
HOPS/TMUB1 Enhances Apoptosis in TP53 Mutation-Independent Setting in Human Cancers
 , , , ,
, , , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. DBD Missense Mutations of TP53 per se Are Not Sufficient to Justify the Deregulation of Its Signaling Pathway
2.2. HOPS Drives Apoptosis of p53 Mutants
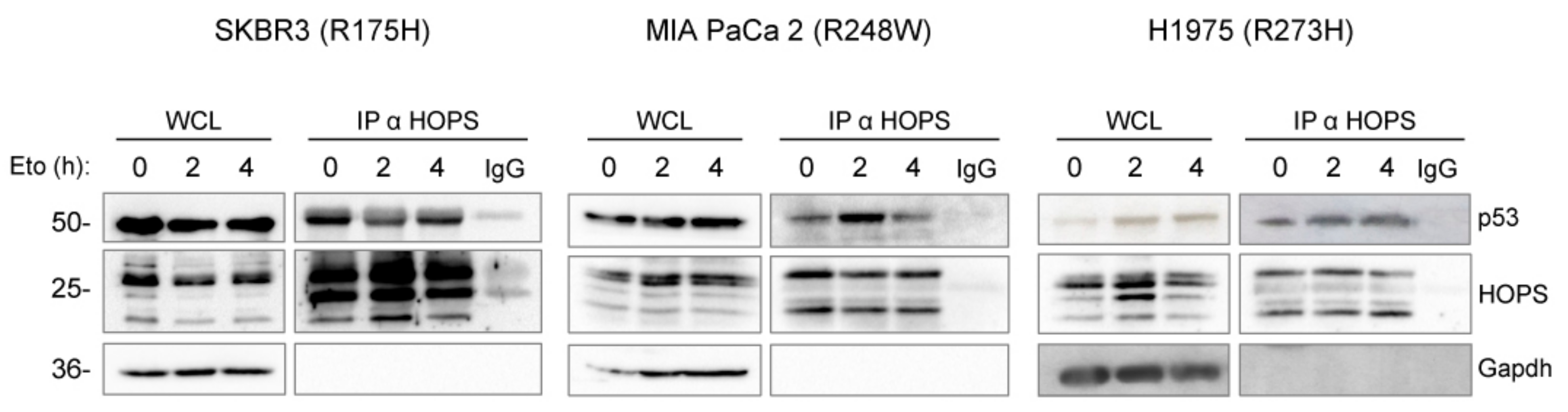
2.3. HOPS Binds TP53 Mutants after Stress Stimuli
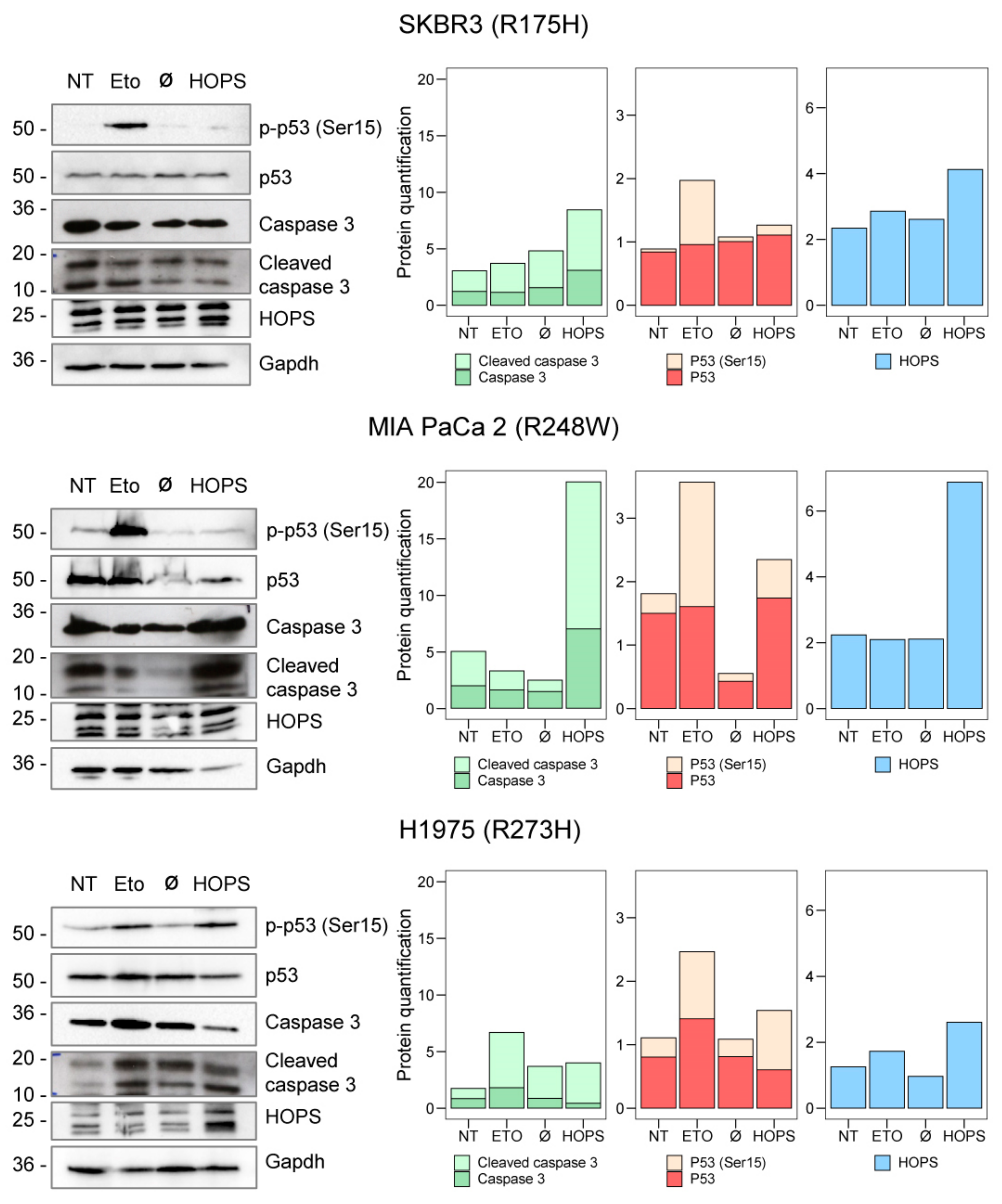
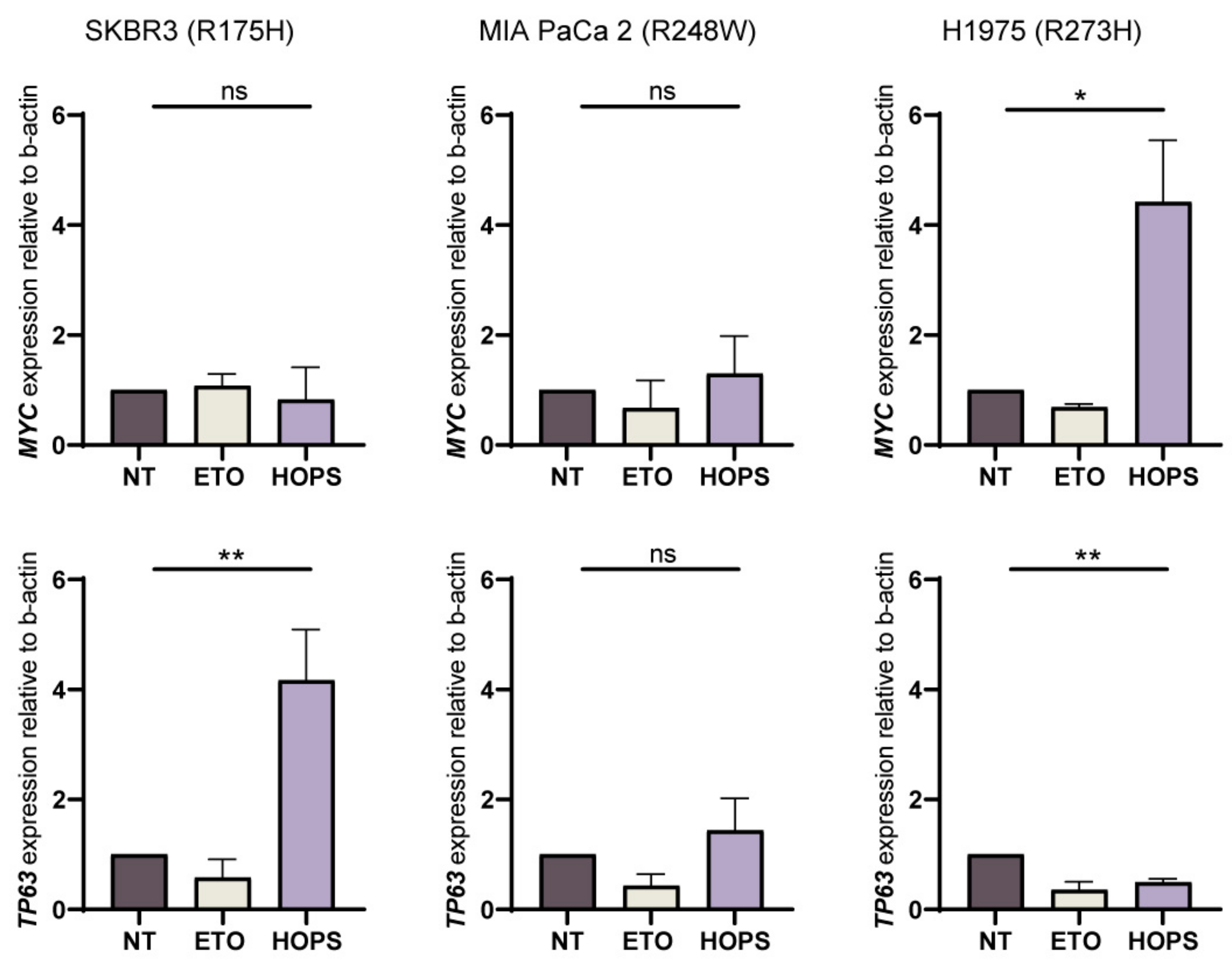
2.4. HOPS Supports Apoptotic Response in TP53 Mutants
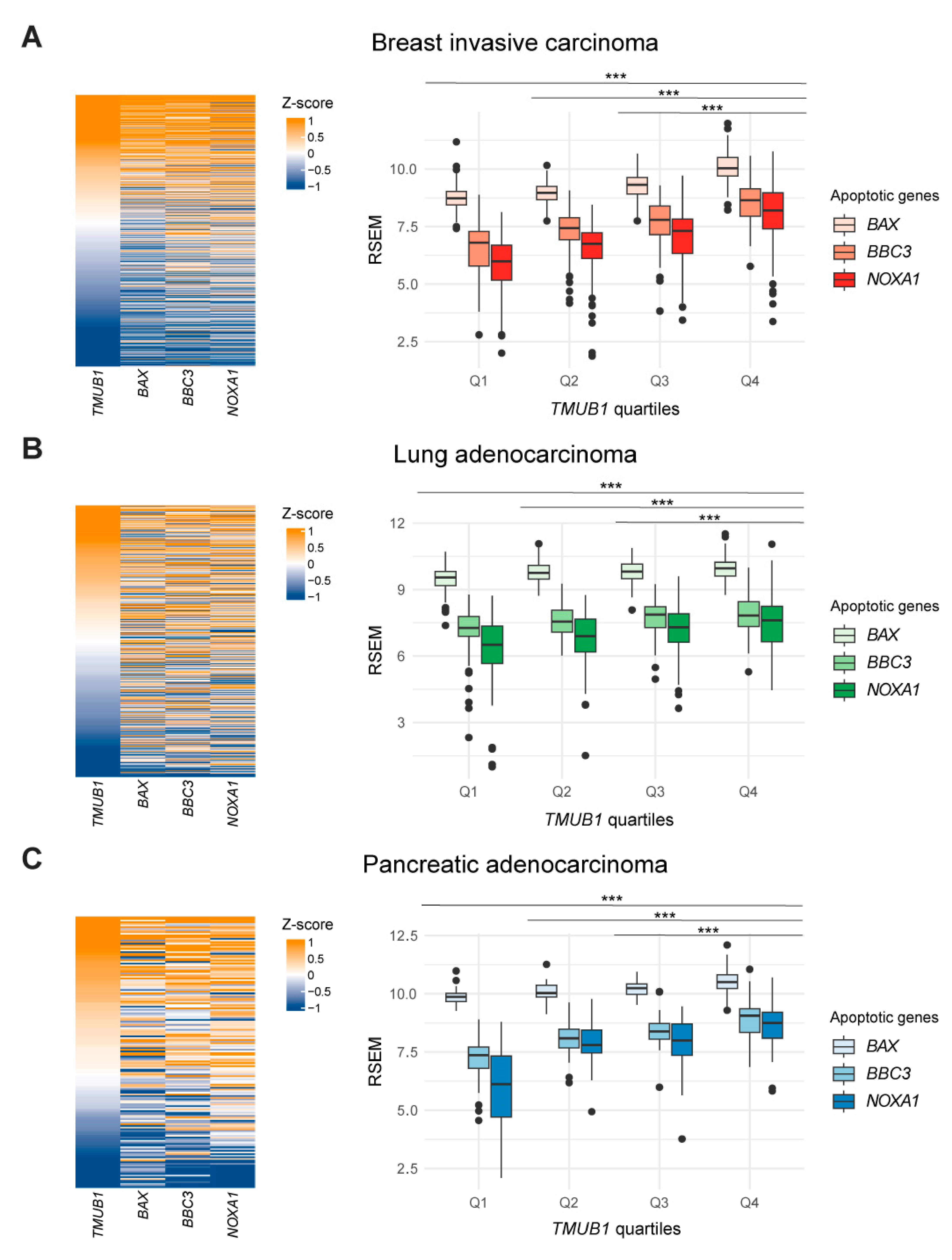
2.5. High Expression Levels of HOPS Positively Correlate with Apoptotic Genes in TCGA Patient Cohort
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Cloning and Plasmid
4.3. Western Blot, Immunoprecipitation, and Antibodies
4.4. Apoptosis Analysis by FACS
4.5. RNA Extraction and Real-Time PCR
4.6. TCGA Database and Data Collection
4.7. DepMap Portale and Data Collection
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levine, A.J.; Oren, M. The First 30 Years of P53: Growing Ever More Complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of P53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. P53, the Cellular Gatekeeper for Growth and Division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Oren, M. Decision Making by P53: Life, Death and Cancer. Cell Death Differ. 2003, 10, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor Suppression in the Absence of P53-Mediated Cell-Cycle Arrest, Apoptosis, and Senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. P53 in Survival, Death and Metabolic Health: A Lifeguard with a Licence to Kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Galluzzi, L.; Morselli, E.; Kepp, O.; Malik, S.A.; Kroemer, G. Autophagy Regulation by P53. Curr. Opin. Cell Biol. 2010, 22, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Ryan, K.M. P53 and Metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, C.; Xu, Z.; Scuoppo, C.; Rillahan, C.D.; Gao, J.; Spitzer, B.; Bosbach, B.; Kastenhuber, E.R.; Baslan, T.; et al. Deletions Linked to TP53 Loss Drive Cancer through P53-Independent Mechanisms. Nature 2016, 531, 471–475. [Google Scholar] [CrossRef]
- Stein, Y.; Aloni-Grinstein, R.; Rotter, V. Mutant P53 Oncogenicity: Dominant-Negative or Gain-of-Function? Carcinogenesis 2020, 41, 1635–1647. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Janic, A.; Chen, Y.; Chang, C.; Lieschke, E.C.; Diepstraten, S.T.; Kueh, A.J.; Bernardini, J.P.; Dewson, G.; O’Reilly, L.A.; et al. Mutant TRP53 Exerts a Target Gene-Selective Dominant-Negative Effect to Drive Tumor Development. Genes Dev. 2018, 32, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Giacomelli, A.O.; Yang, X.; Lintner, R.E.; McFarland, J.M.; Duby, M.; Kim, J.; Howard, T.P.; Takeda, D.Y.; Ly, S.H.; Kim, E.; et al. Mutational Processes Shape the Landscape of TP53 Mutations in Human Cancer. Nat. Genet. 2018, 50, 1381–1387. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 Promotes the Rapid Degradation of P53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Kruse, J.P.; Gu, W. Modes of P53 Regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Della-Fazia, M.A.; Castelli, M.; Piobbico, D.; Pieroni, S.; Servillo, G. HOPS and P53: Thick as Thieves in Life and Death. Cell Cycle 2020, 19, 2996–3003. [Google Scholar] [CrossRef] [PubMed]
- Della-Fazia, M.A.; Castelli, M.; Piobbico, D.; Pieroni, S.; Servillo, G. The Ins and Outs of HOPS/TMUB1 in Biology and Pathology. FEBS J. 2021, 288, 2773–2783. [Google Scholar] [CrossRef] [PubMed]
- Della Fazia, M.A.; Castelli, M.; Bartoli, D.; Pieroni, S.; Pettirossi, V.; Piobbico, D.; Viola-Magni, M.; Servillo, G. HOPS: A Novel CAMP-Dependent Shuttling Protein Involved in Protein Synthesis Regulation. J. Cell Sci. 2005, 118, 3185–3194. [Google Scholar] [CrossRef] [PubMed]
- Castelli, M.; Piobbico, D.; Chiacchiaretta, M.; Brunacci, C.; Pieroni, S.; Bartoli, D.; Gargaro, M.; Fallarino, F.; Puccetti, P.; Soddu, S.; et al. HOPS/TMUB1 Retains P53 in the Cytoplasm and Sustains P53-Dependent Mitochondrial Apoptosis. EMBO Rep. 2020, 21, e48073. [Google Scholar] [CrossRef] [PubMed]
- Pieroni, S.; Della Fazia, M.A.; Castelli, M.; Piobbico, D.; Bartoli, D.; Brunacci, C.; Bellet, M.M.; Viola-Magni, M.; Servillo, G. HOPS Is an Essential Constituent of Centrosome Assembly. Cell Cycle 2008, 7, 1462–1466. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Takagi, H.; Konishi, Y.; Ageta, H.; Ikegami, K.; Yao, I.; Sato, S.; Hatanaka, K.; Inokuchi, K.; Seog, D.H.; et al. Transmembrane and Ubiquitin-like Domain-Containing Protein 1 (Tmub1/HOPS) Facilitates Surface Expression of GluR2-Containing AMPA Receptors. PLoS ONE 2008, 3, e2809. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Savelieva, K.V.; Suwanichkul, A.; Small, D.L.; Kirkpatrick, L.L.; Xu, N.; Lanthorn, T.H.; Ye, G.L. Transmembrane and Ubiquitin-like Domain Containing 1 (Tmub1) Regulates Locomotor Activity and Wakefulness in Mice and Interacts with CAMLG. PLoS ONE 2010, 5, e11261. [Google Scholar] [CrossRef] [PubMed]
- Castelli, M.; Pieroni, S.; Brunacci, C.; Piobbico, D.; Bartoli, D.; Bellet, M.M.; Colombo, E.; Pelicci, P.G.; Della Fazia, M.A.; Servillo, G. Hepatocyte Odd Protein Shuttling (HOPS) Is a Bridging Protein in the Nucleophosmin-P19Arf Network. Oncogene 2013, 32, 3350–3358. [Google Scholar] [CrossRef]
- Bellet, M.M.; Pieroni, S.; Castelli, M.; Piobbico, D.; Fallarino, F.; Romani, L.; Della-Fazia, M.A.; Servillo, G. HOPS/Tmub1 Involvement in the NF-KB-Mediated Inflammatory Response through the Modulation of TRAF6. Cell Death Dis. 2020, 11, 865. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Wang, Y.; Wu, M.; Chen, Y.; Liu, F.; Shen, Z.; Wang, Y.; Xie, S.; Shen, Y.; Sang, L.; et al. Promoting Anti-Tumor Immunity by Targeting TMUB1 to Modulate PD-L1 Polyubiquitination and Glycosylation. Nat. Commun. 2022, 13, 1–18. [Google Scholar] [CrossRef]
- Fu, H.; Zhang, Y.; Chen, J.; Zhou, B.; Chen, G.; Chen, P. Tmub1 Suppresses Hepatocellular Carcinoma by Promoting the Ubiquitination of ΔNp63 Isoforms. Mol. Ther. Oncolytics 2020, 18, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fu, H.; Zhang, Y.; Chen, P. Transmembrane and Ubiquitin-Like Domain Containing 1 Protein (TMUB1) Negatively Regulates Hepatocellular Carcinoma Proliferation via Regulating Signal Transducer and Activator of Transcription 1 (STAT1). Med. Sci. Monit. 2019, 25, 9471. [Google Scholar] [CrossRef] [PubMed]
- Hao, T.; Yu, H.; Huang, D.; Liu, Q. TMUB1 Correlated with Immune Infiltration Is a Prognostic Marker for Colorectal Cancer. Dis. Markers 2022, 2022, 5884531. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, K.; Peng, Y.; Zhang, J.; Ju, Q.; Xu, Q.; Ouyang, M.; He, Z. TMUB1 Expression Is Associated with the Prognosis of Colon Cancer and Immune Cell Infiltration. PeerJ 2023, 11, e16334. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cai, C.; Wu, K.; Hu, L. Transmembrane and Ubiquitin-Like Domain-Containing 1 Promotes Glioma Growth and Indicates Unfavorable Prognosis. Int. J. Genom. 2023, 2023, 3318171. [Google Scholar] [CrossRef]
- Ferracchiato, S.; Di-Iacovo, N.; Scopetti, D.; Piobbico, D.; Castelli, M.; Pieroni, S.; Gargaro, M.; Manni, G.; Brancorsini, S.; Della-Fazia, M.A.; et al. Hops/Tmub1 Heterozygous Mouse Shows Haploinsufficiency Effect in Influencing P53-Mediated Apoptosis. Int. J. Mol. Sci. 2021, 22, 7186. [Google Scholar] [CrossRef] [PubMed]
- Vaske, C.J.; Benz, S.C.; Sanborn, J.Z.; Earl, D.; Szeto, C.; Zhu, J.; Haussler, D.; Stuart, J.M. Inference of Patient-Specific Pathway Activities from Multi-Dimensional Cancer Genomics Data Using PARADIGM. Bioinformatics 2010, 26, i237. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304. [Google Scholar] [CrossRef]
- Schaefer, C.F.; Anthony, K.; Krupa, S.; Buchoff, J.; Day, M.; Hannay, T.; Buetow, K.H. PID: The Pathway Interaction Database. Nucleic Acids Res. 2009, 37, D674–D679. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform Analysis of 12 Cancer Types Reveals Molecular Classification within and across Tissues of Origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef] [PubMed]
- Sonego, M.; Schiappacassi, M.; Lovisa, S.; Dall’Acqua, A.; Bagnoli, M.; Lovat, F.; Libra, M.; D’Andrea, S.; Canzonieri, V.; Militello, L.; et al. Stathmin Regulates Mutant P53 Stability and Transcriptional Activity in Ovarian Cancer. EMBO Mol. Med. 2013, 5, 707–722. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liao, P.; Zeng, S.X.; Lu, H. It Takes a Team: A Gain-of-Function Story of P53-R249S. J. Mol. Cell Biol. 2019, 11, 277–283. [Google Scholar] [CrossRef]
- Gaiddon, C.; Lokshin, M.; Ahn, J.; Zhang, T.; Prives, C. A Subset of Tumor-Derived Mutant Forms of P53 down-Regulate P63 and P73 through a Direct Interaction with the P53 Core Domain. Mol. Cell. Biol. 2001, 21, 1874–1887. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and Interpreting Cancer Genomics Data via the Xena Platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2023. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Gu, Z. Complex Heatmap Visualization. iMeta 2022, 1, e43. [Google Scholar] [CrossRef]
- Gu, Z. ColorRamp2: Generate Color Mapping Functions. 2022. Available online: https://cran.r-project.org/web/packages/colorRamp2/colorRamp2.pdf (accessed on 15 January 2024).






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di-Iacovo, N.; Ferracchiato, S.; Pieroni, S.; Scopetti, D.; Castelli, M.; Piobbico, D.; Pierucci, L.; Gargaro, M.; Chiasserini, D.; Servillo, G.; et al. HOPS/TMUB1 Enhances Apoptosis in TP53 Mutation-Independent Setting in Human Cancers. Int. J. Mol. Sci. 2024, 25, 4600. https://doi.org/10.3390/ijms25094600
Di-Iacovo N, Ferracchiato S, Pieroni S, Scopetti D, Castelli M, Piobbico D, Pierucci L, Gargaro M, Chiasserini D, Servillo G, et al. HOPS/TMUB1 Enhances Apoptosis in TP53 Mutation-Independent Setting in Human Cancers. International Journal of Molecular Sciences. 2024; 25(9):4600. https://doi.org/10.3390/ijms25094600
Chicago/Turabian StyleDi-Iacovo, Nicola, Simona Ferracchiato, Stefania Pieroni, Damiano Scopetti, Marilena Castelli, Danilo Piobbico, Luca Pierucci, Marco Gargaro, Davide Chiasserini, Giuseppe Servillo, and et al. 2024. "HOPS/TMUB1 Enhances Apoptosis in TP53 Mutation-Independent Setting in Human Cancers" International Journal of Molecular Sciences 25, no. 9: 4600. https://doi.org/10.3390/ijms25094600