
The Pathophysiological, Genetic, and Hormonal Changes in Preeclampsia: A Systematic Review of the Molecular Mechanisms


Abstract
:1. Introduction
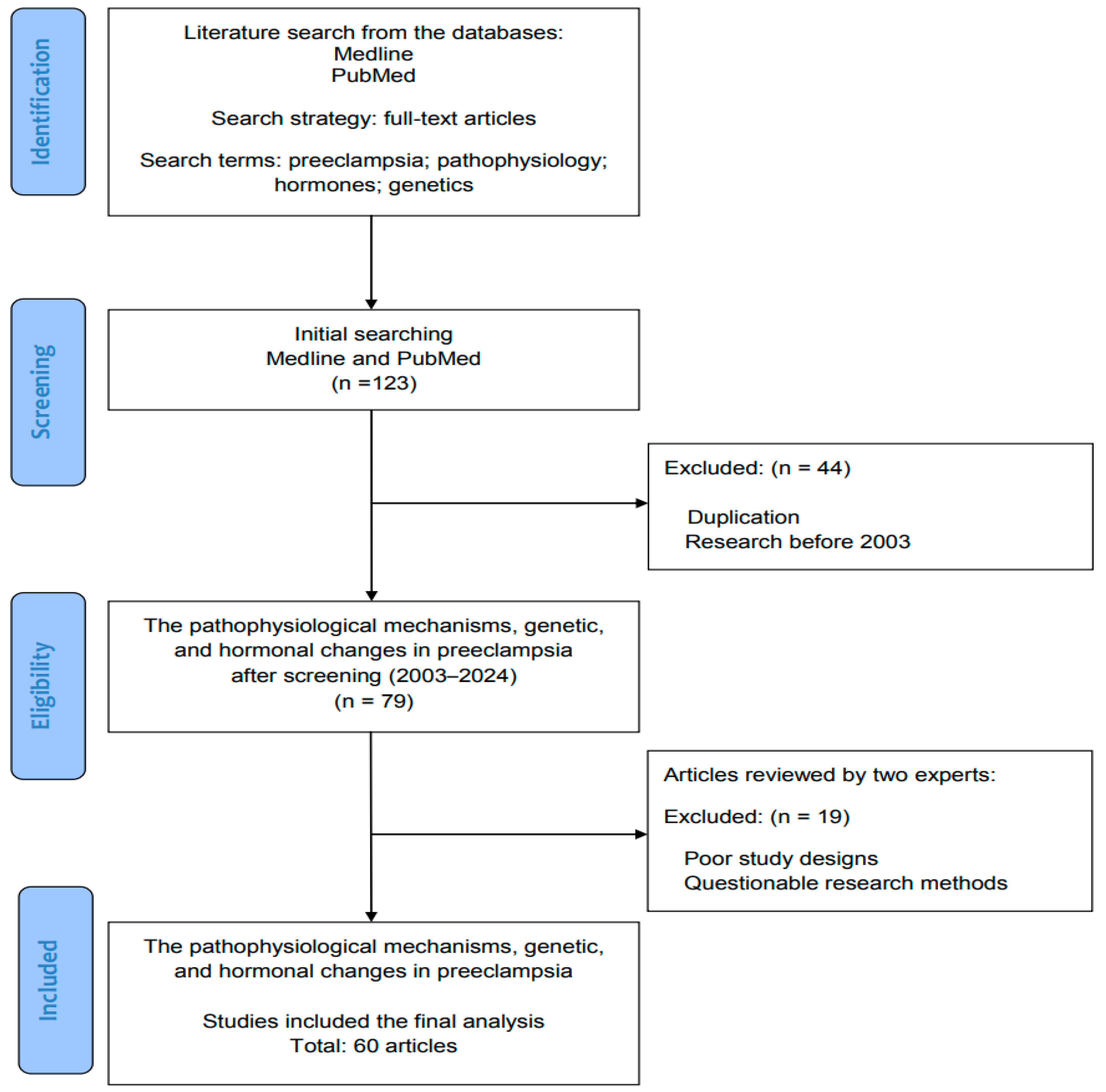
2. Methods: Literature Review and Data Search
Search Terms and Strategies in the Literature
3. The Pathophysiology and Placental Changes in Preeclampsia
3.1. The Two-Stage Model of Preeclampsia
3.2. Placental Findings in Preterm and Term Preeclampsia: The Pathological Changes in the Placenta
3.3. The Roles of Medications in Preeclampsia: Assessment of the Proton Pump Inhibitors (PPIs) Esomeprazole Magnesium Hydrate and Trihydrate
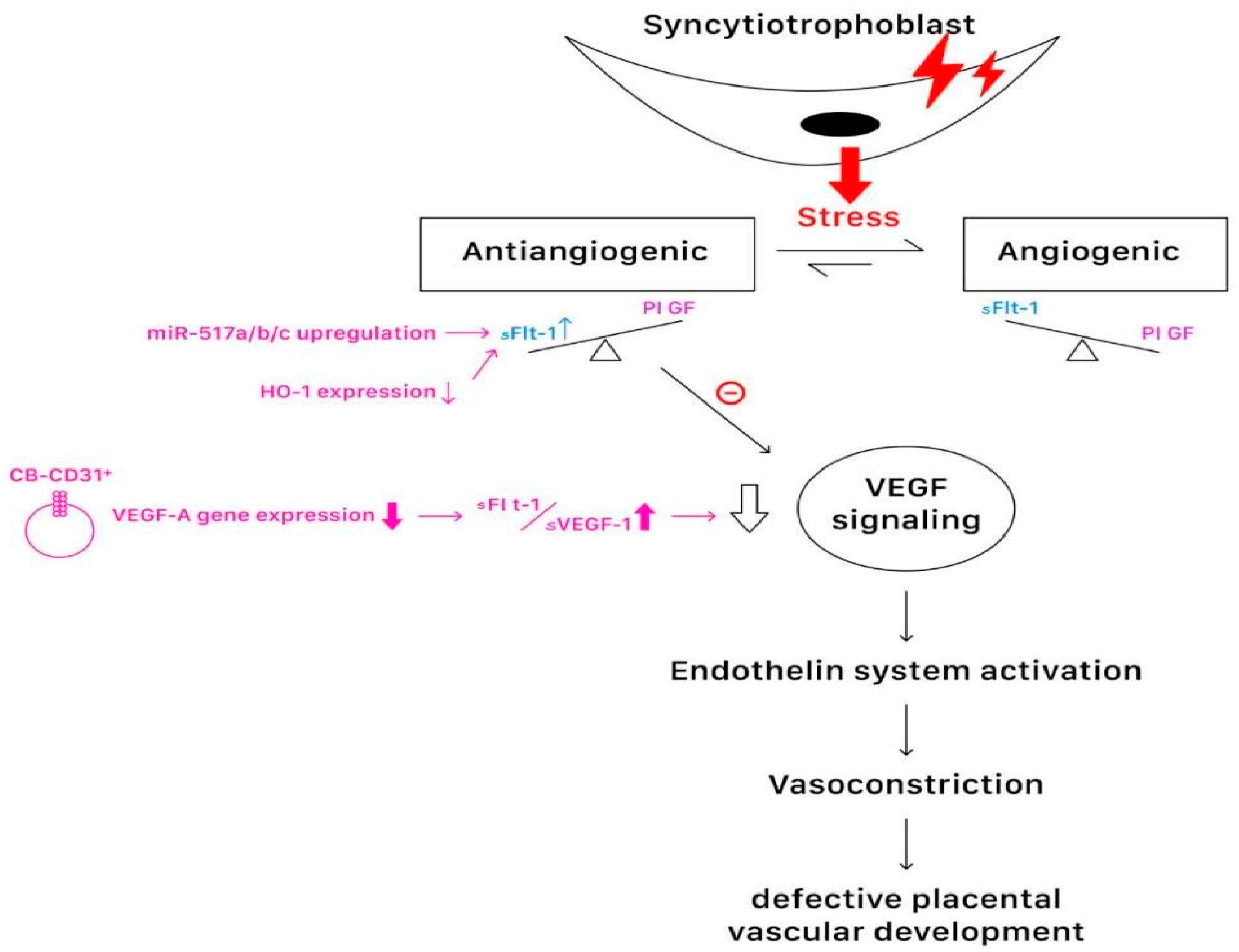
4. Genetic and Hormonal Changes in Preeclampsia
4.1. The Genetic Nature of Preeclampsia
4.1.1. Circular RNA (CircRNA)
4.1.2. microRNA (miRNA)
4.1.3. Long Non-Coding RNA (lncRNA)
4.1.4. CD Genes and Intrauterine Growth Retardation (IUGR)
4.2. The Molecular and Cellular Effects of Hormones, Complements, and Cytokines in Preeclampsia
4.2.1. Fetal Microchimerism in the Two-Stage Model of Preeclampsia
4.2.2. The Roles of Hormones, Complements, and Cytokines in Preeclampsia
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Steegers, E.A.; von Dadelszen, P.; Duvekot, J.J.; Pijnenborg, R. Pre-eclampsia. Lancet 2010, 376, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.; Romero, R.; Yeo, L.; Gomez-Lopez, N.; Chaemsaithong, P.; Jaovisidha, A.; Gotsch, F.; Erez, O. The etiology of preeclampsia. Am. J. Obstet. Gynecol. 2022, 226, S844–S866. [Google Scholar] [CrossRef] [PubMed]
- Ramos, J.G.L.; Sass, N.; Costa, S.H.M. Preeclampsia. Rev. Bras. Ginecol. Obstet. 2017, 39, 496–512. [Google Scholar] [CrossRef] [PubMed]
- Michalczyk, M.; Celewicz, A.; Celewicz, M.; Woźniakowska-Gondek, P.; Rzepka, R. The Role of Inflammation in the Pathogenesis of Preeclampsia. Mediators Inflamm. 2020, 2020, 3864941. [Google Scholar] [CrossRef]
- Inversett, A.; Pivato, C.A.; Cristodoro, M.; Latini, A.C.; Condorelli, G.; Di Simone, N.; Stefanini, G. Update on long-term cardiovascular risk after pre-eclampsia: A systematic review and meta-analysis. Eur. Heart J. Qual. Care Clin. Outcomes 2024, 10, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.J.; Seow, K.M.; Chen, K.H. Preeclampsia: Recent advances in predicting, preventing, and managing the maternal and fetal life-threatening condition. Int. J. Environ. Res. Public. Health 2023, 20, 2994. [Google Scholar] [CrossRef] [PubMed]
- Pankiewicz, K.; Fijałkowska, A.; Issat, T.; Maciejewski, T.M. Insight into the Key Points of Preeclampsia Pathophysiology: Uterine Artery Remodeling and the Role of MicroRNAs. Int. J. Mol. Sci. 2021, 22, 3132. [Google Scholar] [CrossRef]
- Redman, C.W.; Sargent, I.L.; Staff, A.C. IFPA Senior Award Lecture: Making sense of pre-eclampsia—Two placental causes of preeclampsia? Placenta 2014, 35, S20–S25. [Google Scholar] [CrossRef]
- Kornacki, J.; Olejniczak, O.; Sibiak, R.; Gutaj, P.; Wender-Ożegowska, E. Pathophysiology of Pre-Eclampsia-Two Theories of the Development of the Disease. Int. J. Mol. Sci. 2023, 25, 307. [Google Scholar] [CrossRef]
- Tossetta, G.; Fantone, S.; Piani, F.; Crescimanno, C.; Ciavattini, A.; Giannubilo, S.R.; Marzioni, D. Modulation of NRF2/KEAP1 Signaling in Preeclampsia. Cells 2023, 12, 1545. [Google Scholar] [CrossRef]
- Roberts, J.M.; Hubel, C.A. The two stage model of preeclampsia: Variations on the theme. Placenta 2009, 30 (Suppl. A), S32–S37. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.; Rich-Edwards, J.W.; McElrath, T.F.; Garmire, L.; Myatt, L. Subtypes of Preeclampsia: Recognition and Determining Clinical Usefulness. Hypertension 2021, 77, 1430–1441. [Google Scholar] [CrossRef] [PubMed]
- Staff, A.C. The two-stage placental model of preeclampsia: An update. J. Reprod. Immunol. 2019, 134, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, P.; Black, S.; Huppertz, B. Endovascular trophoblast invasion: Implications for the pathogenesis of intrauterine growth retardation and preeclampsia. Biol. Reprod. 2003, 69, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Fujiwara, H.; Konishi, I. Mechanism of maternal vascular remodeling during human pregnancy. Reprod. Med. Biol. 2012, 11, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Ridder, A.; Giorgione, V.; Khalil, A.; Thilaganathan, B. Preeclampsia: The relationship between uterine artery blood flow and trophoblast function. Int. J. Mol. Sci. 2019, 20, 3263. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.; Gammill, H.S. Preeclampsia: Recent insights. Hypertension 2005, 46, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Licini, C.; Avellini, C.; Picchiassi, E.; Mensà, E.; Fantone, S.; Ramini, D.; Tersigni, C.; Tossetta, G.; Castellucci, C.; Tarquini, F.; et al. Pre-eclampsia predictive ability of maternal miR-125b: A clinical and experimental study. Transl. Res. 2021, 228, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Tersigni, C.; Meli, F.; Neri, C.; Iacoangeli, A.; Franco, R.; Lanzone, A.; Scambia, G.; Di Simone, N. Role of Human Leukocyte Antigens at the Feto-Maternal Interface in Normal and Pathological Pregnancy: An Update. Int. J. Mol. Sci. 2020, 21, 4756. [Google Scholar] [CrossRef]
- Tossetta, G.; Fantone, S.; Giannubilo, S.R.; Marinelli-Busilacchi, E.; Ciavattini, A.; Castellucci, M.; Di Simone, N.; Mattioli-Belmonte, M.; Marzioni, D. Pre-eclampsia onset and SPARC: A possible involvement in placenta development. J. Cell Physiol. 2019, 234, 6091–6098. [Google Scholar] [CrossRef]
- Lamarca, B. Endothelial dysfunction: An important mediator in the pathophysiology of hypertension during pre-eclampsia. Minerva Ginecol. 2012, 64, 309–320. [Google Scholar]
- Boeldt, D.S.; Bird, I.M. Vascular adaptation in pregnancy and endothelial dysfunction in preeclampsia. J. Endocrinol. 2017, 232, R27–R44. [Google Scholar] [CrossRef] [PubMed]
- Herse, F.; Dechend, R.; Harsem, N.K.; Wallukat, G.; Janke, J.; Qadri, F.; Hering, L.; Muller, D.N.; Luft, F.C.; Staff, A.C. Dysregulation of the circulating and tissue-based renin-angiotensin system in preeclampsia. Hypertension 2007, 49, 604–611. [Google Scholar] [CrossRef]
- Irani, R.A.; Zhang, Y.; Zhou, C.C.; Blackwell, S.C.; Hicks, M.J.; Ramin, S.M.; Kellems, R.E.; Xia, Y. Autoantibody-mediated angiotensin receptor activation contributes to preeclampsia through tumor necrosis factor-alpha signaling. Hypertension 2010, 55, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Silva, B.R.; Pernomian, L.; Bendhack, L.M. Contribution of oxidative stress to endothelial dysfunction in hypertension. Front. Physiol. 2012, 3, 441. [Google Scholar] [CrossRef] [PubMed]
- Pietro, L.; Guida, J.P.S.; Nobrega, G.M.; Antolini-Tavares, A.; Costa, M.L. Placental Findings in Preterm and Term Preeclampsia: An Integrative Review of the Literature. Rev. Bras. Ginecol. Obstet. 2021, 43, 560–569. [Google Scholar] [CrossRef] [PubMed]
- de Alwis, N.; Fato, B.R.; Beard, S.; Binder, N.K.; Kaitu’u-Lino, T.J.; Onda, K.; Hannan, N.J. Assessment of the Proton Pump Inhibitor, Esomeprazole Magnesium Hydrate and Trihydrate, on Pathophysiological Markers of Preeclampsia in Preclinical Human Models of Disease. Int. J. Mol. Sci. 2022, 23, 9533. [Google Scholar] [CrossRef]
- Gill, S.K.; O’Brien, L.; Einarson, T.R.; Koren, G. The safety of proton pump inhibitors (PPIs) in pregnancy: A meta-analysis. Am. J. Gastroenterol. 2009, 104, 1541–1545. [Google Scholar] [CrossRef]
- Hastie, R.; Bergman, L.; Cluver, C.A.; Wikman, A.; Hannan, N.J.; Walker, S.P.; Wikström, A.K.; Tong, S.; Hesselman, S. Proton Pump Inhibitors and Preeclampsia Risk among 157 720 Women. Hypertension 2019, 73, 1097–1103. [Google Scholar] [CrossRef]
- Hussain, S.; Singh, A.; Antony, B.; Klugarová, J.; Murad, M.H.; Jayraj, A.S.; Langaufová, A.; Klugar, M. Proton Pump Inhibitors Use and Risk of Preeclampsia: A Meta-Analysis. J. Clin. Med. 2022, 11, 4675. [Google Scholar] [CrossRef]
- Matok, I.; Levy, A.; Wiznitzer, A.; Uziel, E.; Koren, G.; Gorodischer, R. The safety of fetal exposure to proton-pump inhibitors during pregnancy. Dig. Dis. Sci. 2012, 57, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, B.; Hviid, A. Use of proton-pump inhibitors in early pregnancy and the risk of birth defects. N. Engl. J. Med. 2010, 363, 2114–2123. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Kaitu’u-Lino, T.J.; Hastie, R.; Brownfoot, F.; Cluver, C.; Hannan, N. Pravastatin, proton-pump inhibitors, metformin, micronutrients, and biologics: New horizons for the prevention or treatment of preeclampsia. Am. J. Obstet. Gynecol. 2022, 226, S1157–S1170. [Google Scholar] [CrossRef] [PubMed]
- Bello, N.A.; Huang, Y.; Syeda, S.K.; Wright, J.D.; D’Alton, M.E.; Friedman, A.M. Receipt of Proton-Pump Inhibitors during Pregnancy and Risk for Preeclampsia. Am. J. Perinatol. 2021, 38, 1519–1525. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.; Noh, Y.; Park, S.H.; Choe, S.A.; Shin, J.Y. Exploration of Proton Pump Inhibitors Use during Pregnancy and Preeclampsia. JAMA Netw. Open 2021, 4, e2124339. [Google Scholar] [CrossRef] [PubMed]
- Cluver, C.A.; Hannan, N.J.; van Papendorp, E.; Hiscock, R.; Beard, S.; Mol, B.W.; Theron, G.B.; Hall, D.R.; Decloedt, E.H.; Stander, M.; et al. Esomeprazole to treat women with preterm preeclampsia: A randomized placebo controlled trial. Am. J. Obstet. Gynecol. 2018, 219, 388.e1–388.e17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, Y.; Yu, F.; Li, X.; Chen, X.; Zhu, D.; Sun, J.; Huang, Q.; Li, M.; Sun, M.; et al. CircRNA_06354 might promote early-onset preeclampsia in humans via hsa-miR-92a-3p/vascular endothelial growth factor-A. J. Hypertens. 2023, 41, 494–507. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Sun, J.; Groome, L.J.; Wang, Y. Differential miRNA expression profiles between the first and third trimester human placentas. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E836–E843. [Google Scholar] [CrossRef] [PubMed]
- Munjas, J.; Sopić, M.; Stefanović, A.; Košir, R.; Ninić, A.; Joksić, I.; Antonić, T.; Spasojević-Kalimanovska, V.; Prosenc Zmrzljak, U. Non-Coding RNAs in Preeclampsia-Molecular Mechanisms and Diagnostic Potential. Int. J. Mol. Sci. 2021, 22, 10652. [Google Scholar] [CrossRef]
- Chen, D.B.; Wang, W. Human placental microRNAs and preeclampsia. Biol. Reprod. 2013, 88, 130. [Google Scholar] [CrossRef]
- Dai, C.; Zhao, C.; Xu, M.; Sui, X.; Sun, L.; Liu, Y.; Su, M.; Wang, H.; Yuan, Y.; Zhang, S.; et al. Serum lncRNAs in early pregnancy as potential biomarkers for the prediction of pregnancy-induced hypertension, including preeclampsia. Mol. Ther. Nucleic Acids 2021, 24, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Medina-Bastidas, D.; Guzmán-Huerta, M.; Borboa-Olivares, H.; Ruiz-Cruz, C.; Parra-Hernández, S.; Flores-Pliego, A.; Salido-Guadarrama, I.; Camargo-Marín, L.; Arambula-Meraz, E.; Estrada-Gutierrez, G. Placental Microarray Profiling Reveals Common mRNA and lncRNA Expression Patterns in Preeclampsia and Intrauterine Growth Restriction. Int. J. Mol. Sci. 2020, 21, 3597. [Google Scholar] [CrossRef] [PubMed]
- Schiessl, B.; Mylonas, I.; Hantschmann, P.; Kuhn, C.; Schulze, S.; Kunze, S.; Friese, K.; Jeschke, U. Expression of endothelial NO synthase, inducible NO synthase, and estrogen receptors alpha and beta in placental tissue of normal, preeclamptic, and intrauterine growth-restricted pregnancies. J. Histochem. Cytochem. 2005, 53, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Sohn, Y.D.; Kim, S.; Rajakumar, A.; Badell, M.L.; Sidell, N.; Yoon, Y.S. Reduced angiovasculogenic and increased inflammatory profiles of cord blood cells in severe but not mild preeclampsia. Sci. Rep. 2021, 11, 3630. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, T.M.; Walker, S.P.; Hannan, N.J.; Tong, S.; Kaitu’u-Lino, T.J. Clinical tools and biomarkers to predict preeclampsia. EBioMedicine 2022, 75, 103780. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Lemoine, E.; Granger, J.P.; Karumanchi, S.A. Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circ. Res. 2019, 124, 1094–1112. [Google Scholar] [CrossRef] [PubMed]
- Tomimatsu, T.; Mimura, K.; Matsuzaki, S.; Endo, M.; Kumasawa, K.; Kimura, T. Preeclampsia: Maternal Systemic Vascular Disorder Caused by Generalized Endothelial Dysfunction due to Placental Antiangiogenic Factors. Int. J. Mol. Sci. 2019, 20, 4246. [Google Scholar] [CrossRef]
- Flint, E.J.; Cerdeira, A.S.; Redman, C.W.; Vatish, M. The role of angiogenic factors in the management of preeclampsia. Acta Obstet. Gynecol. Scand. 2019, 98, 700–707. [Google Scholar] [CrossRef]
- Tomimatsu, T.; Mimura, K.; Endo, M.; Kumasawa, K.; Kimura, T. Pathophysiology of preeclampsia: An angiogenic imbalance and long-lasting systemic vascular dysfunction. Hypertens. Res. 2017, 40, 305–310. [Google Scholar] [CrossRef]
- Jena, M.K.; Sharma, N.R.; Petitt, M.; Maulik, D.; Nayak, N.R. Pathogenesis of Preeclampsia and Therapeutic Approaches Targeting the Placenta. Biomolecules 2020, 10, 953. [Google Scholar] [CrossRef]
- Youssef, L.; Miranda, J.; Blasco, M.; Paules, C.; Crovetto, F.; Palomo, M.; Torramade-Moix, S.; García-Calderó, H.; Tura-Ceide, O.; Dantas, A.P.; et al. Complement and coagulation cascades activation is the main pathophysiological pathway in early-onset severe preeclampsia revealed by maternal proteomics. Sci. Rep. 2021, 11, 3048. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, D.P.; Lekva, T.; Moe, K.; Fjeldstad, H.E.S.; Johnsen, G.M.; Sugulle, M.; Staff, A.C. Pregnancy and postpartum levels of circulating maternal sHLA-G in preeclampsia. J. Reprod. Immunol. 2021, 143, 103249. [Google Scholar] [CrossRef] [PubMed]
- Kenchegowda, D.; Natale, B.; Lemus, M.A.; Natale, D.R.; Fisher, S.A. Inactivation of maternal Hif-1α at mid-pregnancy causes placental defects and deficits in oxygen delivery to the fetal organs under hypoxic stress. Dev. Biol. 2017, 422, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Albers, R.E.; Kaufman, M.R.; Natale, B.V.; Keoni, C.; Kulkarni-Datar, K.; Min, S.; Williams, C.R.; Natale, D.R.C.; Brown, T.L. Trophoblast-Specific Expression of Hif-1α Results in Preeclampsia-Like Symptoms and Fetal Growth Restriction. Sci. Rep. 2019, 9, 2742. [Google Scholar] [CrossRef] [PubMed]
- Küssel, L.; Herkner, H.; Wahrmann, M.; Eskandary, F.; Doberer, K.; Binder, J.; Pateisky, P.; Zeisler, H.; Böhmig, G.A.; Bond, G. Longitudinal assessment of HLA and MIC-A antibodies in uneventful pregnancies and pregnancies complicated by preeclampsia or gestational diabetes. Sci. Rep. 2017, 7, 13524. [Google Scholar] [CrossRef] [PubMed]
- Espino, Y.S.S.; Flores-Pliego, A.; Espejel-Nuñez, A.; Medina-Bastidas, D.; Vadillo-Ortega, F.; Zaga-Clavellina, V.; Estrada-Gutierrez, G. New Insights into the Role of Matrix Metalloproteinases in Preeclampsia. Int. J. Mol. Sci. 2017, 18, 1448. [Google Scholar] [CrossRef] [PubMed]
- Saleh, L.; Danser, J.A.; van den Meiracker, A.H. Role of endothelin in preeclampsia and hypertension following antiangiogenesis treatment. Curr. Opin. Nephrol. Hypertens. 2016, 25, 94–99. [Google Scholar] [CrossRef]
- Weissgerber, T.L.; Milic, N.M.; Milin-Lazovic, J.S.; Garovic, V.D. Impaired Flow-Mediated Dilation before, during, and after Preeclampsia: A Systematic Review and Meta-Analysis. Hypertension 2016, 67, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Gui, S.; Ni, S.; Jia, J.; Gong, Y.; Gao, L.; Zhang, L.; Zhou, R. Inconformity of CXCL3 plasma level and placenta expression in preeclampsia and its effect on trophoblast viability and invasion. PLoS ONE 2014, 9, e114408. [Google Scholar] [CrossRef]
- Asvold, B.O.; Eskild, A.; Vatten, L.J. Human chorionic gonadotropin, angiogenic factors, and preeclampsia risk: A nested case-control study. Acta Obstet. Gynecol. Scand. 2014, 93, 454–462. [Google Scholar] [CrossRef]
- Drost, J.T.; Maas, A.H.; Holewijn, S.; Joosten, L.A.; van Eyck, J.; van der Schouw, Y.T.; de Graaf, J. Novel cardiovascular biomarkers in women with a history of early preeclampsia. Atherosclerosis 2014, 237, 117–122. [Google Scholar] [CrossRef]
- Shomer, E.; Katzenell, S.; Zipori, Y.; Sammour, R.N.; Isermann, B.; Brenner, B.; Aharon, A. Microvesicles of women with gestational hypertension and preeclampsia affect human trophoblast fate and endothelial function. Hypertension 2013, 62, 893–898. [Google Scholar] [CrossRef]
- Ashur-Fabian, O.; Yerushalmi, G.M.; Mazaki-Tovi, S.; Steinberg, D.M.; Goldshtein, I.; Yackobovitch-Gavan, M.; Schiff, E.; Amariglio, N.; Rechavi, G. Cell free expression of hif1α and p21 in maternal peripheral blood as a marker for preeclampsia and fetal growth restriction. PLoS ONE 2012, 7, e37273. [Google Scholar] [CrossRef] [PubMed]
- Carty, D.M.; Anderson, L.A.; Freeman, D.J.; Welsh, P.I.; Brennand, J.E.; Dominiczak, A.F.; Delles, C. Early pregnancy soluble E-selectin concentrations and risk of preeclampsia. J. Hypertens. 2012, 30, 954–959. [Google Scholar] [CrossRef]
- Krönke, G.; Kadl, A.; Ikonomu, E.; Blüml, S.; Fürnkranz, A.; Sarembock, I.J.; Bochkov, V.N.; Exner, M.; Binder, B.R.; Leitinger, N. Expression of heme oxygenase-1 in human vascular cells is regulated by peroxisome proliferator-activated receptors. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, F.P.; Drewlo, S.; Kingdom, J.; Johns, E.J.; Walsh, S.K.; Kenny, L.C. Peroxisome proliferator-activated receptor-γ as a potential therapeutic target in the treatment of preeclampsia. Hypertension 2011, 58, 280–286. [Google Scholar] [CrossRef]
- Kvehaugen, A.S.; Dechend, R.; Ramstad, H.B.; Troisi, R.; Fugelseth, D.; Staff, A.C. Endothelial function and circulating biomarkers are disturbed in women and children after preeclampsia. Hypertension 2011, 58, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Stepanian, A.; Benchenni, S.; Beillat-Lucas, T.; Omnes, S.; Defay, F.; Peynaud-Debayle, E.; Baron, G.; Le Querrec, A.; Dreyfus, M.; Salomon, L.; et al. Search for an association between V249I and T280M CX3CR1 genetic polymorphisms, endothelial injury and preeclampsia: The ECLAXIR study. PLoS ONE 2009, 4, e6192. [Google Scholar] [CrossRef]
- Goldman-Wohl, D.; Yagel, S. Preeclampsia—A placenta developmental biology perspective. J. Reprod. Immunol. 2009, 82, 96–99. [Google Scholar] [CrossRef]
- Xia, Y.; Kellems, R.E. Is preeclampsia an autoimmune disease? Clin. Immunol. 2009, 133, 1–12. [Google Scholar] [CrossRef]
- Meng, T.; Chen, H.; Sun, M.; Wang, H.; Zhao, G.; Wang, X. Identification of differential gene expression profiles in placentas from preeclamptic pregnancies versus normal pregnancies by DNA microarrays. Omics 2012, 16, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Peraçoli, M.T.; Menegon, F.T.; Borges, V.T.; de Araújo Costa, R.A.; Thomazini-Santos, I.A.; Peraçoli, J.C. Platelet aggregation and TGF-beta(1) plasma levels in pregnant women with preeclampsia. J. Reprod. Immunol. 2008, 79, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.J.; Gumaa, K.; Scioscia, M.; Redman, C.W.; Rademacher, T.W. Inositol phosphoglycan P-type in preeclampsia: A novel marker? Hypertension 2007, 49, 84–89. [Google Scholar] [CrossRef]
- Žák, P.; Souček, M. Correlation of tumor necrosis factor alpha, interleukin 6 and interleukin 10 with blood pressure, risk of preeclampsia and low birth weight in gestational diabetes. Physiol. Res. 2019, 68, 395–408. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | Possible Mechanism | Phenomenon and Biological Responses to AT1-AAs in Preeclamptic Women |
|---|---|---|
| Cardiomyocytes in rats | Activate AT1 angiotensin receptors | Increased heart rate |
| Vascular smooth muscle cells | Elevated expression of TF | Hypercoagulation |
| Activation of NADPH oxidase | ROS production | |
| Trophoblast cells | Stimulate the synthesis of plasminogen activator inhibitors (PAI) -1 | Poor trophoblast invasion and impaired spiral artery remodeling |
| Activate AT1 angiotensin receptors | Elevated sFlt1 | |
| Mesangial cells | Stimulate release of IL-6 | Activates proinflammatory response, resulting in renal damage |
| Stimulate release of PAI-1 | Disseminated intravascular coagulation present in the glomerular structures | |
| Monocytes | Produce increased amounts of TF | Monocyte activation and their increased adherence to endothelial cells |
| Platelets, erythrocytes, and lymphocytes | Abnormalities in Ca2+ metabolism |
| Aspect | Key Points of Results |
|---|---|
| Pathophysiology | Fetal microchimerism links placental dysfunction to maternal issues. Key factors include hormones, cytokines, Hif-α dysfunction, MMPs, ETs, and chemokines. |
| Genetics | CircRNA_06354 impacts early-onset preeclampsia. C19MC miRNAs and lncRNAs like IGFBP1 and EGFR-AS1 affect trophoblast function and angiogenesis. |
| Potential biomarkers | CD31+ cells, MMPs, soluble HLA-G, and hCG |
| Therapeutic Interventions | Targeting PPAR-γ and endothelin receptors may mitigate complications. Further studies are needed for clinical validation. |
| Aspect | Key Points of Conclusions and Insights |
|---|---|
| Pathophysiology | Fetal microchimerism acts as a bridge between placental dysfunction and maternal cardiovascular issues, with hormones, cytokines, Hif-α dysfunction, MMPs, ETs, and chemokines playing key roles. ETs and sFlt-1/PlGF imbalance indicate an angiogenic imbalance and endothelial dysfunction, central to the progression of the disease. Flow-mediated dilation, as an endothelial function indicator, is lower in women with preeclampsia, suggesting persistent endothelial dysfunction. CXCL3 elevation suggests its role in severe preeclampsia and shallow trophoblast implantation. |
| Genetics | CircRNA_06354 impacts early-onset preeclampsia by inhibiting trophoblastic cell invasion through the hsa-miR-92a-3p/VEGF-A pathway. C19MC miRNAs and lncRNAs like IGFBP1 and EGFR-AS1 affect trophoblast function and angiogenesis, influencing PE and IUGR. Shared gene dysregulation between PE and IUGR involves immune and inflammatory responses, highlighting the complexity of genetic influences in preeclampsia. |
| Potential biomarkers | CD31+ cells, MMPs, soluble HLA-G, and hCG serve as potential biomarkers for preeclampsia. hCG and PIGF levels across different pregnancy stages have predictive value for preeclampsia, offering early diagnosis opportunities. The elevation of cardiovascular biomarkers, such as SE selectin and PAPPA, highlights an increased risk for cardiovascular disease post-preeclampsia. MVs reflect the pathophysiological state in gestational vascular complications, marking their role in the signaling pathway and their potential as biomarkers. |
| Therapeutic interventions | PPAR-γ activation suggests a protective mechanism against pathophysiological characteristics of preeclampsia, highlighting the importance of targeted therapeutic strategies. Endothelial dysfunction interventions and monitoring are crucial for long-term health, given the persistent endothelial and cardiovascular risks after preeclampsia. This review offers genetic and molecular insights. Understanding the nuances of genetic polymorphisms and molecular interactions, such as CX3CR1 polymorphisms and HLA-G expression, paves the way for precision medicine in preeclampsia prevention and treatment, highlighting the importance of precise therapeutic interventions based on a deep molecular understanding. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiang, Y.-T.; Seow, K.-M.; Chen, K.-H. The Pathophysiological, Genetic, and Hormonal Changes in Preeclampsia: A Systematic Review of the Molecular Mechanisms. Int. J. Mol. Sci. 2024, 25, 4532. https://doi.org/10.3390/ijms25084532
Chiang Y-T, Seow K-M, Chen K-H. The Pathophysiological, Genetic, and Hormonal Changes in Preeclampsia: A Systematic Review of the Molecular Mechanisms. International Journal of Molecular Sciences. 2024; 25(8):4532. https://doi.org/10.3390/ijms25084532
Chicago/Turabian StyleChiang, Yi-Ting, Kok-Min Seow, and Kuo-Hu Chen. 2024. "The Pathophysiological, Genetic, and Hormonal Changes in Preeclampsia: A Systematic Review of the Molecular Mechanisms" International Journal of Molecular Sciences 25, no. 8: 4532. https://doi.org/10.3390/ijms25084532