
Modulating Endoplasmic Reticulum Chaperones and Mutant Protein Degradation in GABRG2(Q390X) Associated with Genetic Epilepsy with Febrile Seizures Plus and Dravet Syndrome

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. The GABRG2(Q390X) Mutation Results in γ2 Dimers and Reduces Expression of the Partnering α1 and β2 Subunits
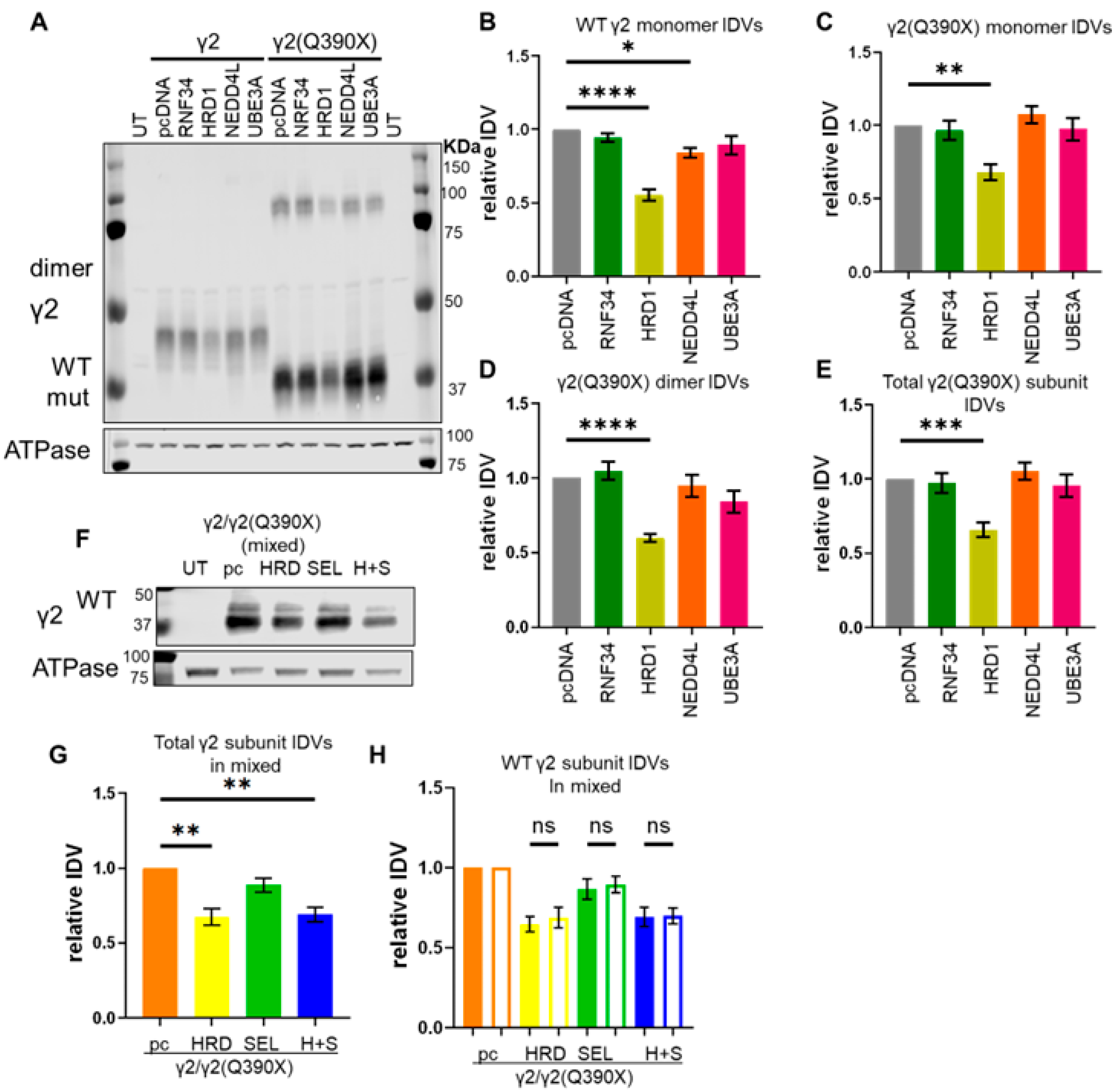
2.2. Overexpression of an E3 Ubiquitin Ligase Increased γ2(Q390X) Subunit Degradation
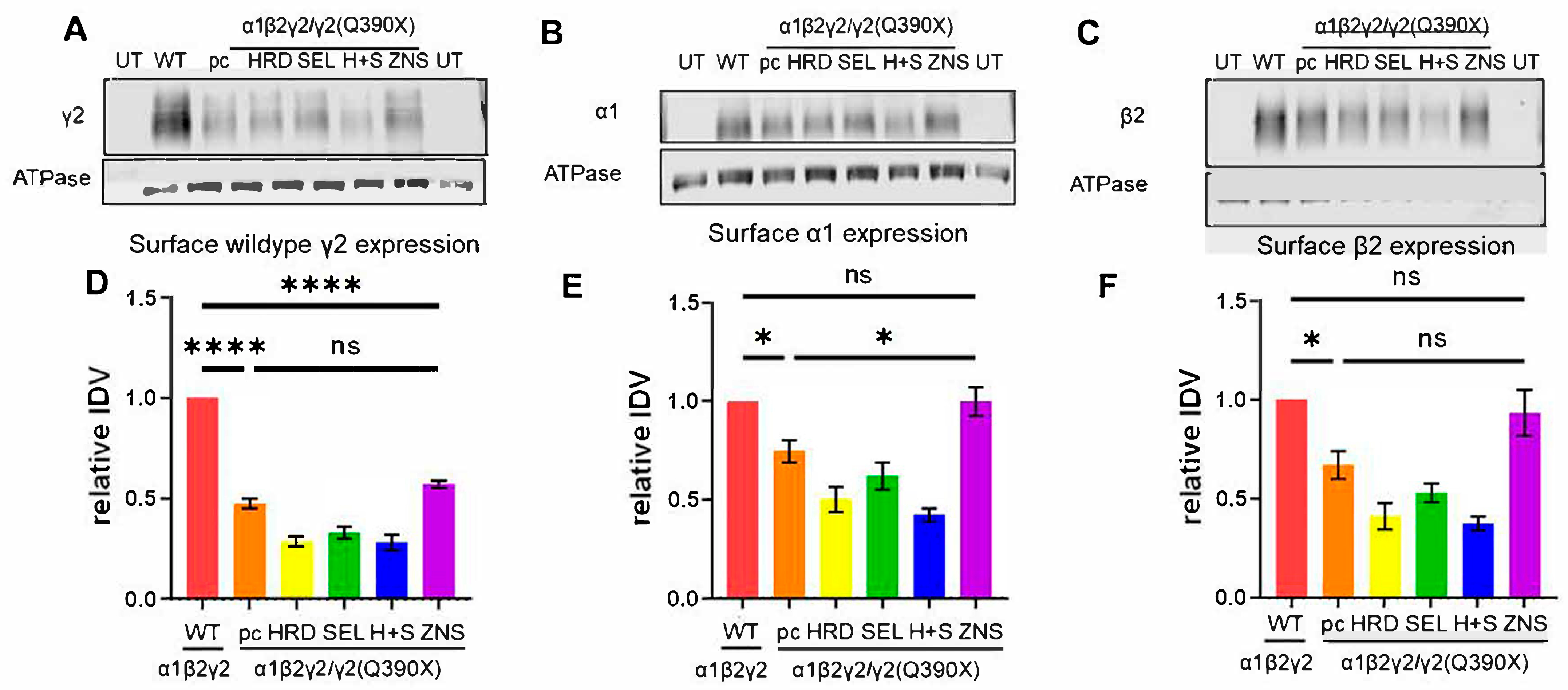
2.3. A Suggested HRD1 Upregulation, ZNS, Increased Surface Expression of GABAAR Subunits In Vitro
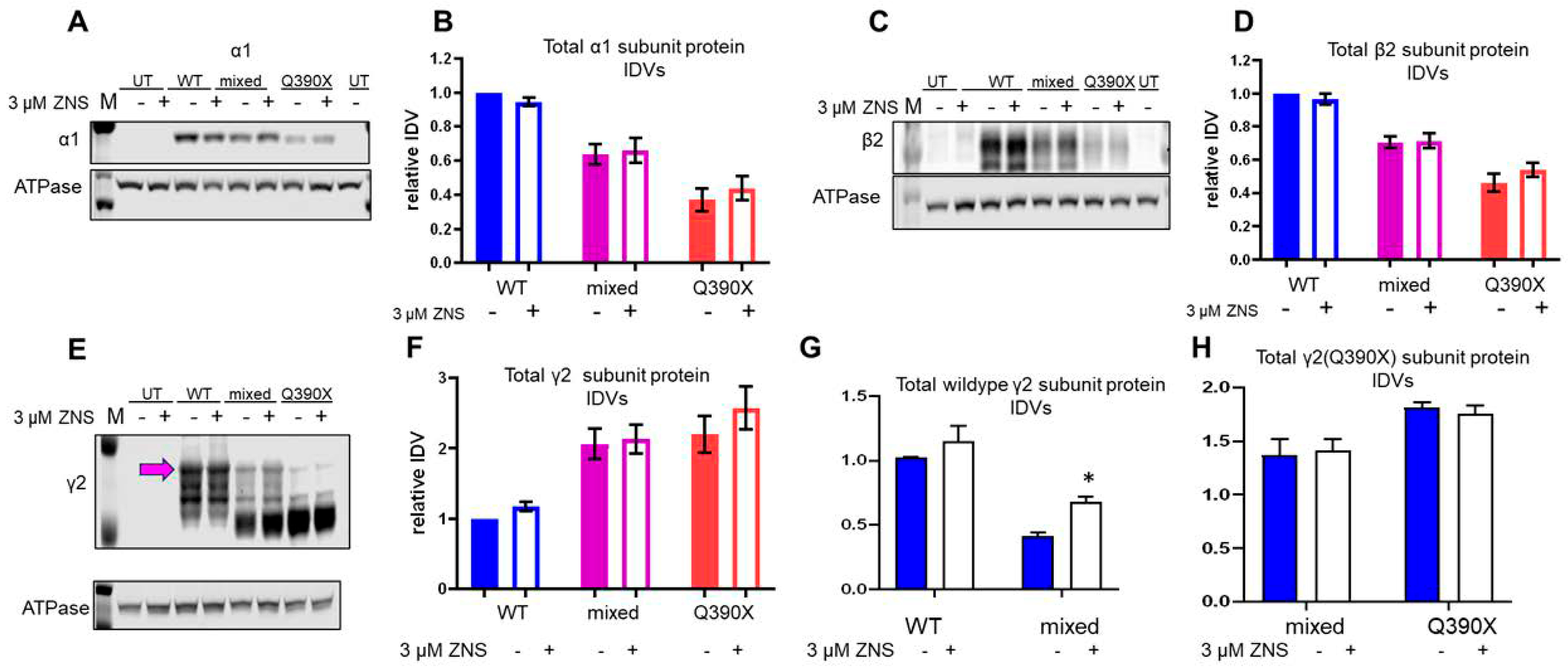
2.4. ZNS Selectively Increased the Wildtype γ2 Subunit but Had No Effect on Total Expression of α1 or β2 Subunits In Vitro
2.5. ZNS Upregulated the Expression of HRD1 Expression
2.5.1. HRD1 Dose-Dependently Degraded the Mutant γ2(Q390X) Subunit Protein in Both Monomers and Dimers
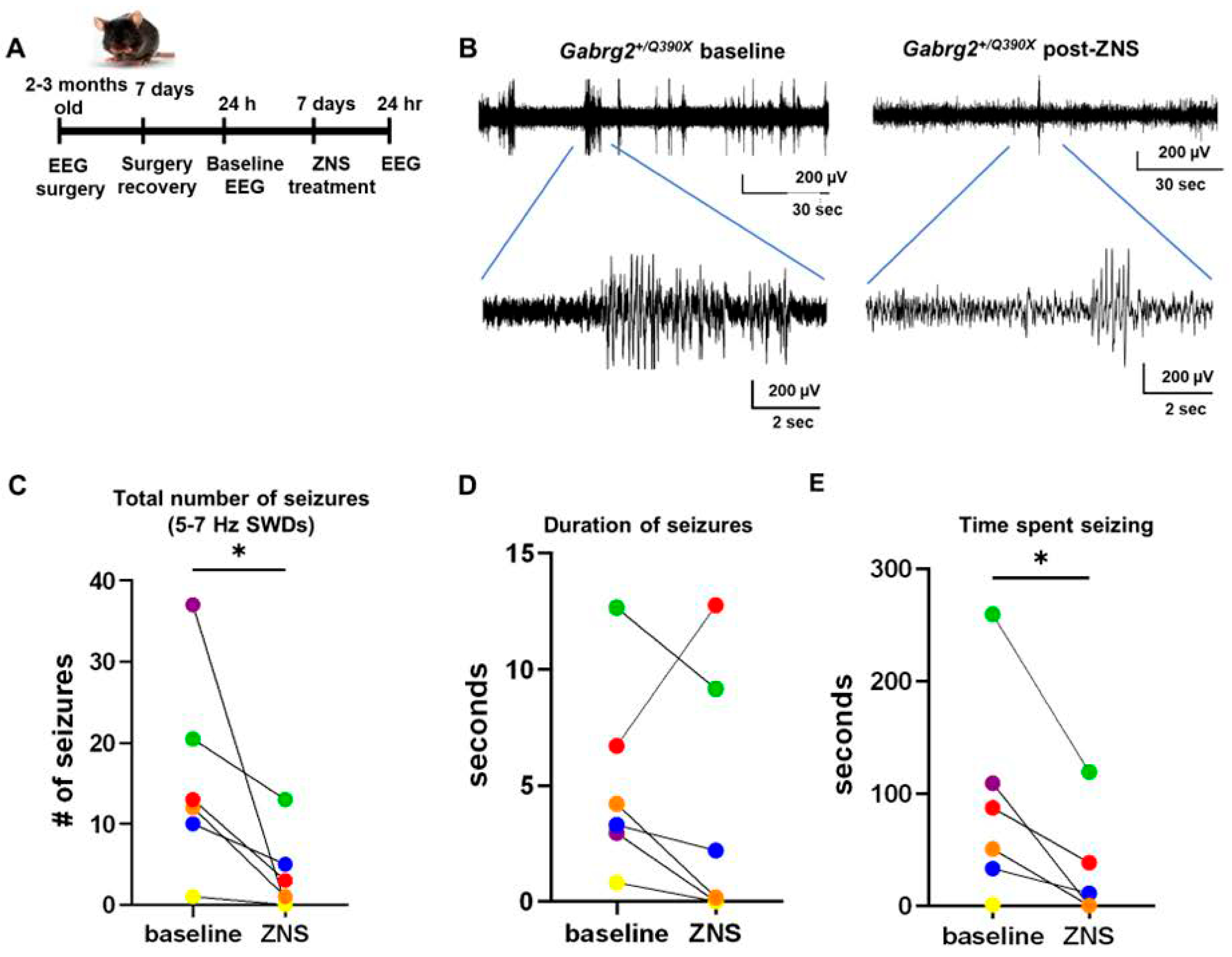
2.5.2. ZNS Reduced Seizures in the Gabrg2+/Q390X Mice
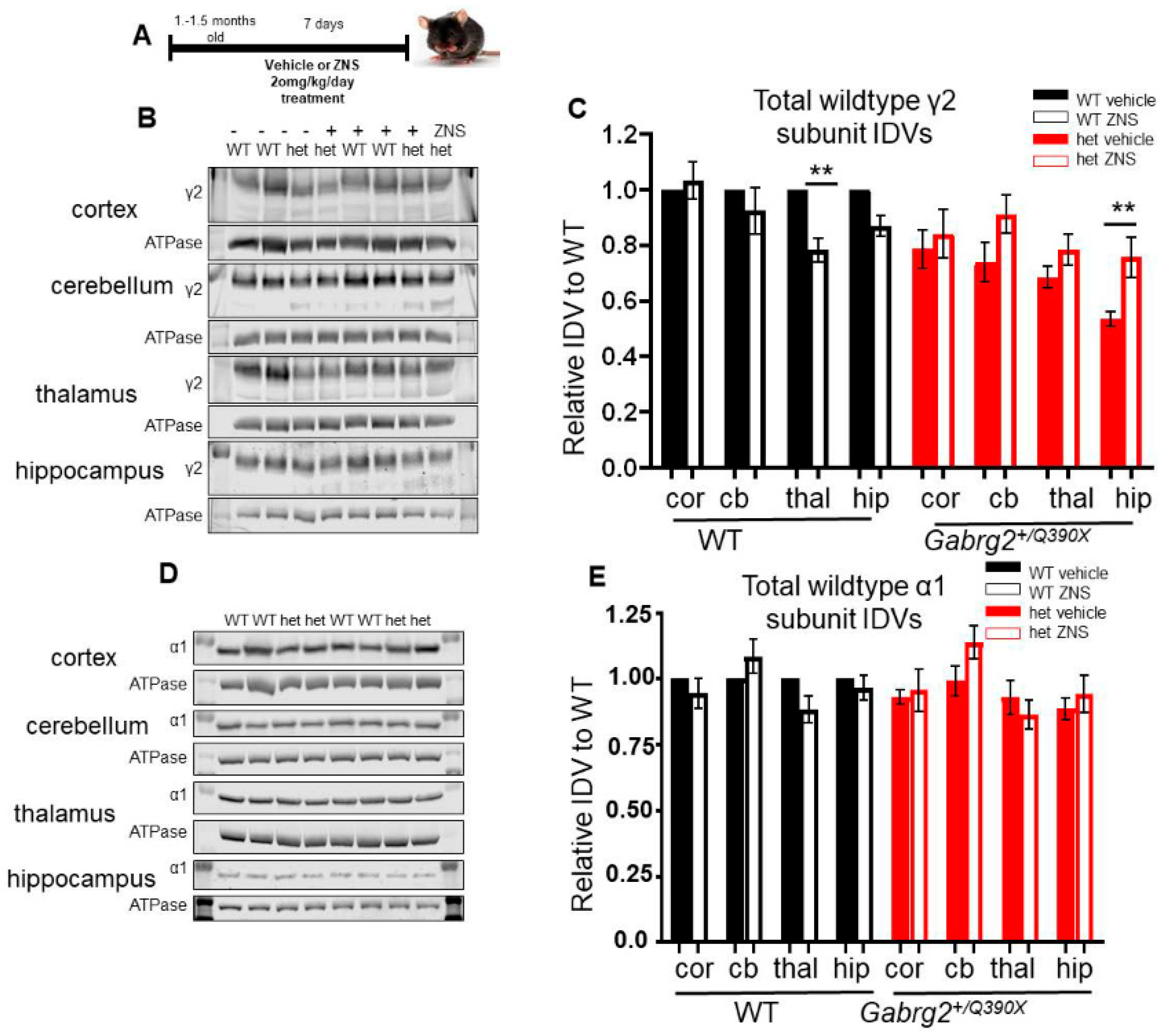
2.6. ZNS Increased the γ2 Subunit of the Wildtype Allele in the Hippocampus of the Mutant Gabrg2+/Q390X Mice
2.7. ZNS Did Not Change Total Expression of α1 or β2 Subunits in Mice
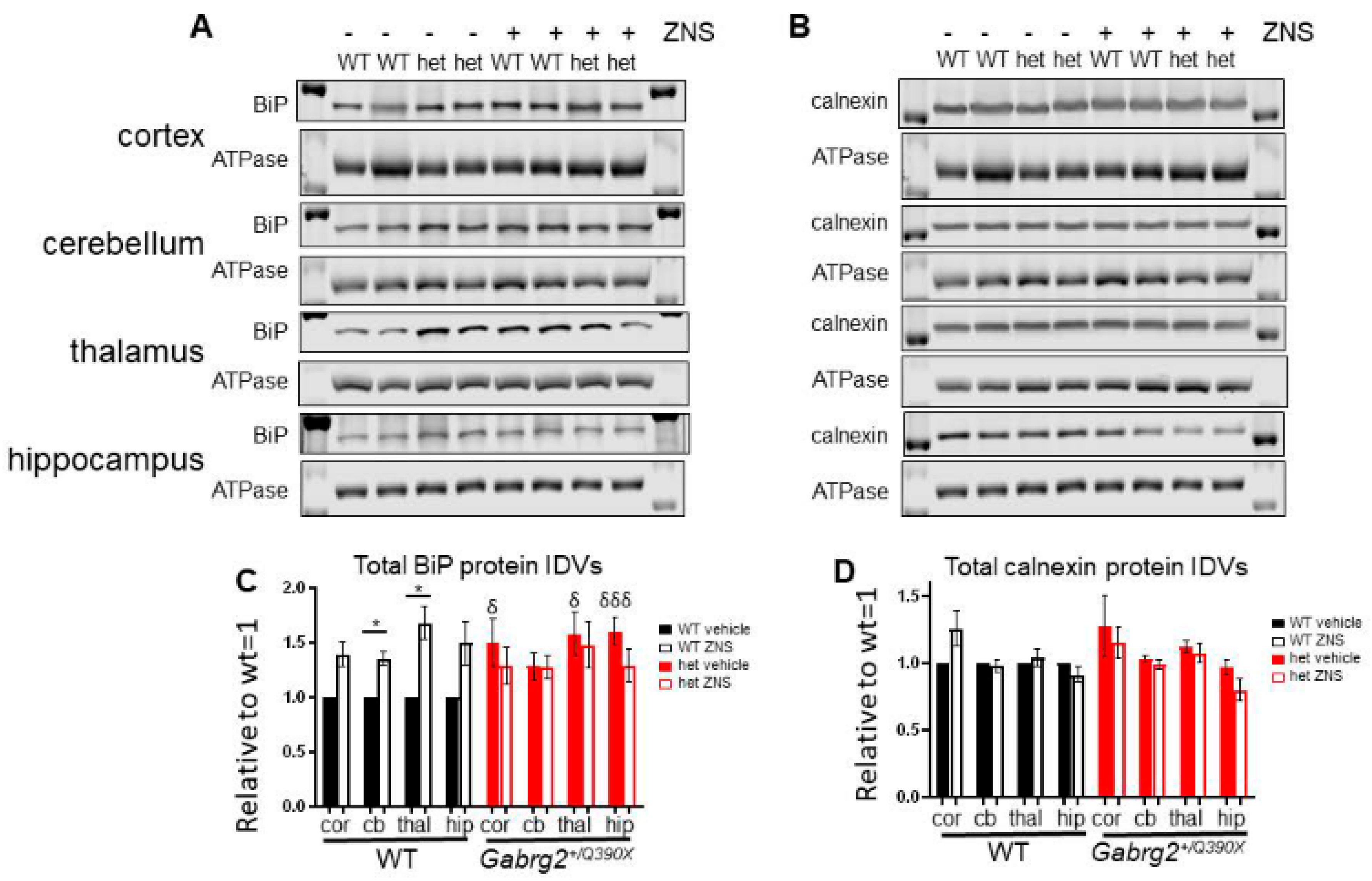
2.8. The Gabrg2+/Q390X Mice Had Increased ER Chaperones Like BiP, which Had Differential Response to ZNS Compared to the Wildtype Mice
3. Discussion
3.1. The γ2(Q390X) Subunit Impairs the Wildtype GABAAR Expression and Disturb ER Protein Homeostasis
3.2. The E3 Ligase HRD1 Facilitates Degradation of the Mutant γ2(Q390X) Subunit
3.3. Enhancing Degradation of the Mutant γ2(Q390X) Subunit Could Relieve the Dominant Negative Suppression on the Wildtype γ2 Subunits
3.4. ZNS Partially Restored Surface Trafficking of the Wildtype GABAAR Subunits and Reduced Seizures in the Gabrg2+/Q390X Mice
3.5. ZNS Treatment Had Differential Effects on the Upregulation of the ER Chaperone BiP
4. Materials and Methods
4.1. Cell Culture and Polyethyleneimine Transfection
4.2. Immunoblot
4.3. Biotinylation
4.4. Gabrg2+/Q390X Mouse Model of GEFS+ and Dravet Syndrome
4.5. Drug Administration and Brain Tissue Preparation in Gabrg2+/Q390X Mice
4.6. EEG Acquisition and Scoring
4.7. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DHEC | dihydroergocristine |
| DNP | dinoprost |
| EEG | electroencephalogram |
| EMG | electromyogram |
| ER | endoplasmic reticulum |
| ERAD | ER-associated degradation |
| FENIB | familial encephalopathy with neuroserpin inclusion bodies |
| GABA | γ-aminobutyric acid |
| GABAAR | GABA receptor type A |
| SEM | standard error of the mean |
| SWD | spike-and-wave discharge |
| ZNS | zonisamide |
References
- Wallace, R.H.; Marini, C.; Petrou, S.; Harkin, L.A.; Bowser, D.N.; Panchal, R.G.; Williams, D.A.; Sutherland, G.R.; Mulley, J.C.; Scheffer, I.E.; et al. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat. Genet. 2001, 28, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Marini, C.; Harkin, L.A.; Wallace, R.H.; Mulley, J.C.; Scheffer, I.E.; Berkovic, S.F. Childhood absence epilepsy and febrile seizures: A family with a GABA(A) receptor mutation. Brain 2003, 126, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Audenaert, D.; Schwartz, E.; Claeys, K.G.; Claes, L.; Deprez, L.; Suls, A.; Van Dyck, T.; Lagae, L.; Van Broeckhoven, C.; Macdonald, R.L.; et al. A novel GABRG2 mutation associated with febrile seizures. Neurology 2006, 67, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Huang, M.-C.; Ishii, A.; Yoshida, S.; Okada, M.; Morita, K.; Nagafuji, H.; Yasumoto, S.; Kaneko, S.; Kojima, T.; et al. Mutational analysis of GABRG2 in a Japanese cohort with childhood epilepsies. J. Hum. Genet. 2010, 55, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Lachance-Touchette, P.; Brown, P.; Meloche, C.; Kinirons, P.; Lapointe, L.; Lacasse, H.; Lortie, A.; Carmant, L.; Bedford, F.; Bowie, D.; et al. Novel α1 and γ2 GABAA receptor subunit mutations in families with idiopathic generalized epilepsy. Eur. J. Neurosci. 2011, 34, 237–249. [Google Scholar] [CrossRef]
- Epi4K Consortium; Epilepsy Phenome/Genome Project. Ultra-rare genetic variation in common epilepsies: A case-control sequencing study. Lancet Neurol. 2017, 16, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Zou, F.; McWalter, K.; Schmidt, L.; Decker, A.; Picker, J.D.; Lincoln, S.; Sweetser, D.A.; Briere, L.C.; Harini, C.; Members of the Undiagnosed Diseases Network; et al. Expanding the phenotypic spectrum of GABRG2 variants: A recurrent GABRG2 missense variant associated with a severe phenotype. J. Neurogenet. 2017, 31, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Komulainen-Ebrahim, J.; Schreiber, J.M.; Kangas, S.M.; Pylkäs, K.; Suo-Palosaari, M.; Rahikkala, E.; Liinamaa, J.; Immonen, E.-V.; Hassinen, I.; Myllynen, P.; et al. Novel variants and phenotypes widen the phenotypic spectrum of GABRG2-related disorders. Seizure 2019, 69, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Saleem, T.; Maqbool, H.; Sheikh, N.; Tayyeb, A.; Mukhtar, M.; Ashfaq, A. GABRG2 C588T Polymorphism Is Associated with Idiopathic Generalized Epilepsy but Not with Antiepileptic Drug Resistance in Pakistani Cohort. BioMed Res. Int. 2022, 2022, 3460792. [Google Scholar] [CrossRef]
- Maillard, P.-Y.; Baer, S.; Schaefer, É.; Desnous, B.; Villeneuve, N.; Lépine, A.; Fabre, A.; Lacoste, C.; El Chehadeh, S.; Piton, A.; et al. Molecular and clinical descriptions of patients with GABAA receptor gene variants (GABRA1, GABRB2, GABRB3, GABRG2): A cohort study, review of literature, and genotype-phenotype correlation. Epilepsia 2022, 63, 2519–2533. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Niu, X.; Cheng, M.; Zeng, Q.; Deng, J.; Tian, X.; Wang, Y.; Yu, J.; Shi, W.; Wu, W.; et al. Phenotypic Spectrum and Prognosis of Epilepsy Patients with GABRG2 Variants. Front. Mol. Neurosci. 2022, 15, 809163. [Google Scholar] [CrossRef] [PubMed]
- Boillot, M.; Morin-Brureau, M.; Picard, F.; Weckhuysen, S.; Lambrecq, V.; Minetti, C.; Striano, P.; Zara, F.; Iacomino, M.; Ishida, S.; et al. Novel GABRG2 mutations cause familial febrile seizures. Neurol. Genet. 2015, 1, e35. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhang, Y.; Liang, J.; Liu, X.; Ma, X.; Wu, H.; Xu, K.; Qin, J.; Qi, Y.; Wu, X. SCN1A, SCN1B, and GABRG2 gene mutation analysis in Chinese families with generalized epilepsy with febrile seizures plus. J. Hum. Genet. 2008, 53, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.-L.; Song, C.-G.; Zhou, H.-M.; Feng, B.; Zhao, J.-J.; Liu, Y.; Man, Y.-L.; Han, J.; Liu, S.-B.; Jiang, W. Rare variants in GABRG2 associated with sleep-related hypermotor epilepsy. J. Neurol. 2022, 269, 4939–4954. [Google Scholar] [CrossRef] [PubMed]
- Harkin, L.A.; Bowser, D.N.; Dibbens, L.M.; Singh, R.; Phillips, F.; Wallace, R.H.; Richards, M.C.; Williams, D.A.; Mulley, J.C.; Berkovic, S.F.; et al. Truncation of the GABAA-receptor gamma2 subunit in a family with generalized epilepsy with febrile seizures plus. Am. J. Hum. Genet. 2002, 70, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-Q.; Shen, W.; Macdonald, R.L. The GABRG2 mutation, Q351X, associated with generalized epilepsy with febrile seizures plus, has both loss of function and dominant-negative suppression. J. Neurosci. 2009, 29, 2845–2856. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Macdonald, R.L. The Intronic GABRG2 Mutation, IVS6+2T→G, Associated with Childhood Absence Epilepsy Altered Subunit mRNA Intron Splicing, Activated Nonsense-Mediated Decay, and Produced a Stable Truncated γ2 Subunit. J. Neurosci. 2012, 32, 5937–5952. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-Q.; Shen, W.; Macdonald, R.L. Trafficking-deficient mutant GABRG2 subunit amount may modify epilepsy phenotype. Ann. Neurol. 2013, 74, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Johnston, A.J.; Kang, J.-Q.; Shen, W.; Pickrell, W.O.; Cushion, T.D.; Davies, J.S.; Baer, K.; Mullins, J.G.L.; Hammond, C.L.; Chung, S.-K.; et al. A Novel GABRG2 Mutation, p.R136*, in a family with GEFS+ and extended phenotypes. Neurobiol. Dis. 2014, 64, 131–141. [Google Scholar] [CrossRef]
- Ishii, A.; Kanaumi, T.; Sohda, M.; Misumi, Y.; Zhang, B.; Kakinuma, N.; Haga, Y.; Watanabe, K.; Takeda, S.; Okada, M.; et al. Association of nonsense mutation in GABRG2 with abnormal trafficking of GABAA receptors in severe epilepsy. Epilepsy Res. 2014, 108, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-Q.; Shen, W.; Zhou, C.; Xu, D.; Macdonald, R.L. The human epilepsy mutation GABRG2(Q390X) causes chronic subunit accumulation and neurodegeneration. Nat. Neurosci. 2015, 18, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Poliquin, S.; Macdonald, R.L.; Dong, M.; Kang, J.-Q. Endoplasmic reticulum stress increases inflammatory cytokines in an epilepsy mouse model Gabrg2+/Q390X knockin: A link between genetic and acquired epilepsy? Epilepsia 2020, 61, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Titus, E.A.; Cooper, A.A. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 2004, 15, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Ghemrawi, R.; Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef] [PubMed]
- Di, X.-J.; Wang, Y.-J.; Han, D.-Y.; Fu, Y.-L.; Duerfeldt, A.S.; Blagg, B.S.J.; Mu, T.-W. Grp94 Protein Delivers γ-Aminobutyric Acid Type A (GABAA) Receptors to Hrd1 Protein-mediated Endoplasmic Reticulum-associated Degradation. J. Biol. Chem. 2016, 291, 9526–9539. [Google Scholar] [CrossRef] [PubMed]
- Whittsette, A.L.; Wang, Y.-J.; Mu, T.-W. The endoplasmic reticulum membrane complex promotes proteostasis of GABAA receptors. iScience 2022, 25, 104754. [Google Scholar] [CrossRef] [PubMed]
- Colombo, S.F.; Mazzo, F.; Pistillo, F.; Gotti, C. Biogenesis, trafficking and up-regulation of nicotinic ACh receptors. Biochem. Pharmacol. 2013, 86, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Jacob, T.C.; Moss, S.J.; Jurd, R. GABAA receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat. Rev. Neurosci. 2008, 9, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Pohl, C.; Dikic, I. Cellular Quality Control by the Ubiquitin-Proteasome System and Autophagy. Available online: https://www.science.org/doi/epdf/10.1126/science.aax3769 (accessed on 7 October 2021).
- Zhu, J.; Tsai, N.-P. Ubiquitination and E3 Ubiquitin Ligases in Rare Neurological Diseases with Comorbid Epilepsy. Neuroscience 2020, 428, 90–99. [Google Scholar] [CrossRef] [PubMed]
- George, A.J.; Hoffiz, Y.C.; Charles, A.J.; Zhu, Y.; Mabb, A.M. A Comprehensive Atlas of E3 Ubiquitin Ligase Mutations in Neurological Disorders. Front. Genet. 2018, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Potjewyd, F.M.; Axtman, A.D. Exploration of Aberrant E3 Ligases Implicated in Alzheimer’s Disease and Development of Chemical Tools to Modulate Their Function. Front. Cell Neurosci. 2021, 15, 768655. [Google Scholar] [CrossRef] [PubMed]
- Lescouzères, L.; Bomont, P. E3 Ubiquitin Ligases in Neurological Diseases: Focus on Gigaxonin and Autophagy. Front. Physiol. 2020, 11, 1022. [Google Scholar] [CrossRef]
- Kang, J.-Q.; Shen, W.; Lee, M.; Gallagher, M.J.; Macdonald, R.L. Slow degradation and aggregation in vitro of mutant GABAA receptor gamma2(Q351X) subunits associated with epilepsy. J. Neurosci. 2010, 30, 13895–13905. [Google Scholar] [CrossRef] [PubMed]
- Warner, T.A.; Shen, W.; Huang, X.; Liu, Z.; Macdonald, R.L.; Kang, J.-Q. Differential molecular and behavioural alterations in mouse models of GABRG2 haploinsufficiency versus dominant negative mutations associated with human epilepsy. Hum. Mol. Genet. 2016, 25, 3192–3207. [Google Scholar] [CrossRef] [PubMed]
- Di, X.-J.; Wang, Y.-J.; Cotter, E.; Wang, M.; Whittsette, A.L.; Han, D.-Y.; Sangwung, P.; Brown, R.; Lynch, J.W.; Keramidas, A.; et al. Proteostasis Regulators Restore Function of Epilepsy-Associated GABAA Receptors. Cell Chem. Biol. 2021, 28, 46–59.e7. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; DeLorey, T.M.; Delgado-Escueta, A.; Olsen, R.W. GABRB3, Epilepsy, and Neurodevelopment. In Jasper’s Basic Mechanisms of the Epilepsies [Internet], 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. Available online: http://www.ncbi.nlm.nih.gov/books/NBK98178/ (accessed on 2 March 2023).
- Saliba, R.S.; Pangalos, M.; Moss, S.J. The ubiquitin-like protein Plic-1 enhances the membrane insertion of GABAA receptors by increasing their stability within the endoplasmic reticulum. J. Biol. Chem. 2008, 283, 18538–18544. [Google Scholar] [CrossRef] [PubMed]
- Kleijnen, M.F.; Shih, A.H.; Zhou, P.; Kumar, S.; Soccio, R.E.; Kedersha, N.L.; Gill, G.; Howley, P.M. The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol. Cell 2000, 6, 409–419. [Google Scholar] [CrossRef]
- Delahanty, R.J.; Zhang, Y.; Bichell, T.J.; Shen, W.; Verdier, K.; Macdonald, R.L.; Xu, L.; Boyd, K.; Williams, J.; Kang, J.-Q. Beyond Epilepsy and Autism: Disruption of GABRB3 Causes Ocular Hypopigmentation. Cell Rep. 2016, 17, 3115–3124. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Jewett, K.A.; Chung, H.J.; Tsai, N.-P. Loss of fragile X protein FMRP impairs homeostatic synaptic downscaling through tumor suppressor p53 and ubiquitin E3 ligase Nedd4-2. Hum. Mol. Genet. 2018, 27, 2805–2816. [Google Scholar] [CrossRef]
- Zhu, J.; Lee, K.Y.; Jewett, K.A.; Man, H.-Y.; Chung, H.J.; Tsai, N.-P. Epilepsy-associated gene Nedd4-2 mediates neuronal activity and seizure susceptibility through AMPA receptors. PLoS Genet. 2017, 13, e1006634. [Google Scholar] [CrossRef] [PubMed]
- Jewett, K.A.; Zhu, J.; Tsai, N.-P. The tumor suppressor p53 guides GluA1 homeostasis through Nedd4-2 during chronic elevation of neuronal activity. J. Neurochem. 2015, 135, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Lee, K.Y.; Jong, T.T.; Tsai, N.-P. C2-lacking isoform of Nedd4-2 regulates excitatory synaptic strength through GluA1 ubiquitination-independent mechanisms. J. Neurochem. 2019, 151, 289–300. [Google Scholar] [CrossRef]
- Jin, H.; Chiou, T.-T.; Serwanski, D.R.; Miralles, C.P.; Pinal, N.; De Blas, A.L. Ring finger protein 34 (RNF34) interacts with and promotes γ-aminobutyric acid type-A receptor degradation via ubiquitination of the γ2 subunit. J. Biol. Chem. 2014, 289, 29420–29436. [Google Scholar] [CrossRef] [PubMed]
- Iida, Y.; Fujimori, T.; Okawa, K.; Nagata, K.; Wada, I.; Hosokawa, N. SEL1L protein critically determines the stability of the HRD1-SEL1L endoplasmic reticulum-associated degradation (ERAD) complex to optimize the degradation kinetics of ERAD substrates. J. Biol. Chem. 2011, 286, 16929–16939. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Tian, J.-H.; He, Y.-X.; Huang, Y.-Y.; Huang, Y.-Q.; Zhang, G.-P.; Luo, J.-D.; Xue, Q.; Yu, X.-Y.; Liu, Y.-H. Zonisamide alleviates cardiac hypertrophy in rats by increasing Hrd1 expression and inhibiting endoplasmic reticulum stress. Acta Pharmacol. Sin. 2021, 42, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Wu, Q.; He, Y.; Shen, Q.; Rekep, M.; Zhang, G.; Luo, J.; Xue, Q.; Liu, Y. Zonisamide, an antiepileptic drug, alleviates diabetic cardiomyopathy by inhibiting endoplasmic reticulum stress. Acta Pharmacol. Sin. 2021, 42, 393–403. [Google Scholar] [CrossRef]
- He, Y.-X.; Shen, Q.-Y.; Tian, J.-H.; Wu, Q.; Xue, Q.; Zhang, G.-P.; Wei, W.; Liu, Y.-H. Zonisamide Ameliorates Cognitive Impairment by Inhibiting ER Stress in a Mouse Model of Type 2 Diabetes Mellitus. Front. Aging Neurosci. 2020, 12, 192. [Google Scholar] [CrossRef] [PubMed]
- Omura, T.; Asari, M.; Yamamoto, J.; Kamiyama, N.; Oka, K.; Hoshina, C.; Maseda, C.; Awaya, T.; Tasaki, Y.; Shiono, H.; et al. HRD1 levels increased by zonisamide prevented cell death and caspase-3 activation caused by endoplasmic reticulum stress in SH-SY5Y cells. J. Mol. Neurosci. 2012, 46, 527–535. [Google Scholar] [CrossRef]
- Tsujii, S.; Ishisaka, M.; Shimazawa, M.; Hashizume, T.; Hara, H. Zonisamide suppresses endoplasmic reticulum stress-induced neuronal cell damage in vitro and in vivo. Eur. J. Pharmacol. 2015, 746, 301–307. [Google Scholar] [CrossRef]
- Omura, T.; Kaneko, M.; Okuma, Y.; Matsubara, K.; Nomura, Y. Endoplasmic reticulum stress and Parkinson’s disease: The role of HRD1 in averting apoptosis in neurodegenerative disease. Oxidative Med. Cell. Longev. 2013, 2013, 239854. [Google Scholar] [CrossRef] [PubMed]
- Nwosu, G.; Mermer, F.; Flamm, C.; Poliquin, S.; Shen, W.; Rigsby, K.; Kang, J.Q. 4-Phenylbutyrate restored γ-aminobutyric acid uptake and reduced seizures in SLC6A1 patient variant-bearing cell and mouse models. Brain Commun. 2022, 4, fcac144. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, M.J.; Shen, W.; Song, L.; Macdonald, R.L. Endoplasmic reticulum retention and associated degradation of a GABAA receptor epilepsy mutation that inserts an aspartate in the M3 transmembrane segment of the alpha1 subunit. J. Biol. Chem. 2005, 280, 37995–38004. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-Q.; Shen, W.; Macdonald, R.L. Two molecular pathways (NMD and ERAD) contribute to a genetic epilepsy associated with the GABAA receptor GABRA1 PTC mutation, 975delC, S326fs328X. J. Neurosci. 2009, 29, 2833–2844. [Google Scholar] [CrossRef] [PubMed]
- Crestani, F.; Lorez, M.; Baer, K.; Essrich, C.; Benke, D.; Laurent, J.P.; Belzung, C.; Fritschy, J.M.; Lüscher, B.; Mohler, H. Decreased GABAA-receptor clustering results in enhanced anxiety and a bias for threat cues. Nat. Neurosci. 1999, 2, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Borowicz, K.K.; Luszczki, J.J.; Sobieszek, G.; Ratnaraj, N.; Patsalos, P.N.; Czuczwar, S.J. Interactions between zonisamide and conventional antiepileptic drugs in the mouse maximal electroshock test model. Eur. Neuropsychopharmacol. 2007, 17, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Sonsalla, P.K.; Wong, L.-Y.; Winnik, B.; Buckley, B. The antiepileptic drug zonisamide inhibits MAO-B and attenuates MPTP toxicity in mice: Clinical relevance. Exp. Neurol. 2010, 221, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Biton, V. Clinical pharmacology and mechanism of action of zonisamide. Clin. Neuropharmacol. 2007, 30, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Ismail, F.S.; Faustmann, P.M.; Förster, E.; Corvace, F.; Faustmann, T.J. Tiagabine and zonisamide differentially regulate the glial properties in an astrocyte-microglia co-culture model of inflammation. Naunyn Schmiedebergs Arch. Pharmacol. 2023, 396, 3253–3267. [Google Scholar] [CrossRef] [PubMed]
- Volnova, A.; Tsytsarev, V.; Ganina, O.; Vélez-Crespo, G.E.; Alves, J.M.; Ignashchenkova, A.; Inyushin, M. The Anti-Epileptic Effects of Carbenoxolone In Vitro and In Vivo. Int. J. Mol. Sci. 2022, 23, 663. [Google Scholar] [CrossRef]
- Kang, J.-Q.; Shen, W.; Macdonald, R.L. Why does fever trigger febrile seizures? GABAA receptor gamma2 subunit mutations associated with idiopathic generalized epilepsies have temperature-dependent trafficking deficiencies. J. Neurosci. 2006, 26, 2590–2597. [Google Scholar] [CrossRef] [PubMed]
- Mermer, F.; Poliquin, S.; Zhou, S.; Wang, X.; Ding, Y.; Yin, F.; Shen, W.; Wang, J.; Rigsby, K.; Xu, D.; et al. Astrocytic GABA transporter 1 deficit in novel SLC6A1 variants mediated epilepsy: Connected from protein destabilization to seizures in mice and humans. Neurobiol. Dis. 2022, 172, 105810. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Wang, H.; Fan, H.; Wang, G. The endoplasmic reticulum (ER)-associated degradation system regulates aggregation and degradation of mutant neuroserpin. J. Biol. Chem. 2011, 286, 20835–20844. [Google Scholar] [CrossRef] [PubMed]
- Edvardson, S.; Nicolae, C.M.; Noh, G.J.; Burton, J.E.; Punzi, G.; Shaag, A.; Bischetsrieder, J.; De Grassi, A.; Pierri, C.L.; Elpeleg, O.; et al. Heterozygous RNF13 Gain-of-Function Variants Are Associated with Congenital Microcephaly, Epileptic Encephalopathy, Blindness, and Failure to Thrive. Am. J. Hum. Genet. 2019, 104, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Carvill, G.L.; McMahon, J.M.; Schneider, A.; Zemel, M.; Myers, C.T.; Saykally, J.; Nguyen, J.; Robbiano, A.; Zara, F.; Specchio, N.; et al. Mutations in the GABA Transporter SLC6A1 Cause Epilepsy with Myoclonic-Atonic Seizures. Am. J. Hum. Genet. 2015, 96, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Poliquin, S.; Mermer, F.; Eissman, J.; Delpire, E.; Wang, J.; Shen, W.; Cai, K.; Li, B.-M.; Li, Z.-Y.; et al. Endoplasmic reticulum retention and degradation of a mutation in SLC6A1 associated with epilepsy and autism. Mol. Brain 2020, 13, 76. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H.; Kato, M.; Mizuguchi, T.; Hamada, K.; Osaka, H.; Tohyama, J.; Uruno, K.; Kumada, S.; Nishiyama, K.; Nishimura, A.; et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat. Genet. 2008, 40, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Guiberson, N.G.L.; Pineda, A.; Abramov, D.; Kharel, P.; Carnazza, K.E.; Wragg, R.T.; Dittman, J.S.; Burré, J. Mechanism-based rescue of Munc18-1 dysfunction in varied encephalopathies by chemical chaperones. Nat. Commun. 2018, 9, 3986. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.C.; Zhang, J.; Pang, W.; Wang, S.; Lee, K.Y.; Cavaretta, J.P.; Walters, J.; Procko, E.; Tsai, N.-P.; Chung, H.J. Reduced axonal surface expression and phosphoinositide sensitivity in Kv7 channels disrupts their function to inhibit neuronal excitability in Kcnq2 epileptic encephalopathy. Neurobiol. Dis. 2018, 118, 76–93. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.-P.; Benske, T.M.; Ahn, L.Y.; Schaffer, A.E.; Paton, J.C.; Paton, A.W.; Mu, T.-W.; Wang, Y.-J. Adapting the endoplasmic reticulum proteostasis rescues epilepsy-associated NMDA receptor variants. Acta Pharmacol. Sin. 2024, 45, 282–297. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poliquin, S.; Nwosu, G.; Randhave, K.; Shen, W.; Flamm, C.; Kang, J.-Q. Modulating Endoplasmic Reticulum Chaperones and Mutant Protein Degradation in GABRG2(Q390X) Associated with Genetic Epilepsy with Febrile Seizures Plus and Dravet Syndrome. Int. J. Mol. Sci. 2024, 25, 4601. https://doi.org/10.3390/ijms25094601
Poliquin S, Nwosu G, Randhave K, Shen W, Flamm C, Kang J-Q. Modulating Endoplasmic Reticulum Chaperones and Mutant Protein Degradation in GABRG2(Q390X) Associated with Genetic Epilepsy with Febrile Seizures Plus and Dravet Syndrome. International Journal of Molecular Sciences. 2024; 25(9):4601. https://doi.org/10.3390/ijms25094601
Chicago/Turabian StylePoliquin, Sarah, Gerald Nwosu, Karishma Randhave, Wangzhen Shen, Carson Flamm, and Jing-Qiong Kang. 2024. "Modulating Endoplasmic Reticulum Chaperones and Mutant Protein Degradation in GABRG2(Q390X) Associated with Genetic Epilepsy with Febrile Seizures Plus and Dravet Syndrome" International Journal of Molecular Sciences 25, no. 9: 4601. https://doi.org/10.3390/ijms25094601