
Digenic Inheritance in Rare Disorders and Mitochondrial Disease—Crossing the Frontier to a More Comprehensive Understanding of Etiology

Abstract
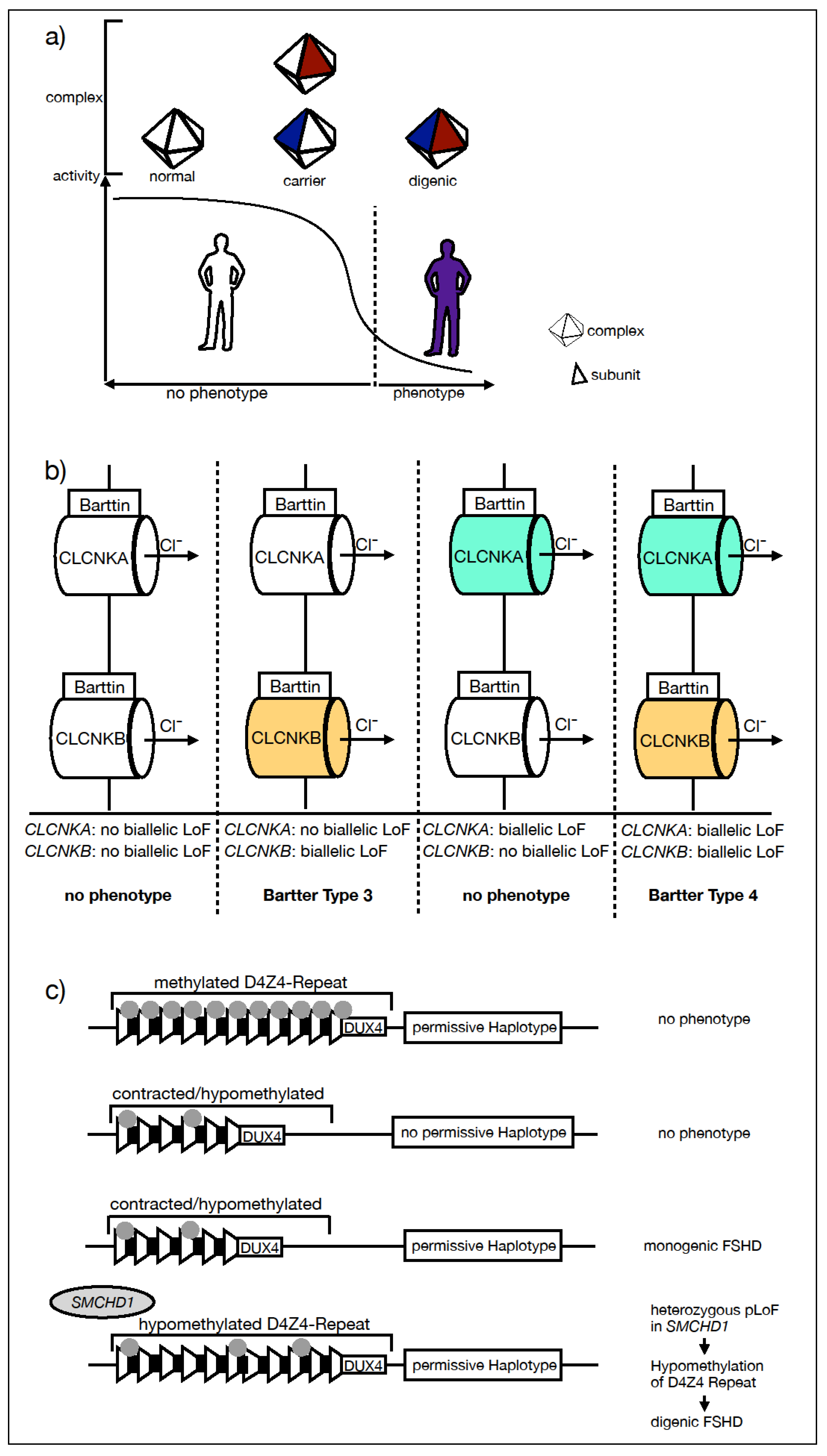
:1. Introduction
2. Pathomechanisms of Digenic Inheritance in Rare Disorders
2.1. Direct Interaction of Gene Products
2.2. Indirect Link of Gene Product Function
2.3. Epigenetic Modification—FSHD
3. Investigating Digenic Inheritance—Approaches and Challenges
3.1. Variant Evaluation in Monogenic Disorders
3.1.1. Population Data
3.1.2. Inheritance Patterns
3.1.3. Functional Data
3.1.4. Curated Knowledge Databases
3.1.5. Computational Prediction Tools
3.2. Statistical Approach in Frequent, Multifactorial Disorders

{kind=link}
{kind=link}
{kind=link}
| Monogenic | Digenic | Multifactorial | |
|---|---|---|---|
| Population Data | rare variants, MAF in keeping with disease incidence | combined MAF of variants in 2 genes in keeping with disease incidence expected to be attributable to digenic cause | common variants (MAF > 1%, >5%) |
| Inheritance Pattern | segregation in pedigree with the disease | segregation analysis is applicable | not applicable due to frequency of the investigated variants and smaller effect sizes |
| Functional Data | functional studies as the gold standard | functional studies challenging to conduct, but plausible mechanism of digenic interaction necessary | usually absent |
| Curated Databases | e.g., OMIM [15], GeneReviews [16] | e.g., OMIM [15], OLIDA [25] | e.g., GWAS Catalog |
| Computational Prediction | computational prediction based on knowledge (e.g., protein structure, sequence features, amino acid conservation) | novel, machine learning powered prediction tools | estimated effect size attributable to a locus |
| Statistical Analysis | burden testing | adaptive burden testing, novel statistical tools | GWAS |
3.3. Variant Evaluation in Digenic Disorders
3.3.1. Population Data
3.3.2. Inheritance Patterns
3.3.3. Statistical Approaches
3.3.4. Functional Data
3.3.5. Curated Knowledge Databases
3.3.6. Computational Prediction Tools
4. Digenic Inheritance in Mitochondrial Disorders
4.1. Phenotypic Spectrum as an Indicator of Genetic Modifying Factors
4.2. Digenic Inheritance in Mitochondrial Disorders with Reduced Penetrance
4.3. Digenic Inheritance in LHON/Leigh Spectrum Disorder
4.4. Approaches and Challenges Investigating Digenic Inheritance in Mitochondriopathies
5. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bamshad, M.J.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome Sequencing as a Tool for Mendelian Disease Gene Discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A. Magnitude of Mendelian Versus Complex Inheritance of Rare Disorders. Am. J. Med. Genet. Part A 2021, 185, 3287–3293. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.M.; Stark, Z.; Farnaes, L.; Tan, T.Y.; White, S.M.; Dimmock, D.; Kingsmore, S.F. Meta-Analysis of the Diagnostic and Clinical Utility of Genome and Exome Sequencing and Chromosomal Microarray in Children with Suspected Genetic Diseases. npj Genom. Med. 2018, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z. Clinical Whole-Exome Sequencing for the Diagnosis of Mendelian Disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef]
- Wright, C.F.; McRae, J.F.; Clayton, S.; Gallone, G.; Aitken, S.; FitzGerald, T.W.; Jones, P.; Prigmore, E.; Rajan, D.; Lord, J. Making New Genetic Diagnoses with Old Data: Iterative Reanalysis and Reporting from Genome-Wide Data in 1,133 Families with Developmental Disorders. Genet. Med. 2018, 20, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Gazzo, A.; Raimondi, D.; Daneels, D.; Moreau, Y.; Smits, G.; Van Dooren, S.; Lenaerts, T. Understanding Mutational Effects in Digenic Diseases. Nucleic Acids Res. 2017, 45, e140. [Google Scholar] [CrossRef] [PubMed]
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef]
- Conley, S.M.; Stuck, M.W.; Watson, J.N.; Zulliger, R.; Burnett, J.L.; Naash, M.I. Prph2 Initiates Outer Segment Morphogenesis but Maturation Requires Prph2/Rom1 Oligomerization. Hum. Mol. Genet. 2019, 28, 459–475. [Google Scholar] [CrossRef] [PubMed]
- Zulliger, R.; Conley, S.M.; Mwoyosvi, M.L.; Al-Ubaidi, M.R.; Naash, M.I. Oligomerization of Prph2 and Rom1 Is Essential for Photoreceptor Outer Segment Formation. Hum. Mol. Genet. 2018, 27, 3507–3518. [Google Scholar] [CrossRef] [PubMed]
- Kajiwara, K.; Berson, E.L.; Dryja, T.P. Digenic Retinitis Pigmentosa Due to Mutations at the Unlinked Peripherin/RDS and ROM1 Loci. Science 1994, 264, 1604–1608. [Google Scholar] [CrossRef]
- Kazmierczak, P.; Sakaguchi, H.; Tokita, J.; Wilson-Kubalek, E.M.; Milligan, R.A.; Müller, U.; Kachar, B. Cadherin 23 and Protocadherin 15 Interact to Form Tip-Link Filaments in Sensory Hair Cells. Nature 2007, 449, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.Y.; Yan, D.; Ouyang, X.M.; Du, L.L.; Yu, H.; Chang, B.; Johnson, K.R.; Liu, X.Z. Digenic Inheritance of Deafness Caused by Mutations in Genes Encoding Cadherin 23 and Protocadherin 15 in Mice and Humans. Hum. Mol. Genet. 2005, 14, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Estévez, R.; Boettger, T.; Stein, V.; Birkenhäger, R.; Otto, E.; Hildebrandt, F.; Jentsch, T.J. Barttin Is a Cl− Channel Beta-Subunit Crucial for Renal Cl− Reabsorption and Inner Ear K+ Secretion. Nature 2001, 414, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Miyamura, N.; Matsumoto, K.; Taguchi, T.; Tokunaga, H.; Nishikawa, T.; Nishida, K.; Toyonaga, T.; Sakakida, M.; Araki, E. Atypical Bartter Syndrome with Sensorineural Deafness with G47R Mutation of the β-Subunit for CLC-Ka and CLC-Kb Chloride Channels, Barttin. J. Clin. Endocrinol. Metab. 2003, 88, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Schlingmann, K.P.; Konrad, M.; Jeck, N.; Waldegger, P.; Reinalter, S.C.; Holder, M.; Seyberth, H.W.; Waldegger, S. Salt Wasting and Deafness Resulting from Mutations in Two Chloride Channels. N. Engl. J. Med. 2004, 350, 1314–1319. [Google Scholar] [CrossRef] [PubMed]
- Nozu, K.; Inagaki, T.; Fu, X.J.; Nozu, Y.; Kaito, H.; Kanda, K.; Sekine, T.; Igarashi, T.; Nakanishi, K.; Yoshikawa, N.; et al. Molecular Analysis of Digenic Inheritance in Bartter Syndrome with Sensorineural Deafness. J. Med. Genet. 2008, 45, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Lemmers, R.J.; Tawil, R.; Petek, L.M.; Balog, J.; Block, G.J.; Santen, G.W.; Amell, A.M.; van der Vliet, P.J.; Almomani, R.; Straasheijm, K.R.; et al. Digenic Inheritance of an SMCHD1 Mutation and an FSHD-Permissive D4Z4 Allele Causes Facioscapulohumeral Muscular Dystrophy Type 2. Nat. Genet. 2012, 44, 1370–1374. [Google Scholar] [CrossRef] [PubMed]
- Van den Boogaard, M.L.; Lemmers, R.; Balog, J.; Wohlgemuth, M.; Auranen, M.; Mitsuhashi, S.; van der Vliet, P.J.; Straasheijm, K.R.; van den Akker, R.F.P.; Kriek, M.; et al. Mutations in Dnmt3b Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am. J. Hum. Genet. 2016, 98, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, K.; Šikrová, D.; Mitsuhashi, S.; Masuda, H.; Sekiguchi, Y.; Sugiyama, A.; Shibuya, K.; Lemmers, R.; Goossens, R.; Ogawa, M.; et al. Homozygous Nonsense Variant in LRIF1 Associated with Facioscapulohumeral Muscular Dystrophy. Neurology 2020, 94, e2441–e2447. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. Clinvar: Improving Access to Variant Interpretations and Supporting Evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Amberger, J.S.; Bocchini, C.A.; Scott, A.F.; Hamosh, A. Omim.Org: Leveraging Knowledge across Phenotype-Gene Relationships. Nucleic Acids Res. 2019, 47, D1038–D1043. [Google Scholar] [CrossRef] [PubMed]
- Adam, M.P.; Feldman, J.; Mirzaa, G.M.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.H.; Gripp, K.W.; Amemiya, A. (Eds.) GeneReviews(®); University of Washington: Seattle, WA, USA, 1993–2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1116/ (accessed on 11 April 2024).
- Gallagher, M.D.; Chen-Plotkin, A.S. The Post-Gwas Era: From Association to Function. Am. J. Hum. Genet. 2018, 102, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Verlouw, J.A.M.; Clemens, E.; de Vries, J.H.; Zolk, O.; Verkerk, A.; Am Zehnhoff-Dinnesen, A.; Medina-Gomez, C.; Lanvers-Kaminsky, C.; Rivadeneira, F.; Langer, T.; et al. A Comparison of Genotyping Arrays. Eur. J. Hum. Genet. 2021, 29, 1611–1624. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the Missing Heritability of Complex Diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef]
- Guo, M.H.; Dauber, A.; Lippincott, M.F.; Chan, Y.M.; Salem, R.M.; Hirschhorn, J.N. Determinants of Power in Gene-Based Burden Testing for Monogenic Disorders. Am. J. Hum. Genet. 2016, 99, 527–539. [Google Scholar] [CrossRef]
- Lee, S.; Abecasis, G.R.; Boehnke, M.; Lin, X. Rare-Variant Association Analysis: Study Designs and Statistical Tests. Am. J. Hum. Genet. 2014, 95, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Kerner, G.; Bouaziz, M.; Cobat, A.; Bigio, B.; Timberlake, A.T.; Bustamante, J.; Lifton, R.P.; Casanova, J.L.; Abel, L. A Genome-Wide Case-Only Test for the Detection of Digenic Inheritance in Human Exomes. Proc. Natl. Acad. Sci. USA 2020, 117, 19367–19375. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhu, Y.; Xiong, M. Genome-Wide Gene-Gene Interaction Analysis for Next-Generation Sequencing. Eur. J. Hum. Genet. 2016, 24, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Gazzo, A.M.; Daneels, D.; Cilia, E.; Bonduelle, M.; Abramowicz, M.; Van Dooren, S.; Smits, G.; Lenaerts, T. Dida: A Curated and Annotated Digenic Diseases Database. Nucleic Acids Res. 2016, 44, D900–D907. [Google Scholar] [CrossRef] [PubMed]
- Nachtegael, C.; Gravel, B.; Dillen, A.; Smits, G.; Nowé, A.; Papadimitriou, S.; Lenaerts, T. Scaling up Oligogenic Diseases Research with Olida: The Oligogenic Diseases Database. Database 2022, 2022, baac023. [Google Scholar] [CrossRef] [PubMed]
- Renaux, A.; Papadimitriou, S.; Versbraegen, N.; Nachtegael, C.; Boutry, S.; Nowé, A.; Smits, G.; Lenaerts, T. Orval: A Novel Platform for the Prediction and Exploration of Disease-Causing Oligogenic Variant Combinations. Nucleic Acids Res. 2019, 47, W93–W98. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, A.; Ott, J. Machine Learning Approaches to Explore Digenic Inheritance. Trends Genet. 2022, 38, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, S.; Gravel, B.; Nachtegael, C.; De Baere, E.; Loeys, B.; Vikkula, M.; Smits, G.; Lenaerts, T. Toward Reporting Standards for the Pathogenicity of Variant Combinations Involved in Multilocus/Oligogenic Diseases. HGG Adv. 2023, 4, 100165. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Cogan, J.D.; Newman, J.H.; Phillips, J.A., 3rd; Hamid, R.; Meiler, J.; Capra, J.A. Identifying Digenic Disease Genes Via Machine Learning in the Undiagnosed Diseases Network. Am. J. Hum. Genet. 2021, 108, 1946–1963. [Google Scholar] [CrossRef]
- Boudellioua, I.; Kulmanov, M.; Schofield, P.N.; Gkoutos, G.V.; Hoehndorf, R. Oligopvp: Phenotype-Driven Analysis of Individual Genomic Information to Prioritize Oligogenic Disease Variants. Sci. Rep. 2018, 8, 14681. [Google Scholar] [CrossRef] [PubMed]
- Versbraegen, N.; Fouché, A.; Nachtegael, C.; Papadimitriou, S.; Gazzo, A.; Smits, G.; Lenaerts, T. Using Game Theory and Decision Decomposition to Effectively Discern and Characterise Bi-Locus Diseases. Artif. Intell. Med. 2019, 99, 101690. [Google Scholar] [CrossRef] [PubMed]
- Schlieben, L.D.; Prokisch, H. The Dimensions of Primary Mitochondrial Disorders. Front. Cell Dev. Biol. 2020, 8, 600079. [Google Scholar] [CrossRef] [PubMed]
- Stendel, C.; Neuhofer, C.; Floride, E.; Yuqing, S.; Ganetzky, R.D.; Park, J.; Freisinger, P.; Kornblum, C.; Kleinle, S.; Schöls, L.; et al. Delineating MT-ATP6-Associated Disease: From Isolated Neuropathy to Early Onset Neurodegeneration. Neurol. Genet. 2020, 6, e393. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.S.; Martikainen, M.H.; Gorman, G.S.; Blain, A.; Bugiardini, E.; Bunting, A.; Schaefer, A.M.; Alston, C.L.; Blakely, E.L.; Sharma, S.; et al. Pathogenic Variants inMT-ATP6: A United Kingdom-Based Mitochondrial Disease Cohort Study. Ann. Neurol. 2019, 86, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Seed, L.M.; Dean, A.; Krishnakumar, D.; Phyu, P.; Horvath, R.; Harijan, P.D. Molecular and Neurological Features of MELAS Syndrome in Paediatric Patients: A Case Series and Review of the Literature. Mol. Genet. Genom. Med. 2022, 10, e1955. [Google Scholar] [CrossRef] [PubMed]
- Cannon, S.J.; Hall, T.; Hawkes, G.; Colclough, K.; Boggan, R.M.; Wright, C.F.; Pickett, S.J.; Hattersley, A.T.; Weedon, M.N.; Patel, K.A. Penetrance and Expressivity of Mitochondrial Variants in a Large Clinically Unselected Population. Hum. Mol. Genet. 2024, 33, 465–474. [Google Scholar] [CrossRef]
- Manwaring, N.; Jones, M.M.; Wang, J.J.; Rochtchina, E.; Howard, C.; Mitchell, P.; Sue, C.M. Population Prevalence of the MELAS A3243G Mutation. Mitochondrion 2007, 7, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Nonaka, I.; Horai, S. A Mutation in the tRNALeu(Uur) Gene Associated with the MELAS Subgroup of Mitochondrial Encephalomyopathies. Nature 1990, 348, 651–653. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Momoi, M.Y.; Tominaga, K.; Momoi, T.; Nihei, K.; Yanagisawa, M.; Kagawa, Y.; Ohta, S. A Point Mutation in the Mitochondrial tRNALeu(Uur) Gene in Melas (Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis and Stroke-Like Episodes). Biochem. Biophys. Res. Commun. 1990, 173, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Donati, A.; Minetti, C.; Moggio, M.; Mongini, T.; et al. The M.3243A>G Mitochondrial DNA Mutation and Related Phenotypes. A Matter of Gender? J. Neurol. 2014, 261, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Nesbitt, V.; Pitceathly, R.D.; Turnbull, D.M.; Taylor, R.W.; Sweeney, M.G.; Mudanohwo, E.E.; Rahman, S.; Hanna, M.G.; McFarland, R. The Uk Mrc Mitochondrial Disease Patient Cohort Study: Clinical Phenotypes Associated with the M.3243A>G Mutation--Implications for Diagnosis and Management. J. Neurol. Neurosurg. Psychiatry 2013, 84, 936–938. [Google Scholar] [CrossRef] [PubMed]
- Pickett, S.J.; Grady, J.P.; Ng, Y.S.; Gorman, G.S.; Schaefer, A.M.; Wilson, I.J.; Cordell, H.J.; Turnbull, D.M.; Taylor, R.W.; McFarland, R. Phenotypic Heterogeneity in M.3243A>G Mitochondrial Disease: The Role of Nuclear Factors. Ann. Clin. Transl. Neurol. 2018, 5, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Boggan, R.M.; Ng, Y.S.; Franklin, I.G.; Alston, C.L.; Blakely, E.L.; Büchner, B.; Bugiardini, E.; Colclough, K.; Feeney, C.; Hanna, M.G.; et al. Defining the Nuclear Genetic Architecture of a Common Maternally Inherited Mitochondrial Disorder. medRxiv 2022. [Google Scholar] [CrossRef]
- Horvath, R.; Kemp, J.P.; Tuppen, H.A.; Hudson, G.; Oldfors, A.; Marie, S.K.; Moslemi, A.R.; Servidei, S.; Holme, E.; Shanske, S.; et al. Molecular Basis of Infantile Reversible Cytochrome C Oxidase Deficiency Myopathy. Brain 2009, 132 Pt 11, 3165–3174. [Google Scholar] [CrossRef] [PubMed]
- Hathazi, D.; Griffin, H.; Jennings, M.J.; Giunta, M.; Powell, C.; Pearce, S.F.; Munro, B.; Wei, W.; Boczonadi, V.; Poulton, J.; et al. Metabolic Shift Underlies Recovery in Reversible Infantile Respiratory Chain Deficiency. EMBO J. 2020, 39, e105364. [Google Scholar] [CrossRef]
- Mackey, D.A.; Ong, J.S.; MacGregor, S.; Whiteman, D.C.; Craig, J.E.; Lopez Sanchez, M.I.G.; Kearns, L.S.; Staffieri, S.E.; Clarke, L.; McGuinness, M.B.; et al. Is the Disease Risk and Penetrance in Leber Hereditary Optic Neuropathy Actually Low? Am. J. Hum. Genet. 2023, 110, 170–176. [Google Scholar] [CrossRef]
- Lopez Sanchez, M.I.G.; Kearns, L.S.; Staffieri, S.E.; Clarke, L.; McGuinness, M.B.; Meteoukki, W.; Samuel, S.; Ruddle, J.B.; Chen, C.; Fraser, C.L.; et al. Establishing Risk of Vision Loss in Leber Hereditary Optic Neuropathy. Am. J. Hum. Genet. 2021, 108, 2159–2170. [Google Scholar] [CrossRef] [PubMed]
- Watson, E.C.; Davis, R.L.; Ravishankar, S.; Copty, J.; Kummerfeld, S.; Sue, C.M. Low Disease Risk and Penetrance in Leber Hereditary Optic Neuropathy. Am. J. Hum. Genet. 2023, 110, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Rabenstein, A.; Catarino, C.B.; Rampeltshammer, V.; Schindler, D.; Gallenmüller, C.; Priglinger, C.; Pogarell, O.; Rüther, T.; Klopstock, T. Smoking and Alcohol, Health-Related Quality of Life and Psychiatric Comorbidities in Leber’s Hereditary Optic Neuropathy Mutation Carriers: A Prospective Cohort Study. Orphanet J. Rare Dis. 2021, 16, 127. [Google Scholar] [CrossRef]
- Fruhman, G.; Landsverk, M.L.; Lotze, T.E.; Hunter, J.V.; Wangler, M.F.; Adesina, A.M.; Wong, L.J.; Scaglia, F. Atypical Presentation of Leigh Syndrome Associated with a Leber Hereditary Optic Neuropathy Primary Mitochondrial DNA Mutation. Mol. Genet. Metab. 2011, 103, 153–160. [Google Scholar] [CrossRef]
- Miyaue, N.; Yamanishi, Y.; Tada, S.; Ando, R.; Yabe, H.; Nagai, M.; Nomoto, M. Repetitive Brainstem Lesions in Mitochondrial DNA 11778G>A Mutation of Leber Hereditary Optic Neuropathy. eNeurologicalSci 2019, 14, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Paquay, S.; Benoit, V.; Wetzburger, C.; Cordonnier, M.; Meire, F.; Charon, A.; Roland, D.; Van Coster, R.; Nassogne, M.C.; Maystadt, I. Uncommon Leber “Plus” Disease Associated with Mitochondrial Mutation M.11778G>A in a Premature Child. J. Child Neurol. 2014, 29, NP18–NP23. [Google Scholar] [CrossRef]
- Stenton, S.L.; Sheremet, N.L.; Catarino, C.B.; Andreeva, N.A.; Assouline, Z.; Barboni, P.; Barel, O.; Berutti, R.; Bychkov, I.; Caporali, L.; et al. Impaired Complex I Repair Causes Recessive Leber’s Hereditary Optic Neuropathy. J. Clin. Investig. 2021, 131, e138267. [Google Scholar] [CrossRef] [PubMed]
- Stenton, S.L.; Tesarova, M.; Sheremet, N.L.; Catarino, C.B.; Carelli, V.; Ciara, E.; Curry, K.; Engvall, M.; Fleming, L.R.; Freisinger, P.; et al. DNAJC30 Defect: A Frequent Cause of Recessive Leber Hereditary Optic Neuropathy and Leigh Syndrome. Brain 2022, 145, 1624–1631. [Google Scholar] [CrossRef]
- Zawadzka, M.; Krygier, M.; Pawłowicz, M.; Wilke, M.; Rutkowska, K.; Gueguen, N.; Desquiret-Dumas, V.; Klee, E.W.; Schimmenti, L.A.; Sławek, J.; et al. Expanding the Phenotype of DNAJC30-Associated Leigh Syndrome. Clin. Genet. 2022, 102, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Nesti, C.; Ticci, C.; Rubegni, A.; Doccini, S.; Scaturro, G.; Vetro, A.; Guerrini, R.; Santorelli, F.M.; Procopio, E. Additive Effect of DNAJC30 and NDUFA9 Mutations Causing Leigh Syndrome. J. Neurol. 2023, 270, 3266–3269. [Google Scholar] [CrossRef] [PubMed]
- Blickhäuser, B.; Stenton, S.L.; Neuhofer, C.M.; Floride, E.; Nesbitt, V.; Fratter, C.; Koch, J.; Kauffmann, B.; Catarino, C.; Schlieben, L.D.; et al. Digenic Leigh Syndrome on the Background of the M.11778G>A Leber Hereditary Optic Neuropathy Variant. Brain 2024, awae057. [Google Scholar] [CrossRef] [PubMed]
- Uittenbogaard, M.; Wang, H.; Zhang, V.W.; Wong, L.J.; Brantner, C.A.; Gropman, A.; Chiaramello, A. The Nuclear Background Influences the Penetrance of the near-Homoplasmic M.1630 A > G MELAS Variant in a Symptomatic Proband and Asymptomatic Mother. Mol. Genet. Metab. 2019, 126, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Zhang, J.; Yi, Q.; Meng, F.; Yu, J.; Ji, Y.; Mo, J.Q.; Tong, Y.; Jiang, P.; Guan, M.X. Leber’s Hereditary Optic Neuropathy Arising from the Synergy between ND1 3635G>A Mutation and Mitochondrial YARS2 Mutations. Investig. Ophthalmol. Vis. Sci. 2021, 62, 22. [Google Scholar] [CrossRef]
- Lopes, A.F.C. Mitochondrial Metabolism and DNA Methylation: A Review of the Interaction between Two Genomes. Clin. Epigenet. 2020, 12, 182. [Google Scholar] [CrossRef] [PubMed]
- Shock, L.S.; Thakkar, P.V.; Peterson, E.J.; Moran, R.G.; Taylor, S.M. DNA Methyltransferase 1, Cytosine Methylation, and Cytosine Hydroxymethylation in Mammalian Mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 3630–3635. [Google Scholar] [CrossRef] [PubMed]
- Chestnut, B.A.; Chang, Q.; Price, A.; Lesuisse, C.; Wong, M.; Martin, L.J. Epigenetic Regulation of Motor Neuron Cell Death through DNA Methylation. J. Neurosci. 2011, 31, 16619–16636. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.K.; Mangalhara, K.C.; Prakasam, G.; Bamezai, R.N.K. DNA Methyltransferase1 (DNMT1) Isoform3 Methylates Mitochondrial Genome and Modulates Its Biology. Sci. Rep. 2017, 7, 1525. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Zhang, J.; Liu, B.; Xu, J.; Cai, B.; Yang, H.; Straube, J.; Yu, X.; Ma, T. Biological Roles of Rna M5C Modification and Its Implications in Cancer Immunotherapy. Biomark. Res. 2022, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Van Haute, L.; Dietmann, S.; Kremer, L.; Hussain, S.; Pearce, S.F.; Powell, C.A.; Rorbach, J.; Lantaff, R.; Blanco, S.; Sauer, S.; et al. Deficient Methylation and Formylation of mt-tRNAMet Wobble Cytosine in a Patient Carrying Mutations in NSUN3. Nat. Commun. 2016, 7, 12039. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neuhofer, C.M.; Prokisch, H. Digenic Inheritance in Rare Disorders and Mitochondrial Disease—Crossing the Frontier to a More Comprehensive Understanding of Etiology. Int. J. Mol. Sci. 2024, 25, 4602. https://doi.org/10.3390/ijms25094602
Neuhofer CM, Prokisch H. Digenic Inheritance in Rare Disorders and Mitochondrial Disease—Crossing the Frontier to a More Comprehensive Understanding of Etiology. International Journal of Molecular Sciences. 2024; 25(9):4602. https://doi.org/10.3390/ijms25094602
Chicago/Turabian StyleNeuhofer, Christiane M., and Holger Prokisch. 2024. "Digenic Inheritance in Rare Disorders and Mitochondrial Disease—Crossing the Frontier to a More Comprehensive Understanding of Etiology" International Journal of Molecular Sciences 25, no. 9: 4602. https://doi.org/10.3390/ijms25094602