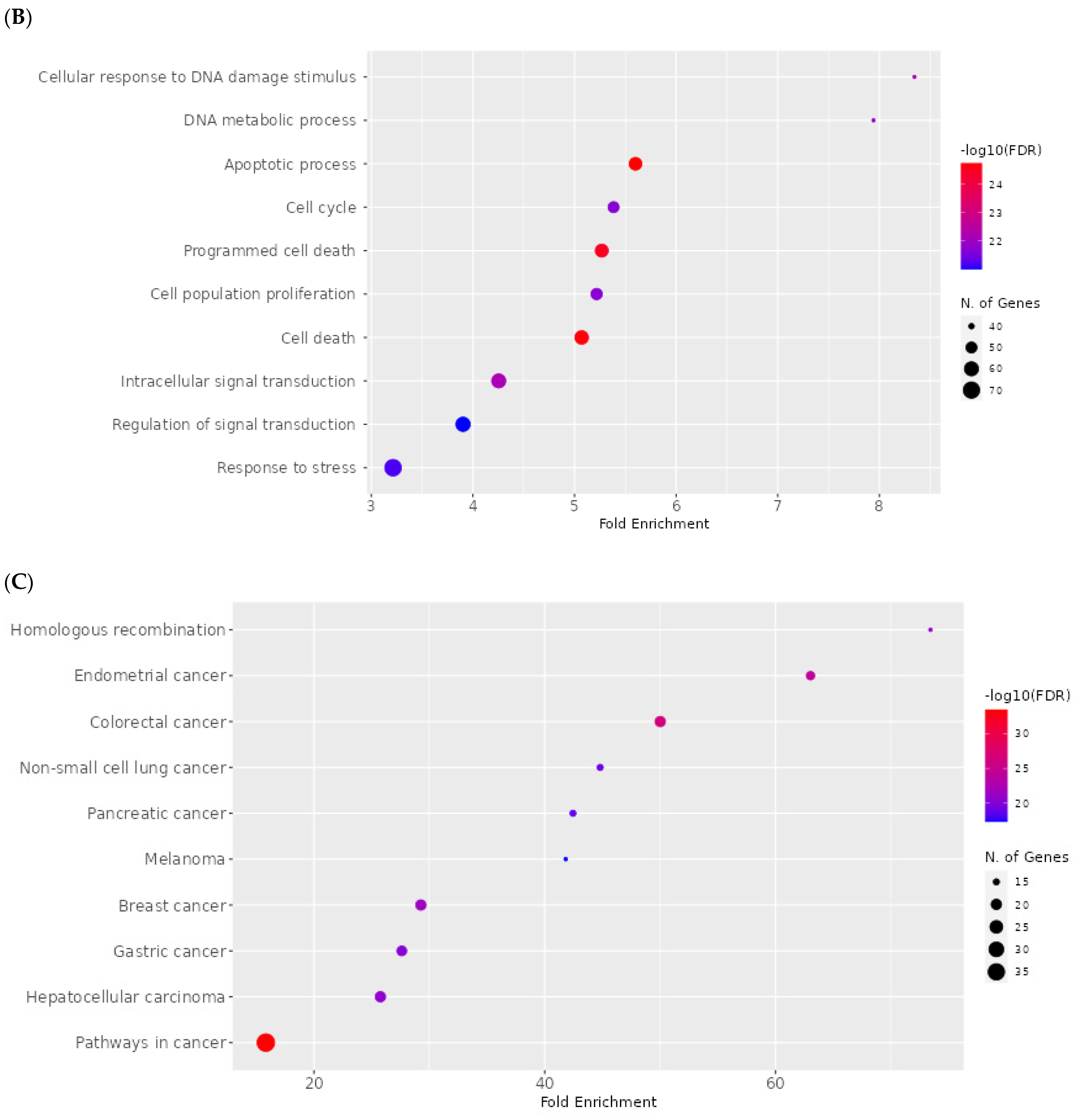
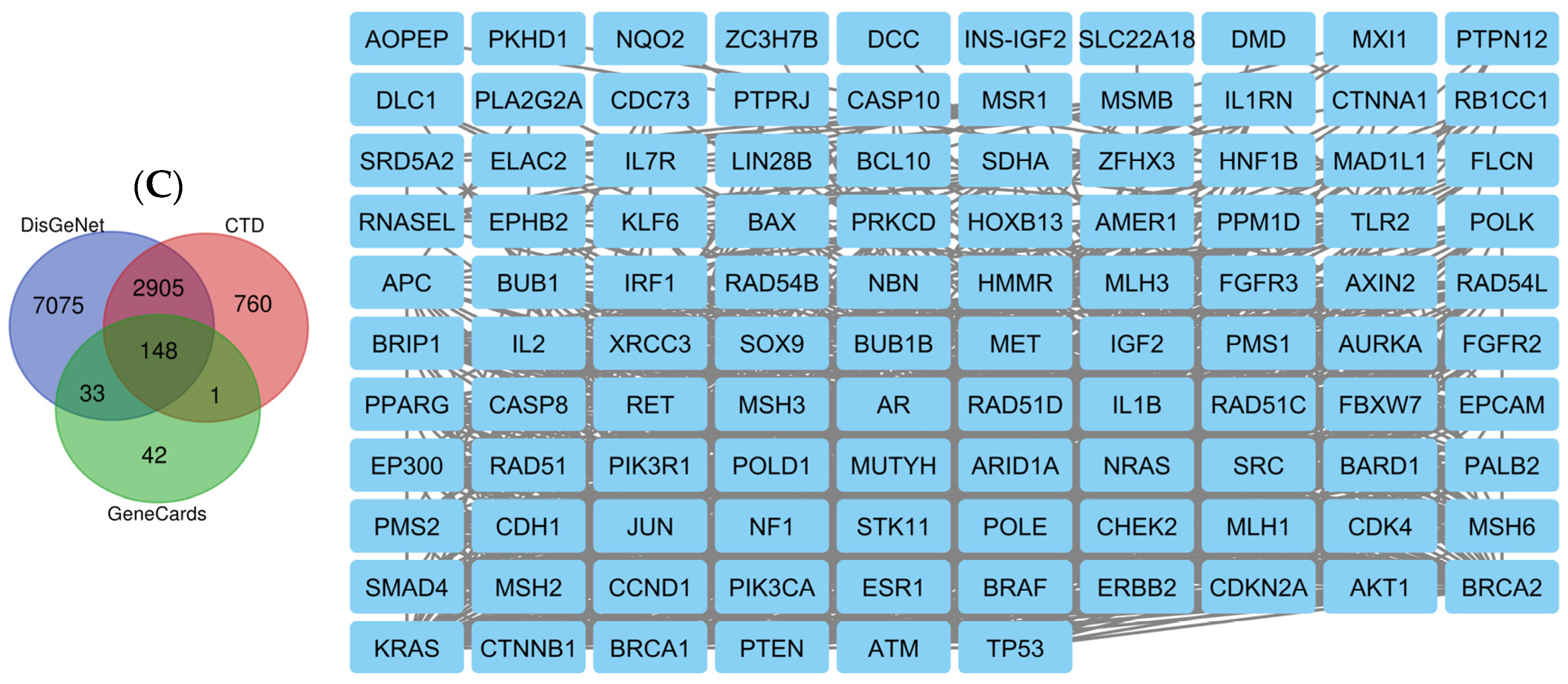
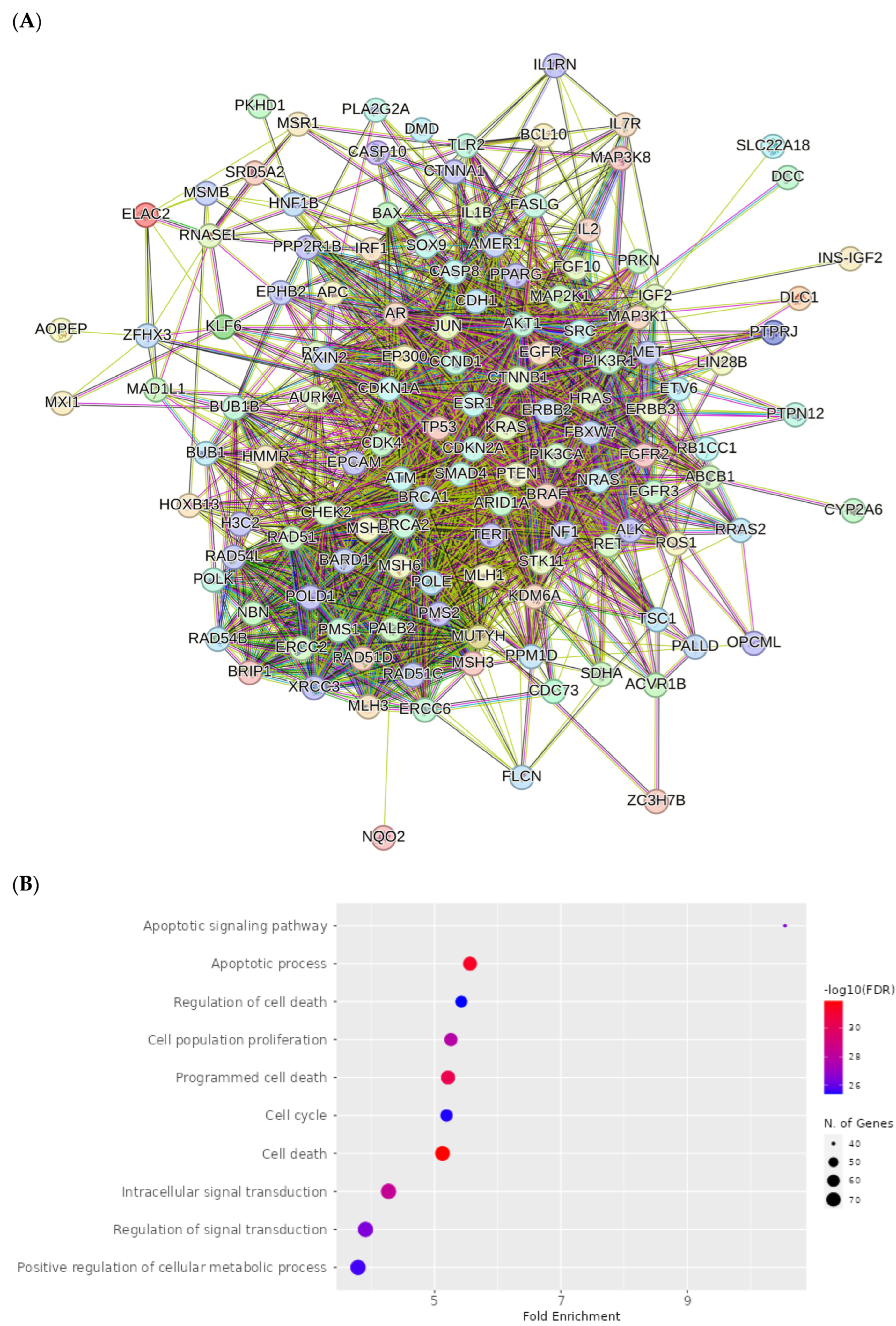
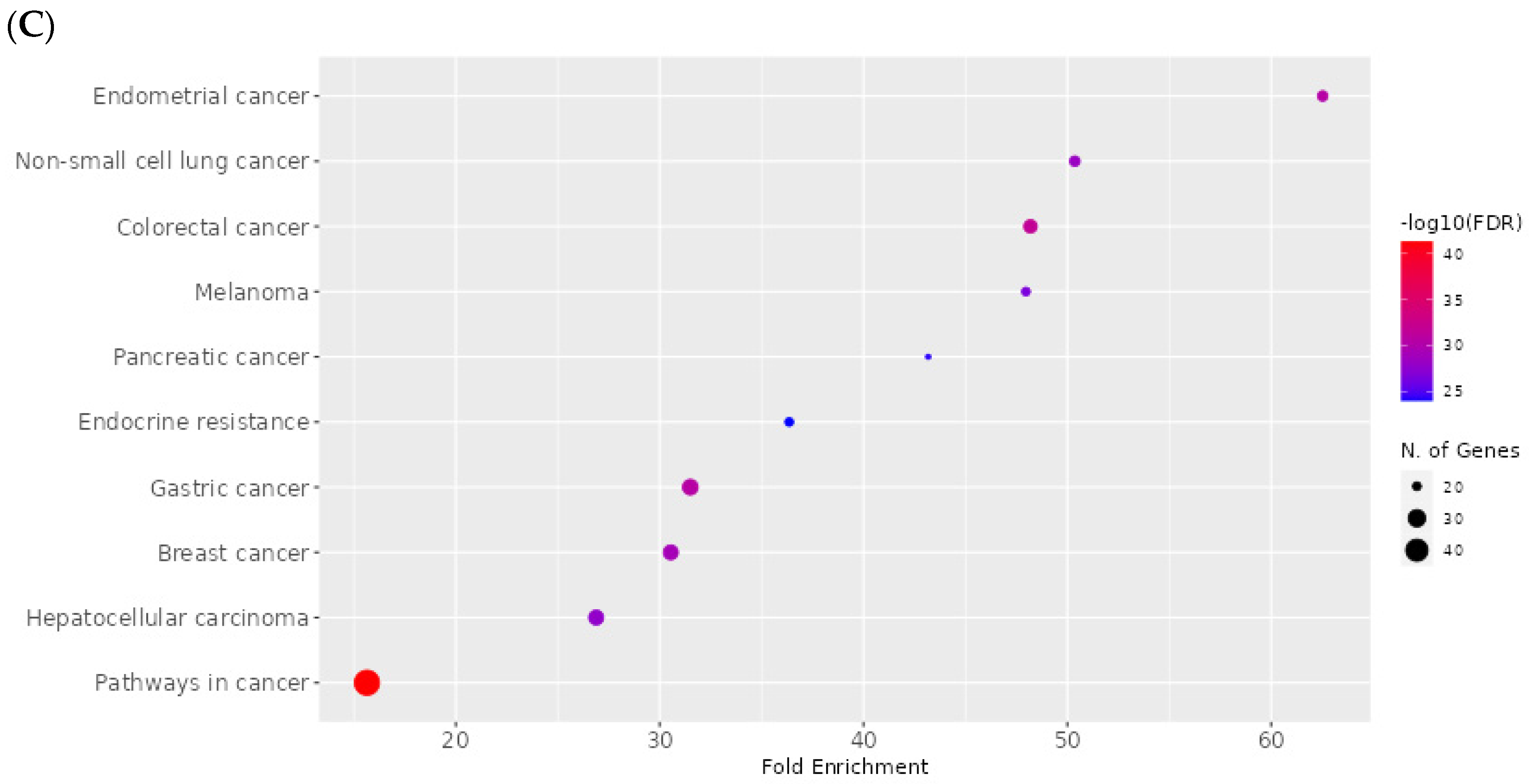
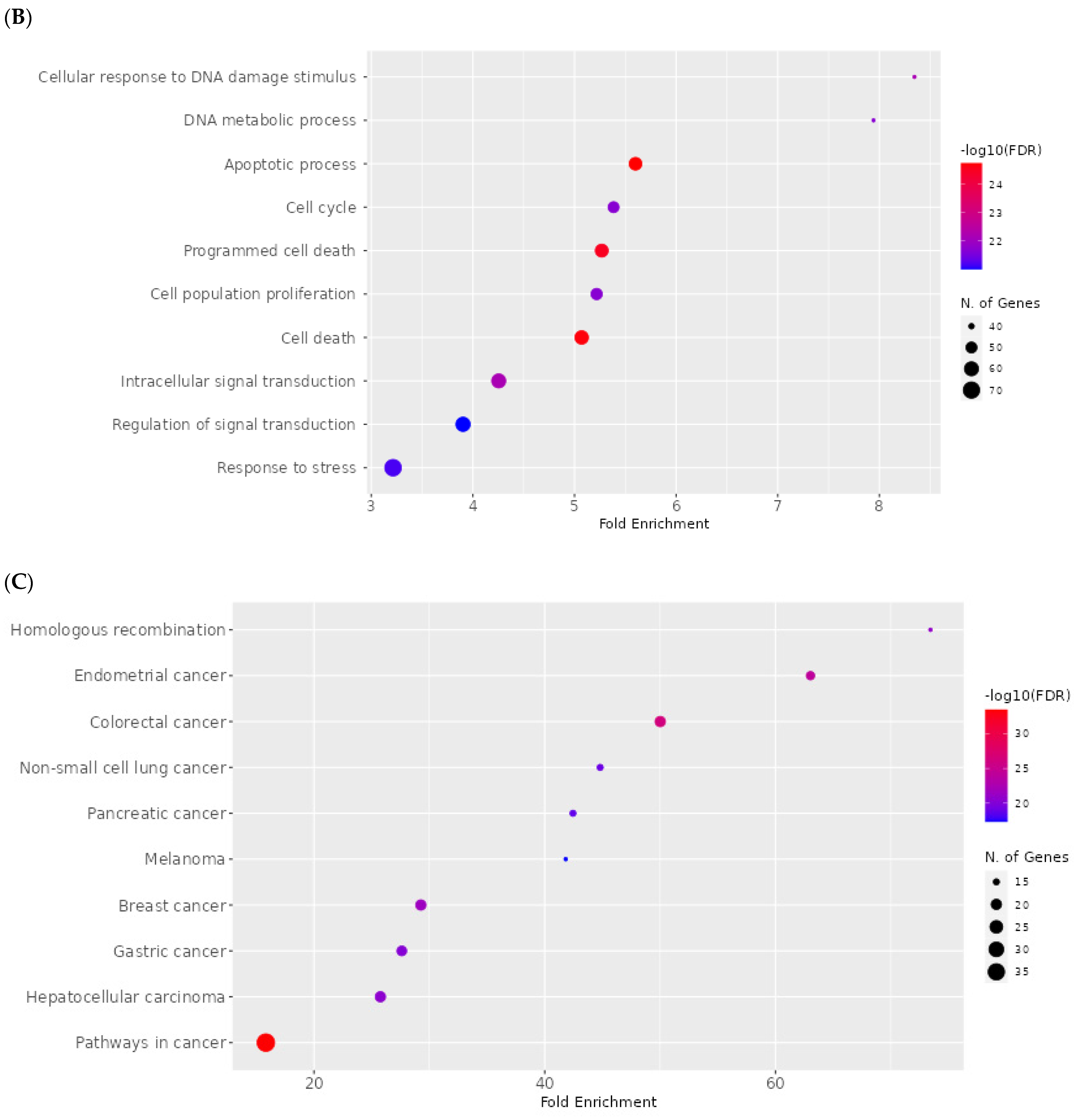
2.8. Network Pharmacology Results
3. Discussion
4. Materials and Methods
4.1. Compounds
4.1.1. General Experimental Procedures
4.1.2. Plant Material
4.1.3. Extraction and Isolation
4.2. ADMET and Drug-Likeness Analysis
4.3. Toxicity Prediction and Molecular Properties
4.4. Anticarcinogenic Activity
4.5. DFT Calculations
4.6. Molecular Docking
4.6.1. Protein and Ligand Preparation
4.6.2. Active Site Prediction
4.6.3. Receptor–Ligand Docking
4.7. Molecular Dynamics Simulations
4.8. Network Pharmacology
4.8.1. Identification of Potential Targets of Analyzed Compounds
4.8.2. Associated Targets of Cancer Diseases
4.8.3. Visualization and Analysis of the Network of the Protein–Protein Interactions
4.8.4. Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mattiuzzi, C.; Lippi, G. Current cancer epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Mathers, C.D.; Loncar, D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006, 3, 2011–2030. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Cancer; WHO: Geneva, Switzerland, 2021.
- Brennan, P.; Davey-Smith, G. Identifying Novel Causes of Cancers to Enhance Cancer Prevention: New Strategies Are Needed. J. Natl. Cancer Inst. 2022, 114, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Modern Cancer Therapies and Traditional Medicine: An Integrative Approach to Combat Cancers; Bentham Science Publishers: Karachi, Pakistan, 2021.
- Elshafie, H.S.; Camele, I.; Mohamed, A.A. A Comprehensive Review on the Biological, Agricultural and Pharmaceutical Properties of Secondary Metabolites Based-Plant Origin. Int. J. Mol. Sci. 2023, 24, 3266. [Google Scholar] [CrossRef]
- Twaij, B.M.; Hasan, M.N. Bioactive Secondary Metabolites from Plant Sources: Types, Synthesis, and Their Therapeutic Uses. Int. J. Plant Biol. 2022, 13, 4–14. [Google Scholar] [CrossRef]
- D’Amelia, V.; Docimo, T.; Crocoll, C.; Rigano, M.M. Specialized metabolites and valuable molecules in crop and medicinal plants: The evolution of their use and strategies for their production. Genes 2021, 12, 936. [Google Scholar] [CrossRef] [PubMed]
- Ascensao, L.; Figueiredo, A.C.; Barroso, J.G.; Pedro, L.G.; Schripsema, J.; Deans, S.G.; Scheffer, J.J.C. Plectranthus madagascariensis: Morphology of the glandular trichomes, essential oil composition, and its biological activity. Int. J. Plant Sci. 1998, 159, 31–38. [Google Scholar] [CrossRef]
- Waldia, S.; Joshi, B.C.; Pathak, U.; Joshi, M.C. The genus plectranthus in India and its chemistry. Chem. Biodivers. 2011, 8, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Lukhoba, C.W.; Simmonds, M.S.J.; Paton, A.J. Plectranthus: A review of ethnobotanical uses. J. Ethnopharmacol. 2006, 103, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, I.A.; Lall, N. Traditional usage and biological activity of Plectranthus madagascariensis and its varieties: A review. J. Ethnopharmacol. 2021, 269, 113663. [Google Scholar] [CrossRef]
- Gaspar-Marques, C.; Simões, M.F.; Valdeira, M.L.; Rodríguez, B. Terpenoids and phenolics from Plectranthus strigosus, bioactivity screening. Nat. Prod. Res. 2008, 22, 167–177. [Google Scholar] [CrossRef]
- Sindhu, M.S.; Poonkothai, M.; Thirumalaisamy, R. Phenolic and terpene compounds from Plectranthus amboinicus (Lour.) Spreng act as promising hepatic anticancer agents screened through in silico and in vitro approaches. South Afr. J. Bot. 2022, 149, 145–159. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, M.; Hou, Y.; Fan, C.; Wei, H.; Shi, L.; Ma, G.; Zhang, J. Inflammatory and cytotoxic activities of abietane terpenoids from nepeta bracteata benth. Molecules 2021, 26, 5603. [Google Scholar] [CrossRef] [PubMed]
- Gáborová, M.; Šmejkal, K.; Kubínová, R. Abietane diterpenes of the genus plectranthus sensu lato. Molecules 2022, 27, 166. [Google Scholar] [CrossRef] [PubMed]
- Helfenstein, A.; Vahermo, M.; Nawrot, D.A.; Demirci, F.; İşcan, G.; Krogerus, S.; Yli-Kauhaluoma, J.; Moreira, V.M.; Tammela, P. Antibacterial profiling of abietane-type diterpenoids. Bioorganic Med. Chem. 2017, 25, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Saijo, H.; Kofujita, H.; Takahashi, K.; Ashitani, T. Antioxidant activity and mechanism of the abietane-type diterpene ferruginol. Nat. Prod. Res. 2015, 29, 1739–1743. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Mogib, M.; Albar, H.A.; Batterjee, S.M. Chemistry of the Genus Plectranthus. Molecules 2002, 7, 271–301. [Google Scholar] [CrossRef]
- Cragg, G.M.; Pezzuto, J.M. Natural Products as a Vital Source for the Discovery of Cancer Chemotherapeutic and Chemopreventive Agents. Med. Princ. Pract. 2016, 25, 41–59. [Google Scholar] [CrossRef]
- Beghyn, T.; Deprez-Poulain, R.; Willand, N.; Folleas, B.; Deprez, B. Natural compounds: Leads or ideas? Bioinspired molecules for drug discovery. Chem. Biol. Drug Des. 2008, 72, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Vibala, B.V.; Praseetha, P.K.; Vijayakumar, S. Evaluating new strategies for anticancer molecules from ethnic medicinal plants through in silico and biological approach—A review. Gene Reports 2020, 18, 1000553. [Google Scholar] [CrossRef]
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Methods for virtual ligand screening and profiling. Br. J. Pharmacol. 2007, 152, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Applications to targets and beyond. Br. J. Pharmacol. 2007, 152, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Shaker, B.; Ahmad, S.; Lee, J.; Jung, C.; Na, D. In silico methods and tools for drug discovery. Comput. Biol. Med. 2021, 137, 104851. [Google Scholar] [CrossRef] [PubMed]
- Filimonov, D.A.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Druzhilovskii, D.S.; Pogodin, P.V.; Poroikov, V.V. Prediction of the biological activity spectra of organic compounds using the pass online web resource. Chem. Heterocycl. Compd. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed]
- Janumyan, Y.M.; Sansam, C.G.; Chattopadhyay, A.; Cheng, N.; Soucie, E.L.; Penn, L.Z.; Andrews, D.; Knudson, C.M.; Yang, E. Bcl-xL/Bcl-2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. EMBO J. 2003, 22, 5459–5470. [Google Scholar] [CrossRef] [PubMed]
- Brentnall, M.; Rodriguez-Menocal, L.; De Guevara, R.L.; Cepero, E.; Boise, L.H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Marei, H.E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Pozzoli, G.; Morrione, A.; Giordano, A.; Cenciarelli, C. p53 signaling in cancer progression and therapy. Cancer Cell Int. 2021, 21, 703. [Google Scholar] [CrossRef] [PubMed]
- Pazzaglia, S.; Pioli, C. Multifaceted role of parp-1 in dna repair and inflammation: Pathological and therapeutic implications in cancer and non-cancer diseases. Cells 2020, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Samtiya, M.; Aluko, R.E.; Dhewa, T.; Moreno-Rojas, J.M. Potential health benefits of plant food-derived bioactive components: An overview. Foods 2021, 10, 839. [Google Scholar] [CrossRef] [PubMed]
- Riaz, M.; Khalid, R.; Afzal, M.; Anjum, F.; Fatima, H.; Zia, S.; Rasool, G.; Egbuna, C.; Mtewa, A.G.; Uche, C.Z.; et al. Phytobioactive compounds as therapeutic agents for human diseases: A review. Food Sci. Nutr. 2023, 11, 2500–2529. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.P.; Mailloux, B.; Kulkarni, S.; Gagné, M.; Long, A.S.; Barton-Maclaren, T.S. Development and application of consensus in silico models for advancing high-throughput toxicological predictions. Front. Pharmacol. 2024, 15, 1307905. [Google Scholar] [CrossRef] [PubMed]
- Burmistrova, O.; Perdomo, J.; Simões, M.F.; Rijo, P.; Quintana, J.; Estévez, F. The abietane diterpenoid parvifloron D from Plectranthus ecklonii is a potent apoptotic inducer in human leukemia cells. Phytomedicine 2015, 22, 1009–1016. [Google Scholar] [CrossRef]
- Abdissa, N.; Frese, M.; Sewald, N. Antimicrobial abietane-type diterpenoids from plectranthus punctatus. Molecules 2017, 22, 1919. [Google Scholar] [CrossRef] [PubMed]
- Napagoda, M.; Gerstmeier, J.; Butschek, H.; Lorenz, S.; De Soyza, S.; Qader, M.; Nagahawatte, A.; Wijayaratne, G.B.; Schneider, B.; Svatoš, A.; et al. Plectranthus Zeylanicus: A Rich Source of Secondary Metabolites with Antimicrobial, Disinfectant and Anti-Inflammatory Activities. Pharmaceuticals 2022, 15, 436. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Figueiredo, T.; de Almeida, R.F.M.; Antunes, C.A.C.; Garcia, C.; Reis, C.P.; Ascensão, L.; Sobral, R.G.; Rijo, P. Unveiling the mechanism of action of 7α-acetoxy-6β-hydroxyroyleanone on an mrsa/visa strain: Membrane and cell wall interactions. Biomolecules 2020, 10, 983. [Google Scholar] [CrossRef] [PubMed]
- Isca, V.M.S.; Sencanski, M.; Filipovic, N.; Dos Santos, D.J.V.A.; Gašparović, A.Č.; Saraíva, L.; Afonso, C.A.M.; Rijo, P.; García-Sosa, A.T. Activity to breast cancer cell lines of different malignancy and predicted interaction with protein kinase C isoforms of royleanones. Int. J. Mol. Sci. 2020, 21, 3671. [Google Scholar] [CrossRef] [PubMed]
- Ntungwe, E.; Domínguez-Martín, E.M.; Teodósio, C.; Teixidó-Trujillo, S.; Armas Capote, N.; Saraiva, L.; Díaz-Lanza, A.M.; Duarte, N.; Rijo, P. Preliminary biological activity screening of plectranthus spp. Extracts for the search of anticancer lead molecules. Pharmaceuticals 2021, 14, 402. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Martín, E.M.; Magalhães, M.; Díaz-Lanza, A.M.; Marques, M.P.; Princiotto, S.; Gómez, A.M.; Efferth, T.; Cabral, C.; Rijo, P. Phytochemical Study and Antiglioblastoma Activity Assessment of Plectranthus hadiensis (Forssk.) Schweinf. ex Sprenger var. hadiensis Stems. Molecules 2022, 27, 3813. [Google Scholar] [CrossRef] [PubMed]
- Matias, D.; Nicolai, M.; Saraiva, L.; Pinheiro, R.; Faustino, C.; Diaz Lanza, A.; Pinto Reis, C.; Stankovic, T.; Dinic, J.; Pesic, M.; et al. Cytotoxic Activity of Royleanone Diterpenes from Plectranthus madagascariensis Benth. ACS Omega 2019, 4, 8094–8103. [Google Scholar] [CrossRef] [PubMed]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Tsuji, T.; Chen, C. Roles of PKC isoforms in the induction of apoptosis elicited by aberrant Ras. Oncogene 2010, 29, 1050–1061. [Google Scholar] [CrossRef] [PubMed]
- Yogosawa, S.; Yoshida, K. Tumor suppressive role for kinases phosphorylating p53 in DNA damage-induced apoptosis. Cancer Sci. 2018, 109, 3376–3382. [Google Scholar] [CrossRef] [PubMed]
- Sitailo, L.A.; Tibudan, S.S.; Denning, M.F. Bax activation and induction of apoptosis in human keratinocytes by the protein kinase C δ catalytic domain. J. Investig. Dermatol. 2004, 123, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Langston, J.C.; Tang, Y.; Kiani, M.F.; Kilpatrick, L.E. The role of tyrosine phosphorylation of protein kinase C delta in infection and inflammation. Int. J. Mol. Sci. 2019, 20, 1498. [Google Scholar] [CrossRef] [PubMed]
- Moscat, J.; Rennert, P.; Diaz-Meco, M.T. PKCζ at the crossroad of NF-κB and Jak1/Stat6 signaling pathways. Cell Death Differ. 2006, 13, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, C.E.S.; Garcia, C.; Pereira, F.; Mota, J.; Pereira, P.; Cebola, M.J.; Reis, C.P.; Correia, I.; Piedade, M.F.M.; Minas Da Piedade, M.E.; et al. Extraction Optimization and Structural and Thermal Characterization of the Antimicrobial Abietane 7α-Acetoxy-6β-hydroxyroyleanone. Mol. Pharm. 2018, 15, 1412–1419. [Google Scholar] [CrossRef] [PubMed]
- Simões, M.F.; Rijo, P.; Duarte, A.; Barbosa, D.; Matias, D.; Delgado, J.; Cirilo, N.; Rodríguez, B. Two new diterpenoids from Plectranthus species. Phytochem. Lett. 2010, 3, 221–225. [Google Scholar] [CrossRef]
- Hensch, M.; Rüedi, P.; Eugster, C.H. Horminon, Taxochinon und weitere Royleanone aus 2 abessinischen Plectranthus-Spezies (Labiatae). Helv. Chim. Acta 1975, 58, 1921–1934. [Google Scholar] [CrossRef]
- Isca, V.; Bangay, G.; Princiotto, S.; Saraíva, L.; Santos, D.; García-Sosa, A.; RIJO, P. Reactivity of 7α-acetoxy-6β-hydroxyroyleanone and ability of its derivatives to modulate PKC isoforms. Res. Sq. 2023, 2023, 1–18. [Google Scholar] [CrossRef]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef] [PubMed]
- Ilardi, E.A.; Vitaku, E.; Njardarson, J.T. Data-mining for sulfur and fluorine: An evaluation of pharmaceuticals to reveal opportunities for drug design and discovery. J. Med. Chem. 2014, 57, 2832–2842. [Google Scholar] [CrossRef]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef] [PubMed]
- Zafrani, Y.; Yeffet, D.; Sod-Moriah, G.; Berliner, A.; Amir, D.; Marciano, D.; Gershonov, E.; Saphier, S. Difluoromethyl Bioisostere: Examining the “Lipophilic Hydrogen Bond Donor” Concept. J. Med. Chem. 2017, 60, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z. The modification of natural products for medical use. Acta Pharm Sin. B 2017, 7, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Majhi, S.; Das, D. Chemical derivatization of natural products: Semisynthesis and pharmacological aspects—A decade update. Tetrahedron 2021, 78, 131801. [Google Scholar] [CrossRef]
- Garcia, C.; Isca, V.M.S.; Pereira, F.; Monteiro, C.M.; Ntungwe, E.; Sousa, F.; Dinic, J.; Holmstedt, S.; Roberto, A.; Díaz-Lanza, A.; et al. Royleanone Derivatives From Plectranthus spp. as a Novel Class of P-Glycoprotein Inhibitors. Front Pharmacol. 2020, 11, 557789. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Bakchi, B.; Krishna, A.D.; Sreecharan, E.; Ganesh, V.B.J.; Niharika, M.; Maharshi, S.; Puttagunta, S.B.; Sigalapalli, D.K.; Bhandare, R.R.; Shaik, A.B. An overview on applications of SwissADME web tool in the design and development of anticancer, antitubercular and antimicrobial agents: A medicinal chemist’s perspective. J. Mol. Struct. 2022, 1259, 132712. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed]
- Borba, J.V.B.; Alves, V.M.; Braga, R.C.; Korn, D.R.; Overdahl, K.; Silva, A.C.; Hall, S.U.S.; Overdahl, E.; Kleinstreuer, N.; Strickland, J.; et al. STopTox: An in Silico Alternative to Animal Testing for Acute Systemic and Topical Toxicity. Environ. Health Perspect. 2022, 130, 27012. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; Von Korff, M.; Reich, J.R.; Rufener, C. OSIRIS, an entirely in-house developed drug discovery informatics system. J. Chem. Inf. Model. 2009, 49, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of activity spectra for biologically active substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Olson, A.J. Using AutoDock for ligand-receptor docking. Curr. Protoc. Bioinform. 2008, 24, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Jejurikar, B.L.; Rohane, S.H. Drug Designing in Discovery Studio. Asian J. Res. Chem. 2021, 14, 135–138. [Google Scholar]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Majeed, A.; Mukhtar, S. Protein–Protein Interaction Network Exploration Using Cytoscape. Methods Mol. Biol. 2023, 2690, 419–427. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Predicted LD50 (mg/kg) | Predicted Toxicity Class | Prediction Accuracy (%) |
|---|---|---|---|
| 1 | 1000 | 4 | 69.26 |
| 2 | 100 | 3 | 67.38 |
| 3 | 100 | 3 | 67.38 |
| 4 | 75 | 3 | 68.07 |
| 5 | 100 | 3 | 54.26 |
| 6 | 100 | 3 | 67.38 |
| Compounds | Antineoplastic Activity | Anticarcinogenic Activity | ||
|---|---|---|---|---|
| Pa Value | Pi Value | Pa Value | Pi Value | |
| 1 | 0.879 | 0.005 | 0.419 | 0.028 |
| 2 | 0.810 | 0.010 | 0.315 | 0.053 |
| 3 | 0.822 | 0.009 | 0.279 | 0.067 |
| 4 | 0.882 | 0.0005 | 0.312 | 0.054 |
| 5 | 0.770 | 0.016 | 0.255 | 0.080 |
| 6 | 0.811 | 0.010 | 0.289 | 0.063 |
| Compound | EHOMO (eV) | ELUMO (eV) | ΔE (eV) | I (eV) | A (eV) | χ (eV) | μ (eV) | η (eV) | S (eV−1) | ω (eV) | ΔNmax |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | −6.885 | −3.433 | 3.45 | 6.89 | 3.43 | 5.16 | −5.16 | 1.73 | 0.29 | 7.71 | 2.99 |
| 2 | −7.279 | −3.572 | 3.71 | 7.28 | 3.57 | 5.43 | −5.43 | 1.85 | 0.27 | 7.94 | 2.93 |
| 3 | −6.229 | −3.599 | 2.63 | 6.23 | 3.60 | 4.91 | −4.91 | 1.32 | 0.38 | 9.18 | 3.74 |
| 4 | −7.226 | −3.488 | 3.74 | 7.23 | 3.49 | 5.36 | −5.36 | 1.87 | 0.27 | 7.68 | 2.87 |
| 5 | −7.081 | −3.429 | 3.65 | 7.08 | 3.43 | 5.26 | −5.26 | 1.83 | 0.27 | 7.56 | 2.88 |
| 6 | −7.035 | −3.493 | 3.54 | 7.04 | 3.49 | 5.26 | −5.26 | 1.77 | 0.28 | 7.82 | 2.97 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merecz-Sadowska, A.; Isca, V.M.S.; Sitarek, P.; Kowalczyk, T.; Małecka, M.; Zajdel, K.; Zielińska-Bliźniewska, H.; Jęcek, M.; Rijo, P.; Zajdel, R. Exploring the Anticancer Potential of Semisynthetic Derivatives of 7α-Acetoxy-6β-hydroxyroyleanone from Plectranthus sp.: An In Silico Approach. Int. J. Mol. Sci. 2024, 25, 4529. https://doi.org/10.3390/ijms25084529
Merecz-Sadowska A, Isca VMS, Sitarek P, Kowalczyk T, Małecka M, Zajdel K, Zielińska-Bliźniewska H, Jęcek M, Rijo P, Zajdel R. Exploring the Anticancer Potential of Semisynthetic Derivatives of 7α-Acetoxy-6β-hydroxyroyleanone from Plectranthus sp.: An In Silico Approach. International Journal of Molecular Sciences. 2024; 25(8):4529. https://doi.org/10.3390/ijms25084529
Chicago/Turabian StyleMerecz-Sadowska, Anna, Vera M. S. Isca, Przemysław Sitarek, Tomasz Kowalczyk, Magdalena Małecka, Karolina Zajdel, Hanna Zielińska-Bliźniewska, Mariusz Jęcek, Patricia Rijo, and Radosław Zajdel. 2024. "Exploring the Anticancer Potential of Semisynthetic Derivatives of 7α-Acetoxy-6β-hydroxyroyleanone from Plectranthus sp.: An In Silico Approach" International Journal of Molecular Sciences 25, no. 8: 4529. https://doi.org/10.3390/ijms25084529