
OMA1-Mediated Mitochondrial Dynamics Balance Organellar Homeostasis Upstream of Cellular Stress Responses

{kind=link}
{kind=link}
Abstract
:1. Mitochondrial Organization Mediates Organellar Function
2. Mitochondrial Fission Is Mediated by Recruitment of Membrane-Shaping Proteins
3. OMA1 Links Mitochondrial Fusion with Bioenergetic Function
4. OMA1 Impacts on the Cell at Large: Apoptosis, Autophagy, and Integrated Stress Response
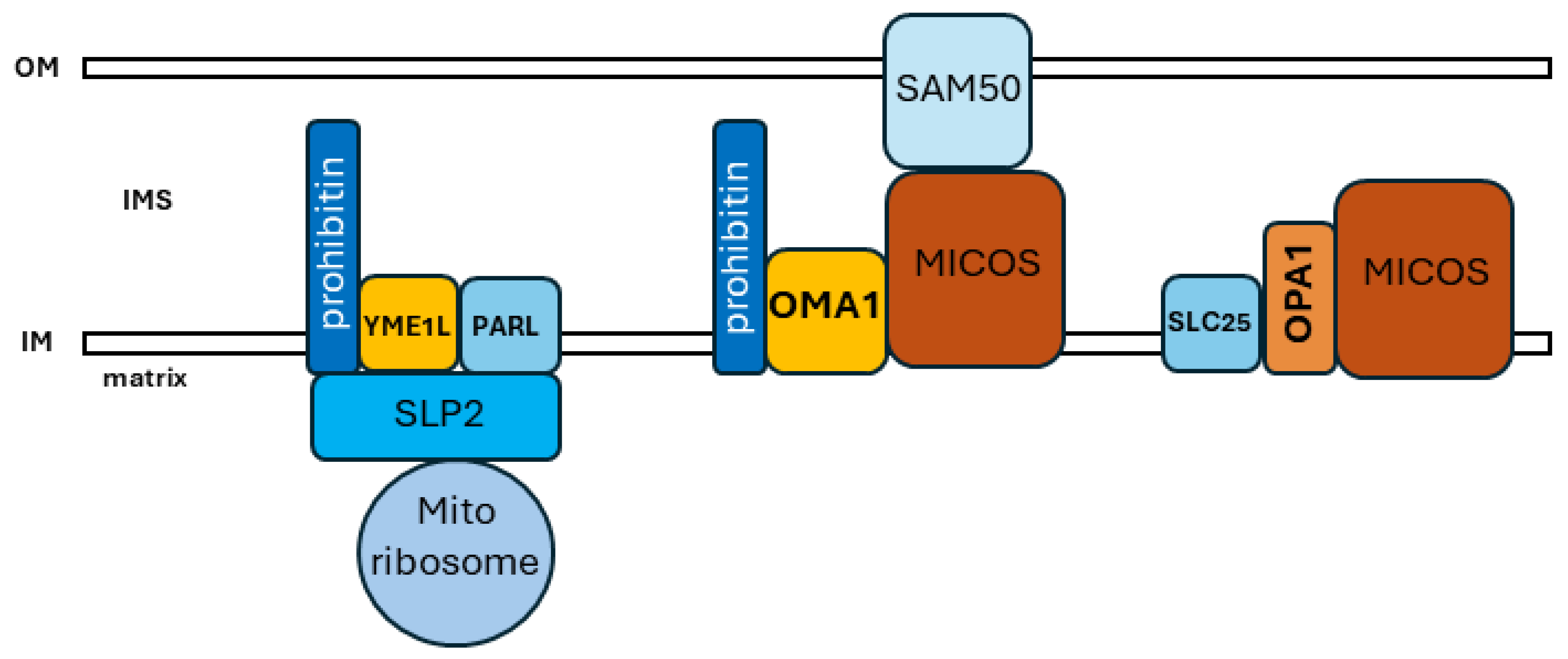
5. Interactions, Domains, and Physiology within the Organellar Network
Funding
Acknowledgments
Conflicts of Interest
References
- Sjostrand, F.S. Electron microscopy of mitochondria and cytoplasmic double membranes. Nature 1953, 171, 30–32. [Google Scholar] [CrossRef] [PubMed]
- Palade, G.E. The fine structure of mitochondria. Anat. Rec. 1952, 114, 427–451. [Google Scholar] [CrossRef] [PubMed]
- Frey, T.G.; Mannella, C.A. The internal structure of mitochondria. Trends Biochem. Sci. 2000, 25, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Perkins, G.; Renken, C.; Martone, M.E.; Young, S.J.; Ellisman, M.; Frey, T. Electron tomography of neuronal mitochondria: Three-dimensional structure and organization of cristae and membrane contacts. J. Struct. Biol. 1997, 119, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Elstner, M.; Andreoli, C.; Klopstock, T.; Meitinger, T.; Prokisch, H. The mitochondrial proteome database: MitoP2. Methods Enzymol. 2009, 457, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.W.; Fahy, E.; Zhang, B.; Glenn, G.M.; Warnock, D.E.; Wiley, S.; Murphy, A.N.; Gaucher, S.P.; Capaldi, R.A.; Gibson, B.W.; et al. Characterization of the human heart mitochondrial proteome. Nat. Biotechnol. 2003, 21, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Gibson, B.W. The human mitochondrial proteome: Oxidative stress, protein modifications and oxidative phosphorylation. Int. J. Biochem. Cell Biol. 2005, 37, 927–934. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Schon, E.A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A. Bioenergetics through thick and thin. Science 2018, 362, 1114–1115. [Google Scholar] [CrossRef] [PubMed]
- Letts, J.A.; Fiedorczuk, K.; Sazanov, L.A. The architecture of respiratory supercomplexes. Nature 2016, 537, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Dudkina, N.V.; Kudryashev, M.; Stahlberg, H.; Boekema, E.J. Interaction of complexes I, III, and IV within the bovine respirasome by single particle cryoelectron tomography. Proc. Natl. Acad. Sci. USA 2011, 108, 15196–15200. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Gu, J.; Guo, R.; Huang, Y.; Yang, M. Structure of Mammalian Respiratory Supercomplex I(1)III(2)IV(1). Cell 2016, 167, 1598–1609.e10. [Google Scholar] [CrossRef] [PubMed]
- Minauro-Sanmiguel, F.; Wilkens, S.; Garcia, J.J. Structure of dimeric mitochondrial ATP synthase: Novel F0 bridging features and the structural basis of mitochondrial cristae biogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 12356–12358. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Parey, K.; Bublitz, M.; Mills, D.J.; Zickermann, V.; Vonck, J.; Kuhlbrandt, W.; Meier, T. Structure of a Complete ATP Synthase Dimer Reveals the Molecular Basis of Inner Mitochondrial Membrane Morphology. Mol. Cell 2016, 63, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Amchenkova, A.A.; Bakeeva, L.E.; Chentsov, Y.S.; Skulachev, V.P.; Zorov, D.B. Coupling membranes as energy-transmitting cables. I. Filamentous mitochondria in fibroblasts and mitochondrial clusters in cardiomyocytes. J. Cell Biol. 1988, 107, 481–495. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Kukat, C.; Wurm, C.A.; Spahr, H.; Falkenberg, M.; Larsson, N.G.; Jakobs, S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc. Natl. Acad. Sci. USA 2011, 108, 13534–13539. [Google Scholar] [CrossRef]
- Legros, F.; Malka, F.; Frachon, P.; Lombes, A.; Rojo, M. Organization and dynamics of human mitochondrial DNA. J. Cell Sci. 2004, 117 Pt 13, 2653–2662. [Google Scholar] [CrossRef]
- He, J.; Cooper, H.M.; Reyes, A.; Di Re, M.; Sembongi, H.; Litwin, T.R.; Gao, J.; Neuman, K.C.; Fearnley, I.M.; Spinazzola, A.; et al. Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucleic Acids Res. 2012, 40, 6109–6121. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.E.; White, K.; Davey, T.; Philips, J.; Ogden, R.T.; Lawless, C.; Warren, C.; Hall, M.G.; Ng, Y.S.; Falkous, G.; et al. Quantitative 3D Mapping of the Human Skeletal Muscle Mitochondrial Network. Cell Rep. 2019, 27, 321. [Google Scholar] [CrossRef] [PubMed]
- Martin, O.J.; Lai, L.; Soundarapandian, M.M.; Leone, T.C.; Zorzano, A.; Keller, M.P.; Attie, A.D.; Muoio, D.M.; Kelly, D.P. A role for peroxisome proliferator-activated receptor gamma coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ. Res. 2014, 114, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Soriano, F.X.; Liesa, M.; Bach, D.; Chan, D.C.; Palacin, M.; Zorzano, A. Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator-activated receptor-gamma coactivator-1 alpha, estrogen-related receptor-alpha, and mitofusin 2. Diabetes 2006, 55, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Korobova, F.; Gauvin, T.J.; Higgs, H.N. A role for myosin II in mammalian mitochondrial fission. Curr. Biol. 2014, 24, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.; Krueger, E.W.; Oswald, B.J.; McNiven, M.A. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell. Biol. 2003, 23, 5409–5420. [Google Scholar] [CrossRef] [PubMed]
- Gandre-Babbe, S.; van der Bliek, A.M. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell 2008, 19, 2402–2412. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Singh, A.P.; Stroud, D.A.; Palmer, C.S.; Stojanovski, D.; Ramachandran, R.; Ryan, M.T. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J. Cell Sci. 2016, 129, 2170–2181. [Google Scholar] [CrossRef]
- Palmer, C.S.; Elgass, K.D.; Parton, R.G.; Osellame, L.D.; Stojanovski, D.; Ryan, M.T. Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J. Biol. Chem. 2013, 288, 27584–27593. [Google Scholar] [CrossRef] [PubMed]
- Dickey, A.S.; Strack, S. PKA/AKAP1 and PP2A/Bbeta2 regulate neuronal morphogenesis via Drp1 phosphorylation and mitochondrial bioenergetics. J. Neurosci. 2011, 31, 15716–15726. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef]
- Merrill, R.A.; Dagda, R.K.; Dickey, A.S.; Cribbs, J.T.; Green, S.H.; Usachev, Y.M.; Strack, S. Mechanism of neuroprotective mitochondrial remodeling by PKA/AKAP1. PLoS Biol. 2011, 9, e1000612. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Westrate, L.M.; Wu, H.; Page, C.; Voeltz, G.K. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 2016, 540, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, T.B.; Sanchez-Guerrero, A.; Milosevic, I.; Raimundo, N. Mitochondrial fission requires DRP1 but not dynamins. Nature 2019, 570, E34–E42. [Google Scholar] [CrossRef] [PubMed]
- James, D.I.; Parone, P.A.; Mattenberger, Y.; Martinou, J.C. hFis1, a novel component of the mammalian mitochondrial fission machinery. J. Biol. Chem. 2003, 278, 36373–36379. [Google Scholar] [CrossRef] [PubMed]
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Favaro, G.; Romanello, V.; Varanita, T.; Andrea Desbats, M.; Morbidoni, V.; Tezze, C.; Albiero, M.; Canato, M.; Gherardi, G.; De Stefani, D.; et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 2019, 10, 2576. [Google Scholar] [CrossRef] [PubMed]
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001, 114 Pt 5, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.L.; Meng, S.; Chen, Y.; Feng, J.X.; Gu, D.D.; Yu, B.; Li, Y.J.; Yang, J.Y.; Liao, S.; Chan, D.C.; et al. MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature 2017, 542, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Cao, Y.L.; Feng, J.X.; Qi, Y.; Meng, S.; Yang, J.F.; Zhong, Y.T.; Kang, S.; Chen, X.; Lan, L.; et al. Structural insights of human mitofusin-2 into mitochondrial fusion and CMT2A onset. Nat. Commun. 2019, 10, 4914. [Google Scholar] [CrossRef] [PubMed]
- Olichon, A.; Emorine, L.J.; Descoins, E.; Pelloquin, L.; Brichese, L.; Gas, N.; Guillou, E.; Delettre, C.; Valette, A.; Hamel, C.P.; et al. The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 2002, 523, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Legros, F.; Lombes, A.; Frachon, P.; Rojo, M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol. Biol. Cell 2002, 13, 4343–4354. [Google Scholar] [CrossRef] [PubMed]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 2017, 19, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Baricault, L.; Segui, B.; Guegand, L.; Olichon, A.; Valette, A.; Larminat, F.; Lenaers, G. OPA1 cleavage depends on decreased mitochondrial ATP level and bivalent metals. Exp. Cell Res. 2007, 313, 3800–3808. [Google Scholar] [CrossRef] [PubMed]
- Griparic, L.; Kanazawa, T.; van der Bliek, A.M. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J. Cell Biol. 2007, 178, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Smith, S.B.; Sheu, S.S.; Yoon, Y. The short variant of optic atrophy 1 (OPA1) improves cell survival under oxidative stress. J. Biol. Chem. 2020, 295, 6543–6560. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Smith, S.B.; Yoon, Y. The short variant of the mitochondrial dynamin OPA1 maintains mitochondrial energetics and cristae structure. J. Biol. Chem. 2017, 292, 7115–7130. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, B.; Klec, C.; Leitinger, G.; Bernhart, E.; Rost, R.; Bischof, H.; Madreiter-Sokolowski, C.T.; Radulovic, S.; Eroglu, E.; Sattler, W.; et al. MICU1 controls cristae junction and spatially anchors mitochondrial Ca(2+) uniporter complex. Nat. Commun. 2019, 10, 3732. [Google Scholar] [CrossRef] [PubMed]
- Pfanner, N.; van der Laan, M.; Amati, P.; Capaldi, R.A.; Caudy, A.A.; Chacinska, A.; Darshi, M.; Deckers, M.; Hoppins, S.; Icho, T.; et al. Uniform nomenclature for the mitochondrial contact site and cristae organizing system. J. Cell Biol. 2014, 204, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Stephan, T.; Bruser, C.; Deckers, M.; Steyer, A.M.; Balzarotti, F.; Barbot, M.; Behr, T.S.; Heim, G.; Hubner, W.; Ilgen, P.; et al. MICOS assembly controls mitochondrial inner membrane remodeling and crista junction redistribution to mediate cristae formation. EMBO J. 2020, 39, e104105. [Google Scholar] [CrossRef] [PubMed]
- Glytsou, C.; Calvo, E.; Cogliati, S.; Mehrotra, A.; Anastasia, I.; Rigoni, G.; Raimondi, A.; Shintani, N.; Loureiro, M.; Vazquez, J.; et al. Optic Atrophy 1 Is Epistatic to the Core MICOS Component MIC60 in Mitochondrial Cristae Shape Control. Cell Rep. 2016, 17, 3024–3034. [Google Scholar] [CrossRef] [PubMed]
- Cadena, L.R.; Gahura, O.; Panicucci, B.; Zikova, A.; Hashimi, H. Mitochondrial Contact Site and Cristae Organization System and F(1)F(O)-ATP Synthase Crosstalk Is a Fundamental Property of Mitochondrial Cristae. mSphere 2021, 6, e00327-21. [Google Scholar] [CrossRef]
- Gilkerson, R.W.; Selker, J.M.; Capaldi, R.A. The cristal membrane of mitochondria is the principal site of oxidative phosphorylation. FEBS Lett. 2003, 546, 355–358. [Google Scholar] [CrossRef]
- Wolf, D.M.; Segawa, M.; Kondadi, A.K.; Anand, R.; Bailey, S.T.; Reichert, A.S.; van der Bliek, A.M.; Shackelford, D.B.; Liesa, M.; Shirihai, O.S. Individual cristae within the same mitochondrion display different membrane potentials and are functionally independent. EMBO J. 2019, 38, e101056. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Barrera, M.; Koob, S.; Dikov, D.; Vogel, F.; Reichert, A.S. OPA1 functionally interacts with MIC60 but is dispensable for crista junction formation. FEBS Lett. 2016, 590, 3309–3322. [Google Scholar] [CrossRef] [PubMed]
- Patten, D.A.; Wong, J.; Khacho, M.; Soubannier, V.; Mailloux, R.J.; Pilon-Larose, K.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Trinkle-Mulcahy, L.; et al. OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 2014, 33, 2676–2691. [Google Scholar] [CrossRef] [PubMed]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 2009, 187, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; van der Bliek, A.M. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 2009, 187, 959–966. [Google Scholar] [CrossRef]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.J.; Lampe, P.A.; Stojanovski, D.; Korwitz, A.; Anand, R.; Tatsuta, T.; Langer, T. Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J. 2014, 33, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Quiros, P.M.; Ramsay, A.J.; Sala, D.; Fernandez-Vizarra, E.; Rodriguez, F.; Peinado, J.R.; Fernandez-Garcia, M.S.; Vega, J.A.; Enriquez, J.A.; Zorzano, A.; et al. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J. 2012, 31, 2117–2133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Li, H.; Song, Z. Membrane depolarization activates the mitochondrial protease OMA1 by stimulating self-cleavage. EMBO Rep. 2014, 15, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Rainbolt, T.K.; Lebeau, J.; Puchades, C.; Wiseman, R.L. Reciprocal Degradation of YME1L and OMA1 Adapts Mitochondrial Proteolytic Activity during Stress. Cell Rep. 2016, 14, 2041–2049. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.J.; Kahl, A.; Fruitman, H.; Qian, L.; Zhou, P.; Manfredi, G.; Iadecola, C. Prohibitin levels regulate OMA1 activity and turnover in neurons. Cell Death Differ. 2019, 27, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.; Hu, J.; Che, Y.; Liu, Y.; Luo, Z.; Cheng, J.; Han, Q.; He, H.; Zhou, Q. CHCHD2 and CHCHD10 regulate mitochondrial dynamics and integrated stress response. Cell Death Dis. 2022, 13, 156. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Bossy-Wetzel, E.; Newmeyer, D.D.; Green, D.R. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J. 1998, 17, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Ashiya, M.; Buttle, K.; Weiler, S.; Oakes, S.A.; Mannella, C.A.; Korsmeyer, S.J. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 2002, 2, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Varanita, T.; Soriano, M.E.; Romanello, V.; Zaglia, T.; Quintana-Cabrera, R.; Semenzato, M.; Menabo, R.; Costa, V.; Civiletto, G.; Pesce, P.; et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015, 21, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Viana, M.P.; Levytskyy, R.M.; Anand, R.; Reichert, A.S.; Khalimonchuk, O. Protease OMA1 modulates mitochondrial bioenergetics and ultrastructure through dynamic association with MICOS complex. iScience 2021, 24, 102119. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [PubMed]
- MacVicar, T.D.; Lane, J.D. Impaired OMA1-dependent cleavage of OPA1 and reduced DRP1 fission activity combine to prevent mitophagy in cells that are dependent on oxidative phosphorylation. J. Cell Sci. 2014, 127 Pt 10, 2313–2325. [Google Scholar] [CrossRef]
- Owusu-Ansah, E.; Yavari, A.; Mandal, S.; Banerjee, U. Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat. Genet. 2008, 40, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Higuchi-Sanabria, R.; Frankino, P.A.; Paul, J.W., 3rd; Tronnes, S.U.; Dillin, A. A Futile Battle? Protein Quality Control and the Stress of Aging. Dev. Cell 2018, 44, 139–163. [Google Scholar] [CrossRef] [PubMed]
- Fessler, E.; Eckl, E.M.; Schmitt, S.; Mancilla, I.A.; Meyer-Bender, M.F.; Hanf, M.; Philippou-Massier, J.; Krebs, S.; Zischka, H.; Jae, L.T. A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 2020, 579, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Aviles, G.; Liu, Y.; Tian, R.; Unger, B.A.; Lin, Y.T.; Wiita, A.P.; Xu, K.; Correia, M.A.; Kampmann, M. Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature 2020, 579, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J., 2nd; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.J.; Harding, A.E.; Morgan-Hughes, J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Karamanlidis, G.; Nascimben, L.; Couper, G.S.; Shekar, P.S.; del Monte, F.; Tian, R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ. Res. 2010, 106, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Ritov, V.B.; Menshikova, E.V.; Azuma, K.; Wood, R.; Toledo, F.G.; Goodpaster, B.H.; Ruderman, N.B.; Kelley, D.E. Deficiency of electron transport chain in human skeletal muscle mitochondria in type 2 diabetes mellitus and obesity. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E49–E58. [Google Scholar] [CrossRef] [PubMed]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Garcia-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Ruperez, F.J.; Barbas, C.; Ibanez, B.; Langer, T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350, aad0116. [Google Scholar] [CrossRef] [PubMed]
- Korwitz, A.; Merkwirth, C.; Richter-Dennerlein, R.; Troder, S.E.; Sprenger, H.G.; Quiros, P.M.; Lopez-Otin, C.; Rugarli, E.I.; Langer, T. Loss of OMA1 delays neurodegeneration by preventing stress-induced OPA1 processing in mitochondria. J. Cell Biol. 2016, 212, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, H.; Xu, Z.; Ma, C.; Wang, T.; You, W.; Yu, Z.; Shen, H.; Chen, G. Ischemia-induced cleavage of OPA1 at S1 site aggravates mitochondrial fragmentation and reperfusion injury in neurons. Cell Death Dis. 2022, 13, 321. [Google Scholar] [CrossRef] [PubMed]
- Triolo, M.; Baker, N.; Agarwal, S.; Larionov, N.; Podinic, T.; Khacho, M. Optic atrophy 1 mediates muscle differentiation by promoting a metabolic switch via the supercomplex assembly factor SCAF1. iScience 2024, 27, 109164. [Google Scholar] [CrossRef] [PubMed]
- Senos Demarco, R.; Uyemura, B.S.; D’Alterio, C.; Jones, D.L. Mitochondrial fusion regulates lipid homeostasis and stem cell maintenance in the Drosophila testis. Nat. Cell Biol. 2019, 21, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Caglayan, S.; Hashim, A.; Cieslar-Pobuda, A.; Jensen, V.; Behringer, S.; Talug, B.; Chu, D.T.; Pecquet, C.; Rogne, M.; Brech, A.; et al. Optic Atrophy 1 Controls Human Neuronal Development by Preventing Aberrant Nuclear DNA Methylation. iScience 2020, 23, 101154. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, A.; Cipolat, S.; Chen, Y.; Dorn, G.W., 2nd; Scorrano, L. Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and Notch signaling. Science 2013, 342, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zuo, M.; Zeng, L.; Cui, K.; Liu, B.; Yan, C.; Chen, L.; Dong, J.; Shangguan, F.; Hu, W.; et al. OMA1 reprograms metabolism under hypoxia to promote colorectal cancer development. EMBO Rep. 2021, 22, e50827. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Yu, H.; Kong, Q.; Wang, B.; Shen, L.; Dong, D.; Sun, L. The Mitochondrial PHB2/OMA1/DELE1 Pathway Cooperates with Endoplasmic Reticulum Stress to Facilitate the Response to Chemotherapeutics in Ovarian Cancer. Int. J. Mol. Sci. 2022, 23, 1320. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, T.J.; Galloway, C.A.; Zhi, W.; Xiao, W.; de Mesy Bentley, K.L.; Sharma, A.; Teng, Y.; Sesaki, H.; Yoon, Y. The mitochondrial fusion protein OPA1 is dispensable in the liver and its absence induces mitohormesis to protect liver from drug-induced injury. Nat. Commun. 2023, 14, 6721. [Google Scholar] [CrossRef] [PubMed]
- Strauss, M.; Hofhaus, G.; Schroder, R.R.; Kuhlbrandt, W. Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 2008, 27, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Abrisch, R.G.; Gumbin, S.C.; Wisniewski, B.T.; Lackner, L.L.; Voeltz, G.K. Fission and fusion machineries converge at ER contact sites to regulate mitochondrial morphology. J. Cell Biol. 2020, 219, e201911122. [Google Scholar] [CrossRef] [PubMed]
- Tirrell, P.S.; Nguyen, K.N.; Luby-Phelps, K.; Friedman, J.R. MICOS subcomplexes assemble independently on the mitochondrial inner membrane in proximity to ER contact sites. J. Cell Biol. 2020, 219, e202003024. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Lopez-Domenech, G.; Halff, E.F.; Covill-Cooke, C.; Ivankovic, D.; Melandri, D.; Arancibia-Carcamo, I.L.; Burden, J.J.; Lowe, A.R.; Kittler, J.T. Miro clusters regulate ER-mitochondria contact sites and link cristae organization to the mitochondrial transport machinery. Nat. Commun. 2019, 10, 4399. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Saita, S.; Nolte, H.; Muller, S.; Konig, T.; Richter-Dennerlein, R.; Sprenger, H.G.; Madrenas, J.; Muhlmeister, M.; Brandt, U.; et al. The membrane scaffold SLP2 anchors a proteolytic hub in mitochondria containing PARL and the i-AAA protease YME1L. EMBO Rep. 2016, 17, 1844–1856. [Google Scholar] [CrossRef]
- Mitsopoulos, P.; Lapohos, O.; Weraarpachai, W.; Antonicka, H.; Chang, Y.H.; Madrenas, J. Stomatin-like protein 2 deficiency results in impaired mitochondrial translation. PLoS ONE 2017, 12, e0179967. [Google Scholar] [CrossRef] [PubMed]
- Tondera, D.; Grandemange, S.; Jourdain, A.; Karbowski, M.; Mattenberger, Y.; Herzig, S.; Da Cruz, S.; Clerc, P.; Raschke, I.; Merkwirth, C.; et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009, 28, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Bohovych, I.; Dietz, J.V.; Swenson, S.; Zahayko, N.; Khalimonchuk, O. Redox Regulation of the Mitochondrial Quality Control Protease Oma1. Antioxid. Redox Signal. 2019, 31, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Tatsuta, T.; Model, K.; Langer, T. Formation of membrane-bound ring complexes by prohibitins in mitochondria. Mol. Biol. Cell 2005, 16, 248–259. [Google Scholar] [CrossRef]
- von der Malsburg, A.; Sapp, G.M.; Zuccaro, K.E.; von Appen, A.; Moss, F.R., 3rd; Kalia, R.; Bennett, J.A.; Abriata, L.A.; Dal Peraro, M.; van der Laan, M.; et al. Structural mechanism of mitochondrial membrane remodelling by human OPA1. Nature 2023, 620, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Kondadi, A.K.; Anand, R.; Hansch, S.; Urbach, J.; Zobel, T.; Wolf, D.M.; Segawa, M.; Liesa, M.; Shirihai, O.S.; Weidtkamp-Peters, S.; et al. Cristae undergo continuous cycles of membrane remodelling in a MICOS-dependent manner. EMBO Rep. 2020, 21, e49776. [Google Scholar] [CrossRef] [PubMed]
- Bulthuis, E.P.; Dieteren, C.E.J.; Bergmans, J.; Berkhout, J.; Wagenaars, J.A.; van de Westerlo, E.M.A.; Podhumljak, E.; Hink, M.A.; Hesp, L.F.B.; Rosa, H.S.; et al. Stress-dependent macromolecular crowding in the mitochondrial matrix. EMBO J. 2023, 42, e108533. [Google Scholar] [CrossRef] [PubMed]
- Murata, D.; Yamada, T.; Tokuyama, T.; Arai, K.; Quiros, P.M.; Lopez-Otin, C.; Iijima, M.; Sesaki, H. Mitochondrial Safeguard: A stress response that offsets extreme fusion and protects respiratory function via flickering-induced Oma1 activation. EMBO J. 2020, 39, e105074. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gilkerson, R.; Kaur, H.; Carrillo, O.; Ramos, I. OMA1-Mediated Mitochondrial Dynamics Balance Organellar Homeostasis Upstream of Cellular Stress Responses. Int. J. Mol. Sci. 2024, 25, 4566. https://doi.org/10.3390/ijms25084566
Gilkerson R, Kaur H, Carrillo O, Ramos I. OMA1-Mediated Mitochondrial Dynamics Balance Organellar Homeostasis Upstream of Cellular Stress Responses. International Journal of Molecular Sciences. 2024; 25(8):4566. https://doi.org/10.3390/ijms25084566
Chicago/Turabian StyleGilkerson, Robert, Harpreet Kaur, Omar Carrillo, and Isaiah Ramos. 2024. "OMA1-Mediated Mitochondrial Dynamics Balance Organellar Homeostasis Upstream of Cellular Stress Responses" International Journal of Molecular Sciences 25, no. 8: 4566. https://doi.org/10.3390/ijms25084566