
Strengthening the Evidence for a Causal Link between Type 2 Diabetes Mellitus and Pancreatic Cancer: Insights from Two-Sample and Multivariable Mendelian Randomization

Abstract
:1. Introduction
2. Results
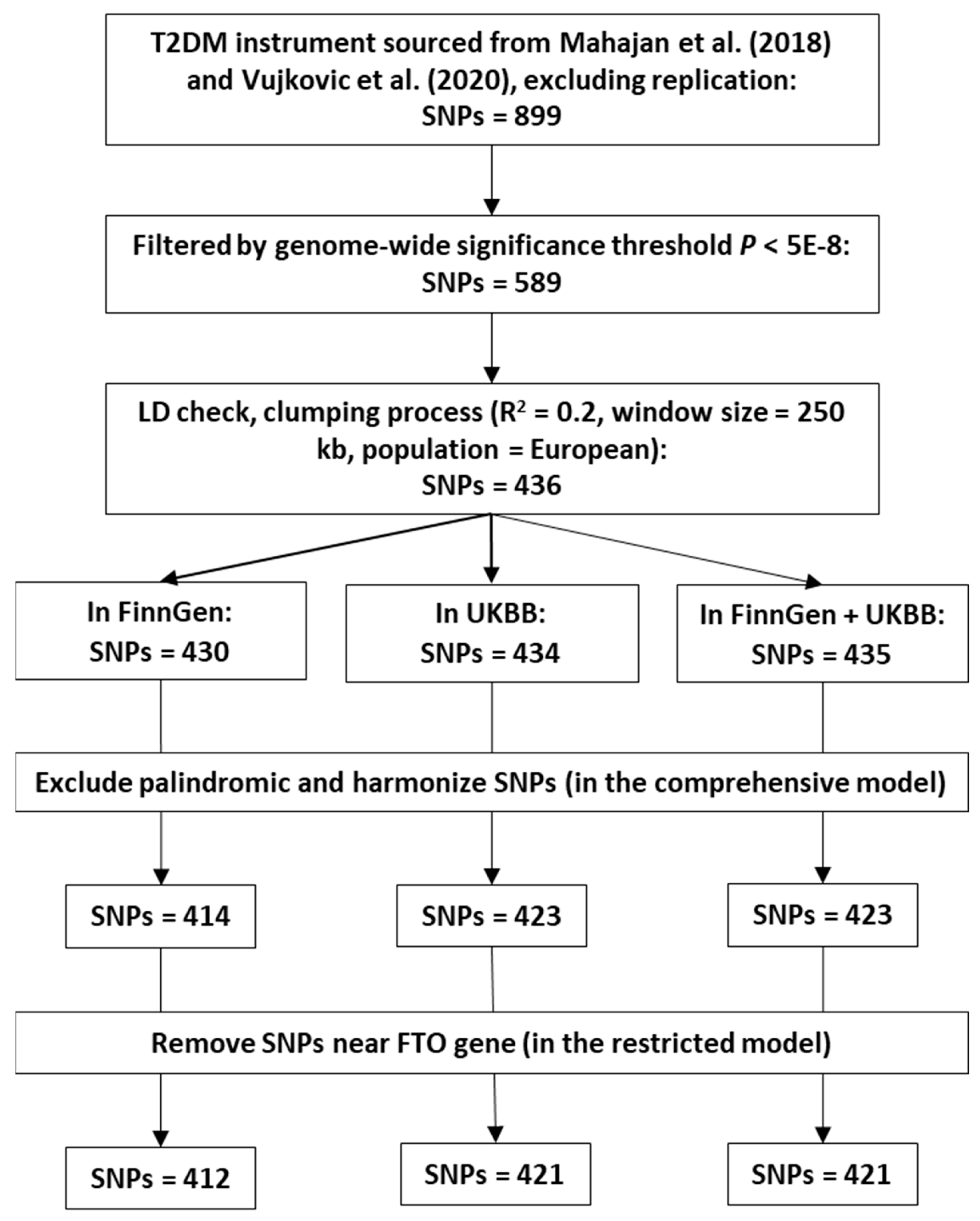
2.1. Selection of Instrumental Variables
2.2. MR Analysis
2.3. Sensitivity Analysis
2.4. Multivariable Mendelian Randomization (MR)
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Genetic Instrument Selection
4.3. Outcome Data Source
4.4. The Strength of the Selection of Instrumental Variables and Power Calculations
4.5. Two-Sample Mendelian Randomization (2SMR) Analysis
4.6. Multivariable Mendelian Randomization (MR)
4.7. Statistical Analysis
4.8. Ethics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.E.M.; Lam, F.; Laversanne, M.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France, 2020; Available online: https://gco.iarc.who.int/today (accessed on 6 February 2024).
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.E.M.; Lam, F.; Colombet, M.; Mery, L.; Pineros, M.; Znaor, A.; Soerjomataram, I. Global Cancer Observatory: Cancer Tomorrow; International Agency for Research on Cancer: Lyon, France, 2020; Available online: http://gco.iarc.fr/tomorrow/graphic-isotype?type=1&population=900&mode=population&sex=0&cancer=39&age_group=value&apc_male=0&apc_female=0 (accessed on 7 February 2024).
- Klein, A.P. Pancreatic cancer epidemiology: Understanding the role of lifestyle and inherited risk factors. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 493–502. [Google Scholar] [CrossRef]
- Hu, J.X.; Zhao, C.F.; Chen, W.B.; Liu, Q.C.; Li, Q.W.; Lin, Y.Y.; Gao, F. Pancreatic cancer: A review of epidemiology, trend, and risk factors. World J. Gastroenterol. 2021, 27, 4298–4321. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Chen, H.; Lu, M.; Zhang, Y.; Lu, B.; You, L.; Zhang, T.; Dai, M.; Zhao, Y. Advances in the epidemiology of pancreatic cancer: Trends, risk factors, screening, and prognosis. Cancer Lett. 2021, 520, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Liu, W. Pancreatic Cancer: A Review of Risk Factors, Diagnosis, and Treatment. Technol. Cancer Res. Treat. 2020, 19, 1533033820962117. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef]
- Khan, M.A.B.; Hashim, M.J.; King, J.K.; Govender, R.D.; Mustafa, H.; Al Kaabi, J. Epidemiology of Type 2 Diabetes—Global Burden of Disease and Forecasted Trends. J. Epidemiol. Glob. Health 2020, 10, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.E.; Dorrance, K.A.; Oxenreiter, M.M.; Yan, K.R.; Close, K.L. The type 2 diabetes ‘modern preventable pandemic’ and replicable lessons from the COVID-19 crisis. Prev. Med. Rep. 2022, 25, 101636. [Google Scholar] [CrossRef]
- Burden of diabetes and hyperglycaemia in adults in the Americas, 1990-2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Diabetes Endocrinol. 2022, 10, 655–667. [CrossRef]
- Ke, T.M.; Lophatananon, A.; Muir, K.R. Risk Factors Associated with Pancreatic Cancer in the UK Biobank Cohort. Cancers 2022, 14, 4991. [Google Scholar] [CrossRef] [PubMed]
- Ke, T.M.; Lophatananon, A.; Muir, K.R. An Integrative Pancreatic Cancer Risk Prediction Model in the UK Biobank. Biomedicines 2023, 11, 3206. [Google Scholar] [CrossRef] [PubMed]
- CDC: Diabetes Fast Facts. Available online: https://www.cdc.gov/diabetes/basics/quick-facts.html#:~:text=Diabetes%20Fast%20Facts,-Diabetes%20Fast%20Facts&text=Type%202%20diabetes%20accounts%20for,for%20approximately%205%2D10%25 (accessed on 14 February 2024).
- Andersen, D.K.; Korc, M.; Petersen, G.M.; Eibl, G.; Li, D.; Rickels, M.R.; Chari, S.T.; Abbruzzese, J.L. Diabetes, Pancreatogenic Diabetes, and Pancreatic Cancer. Diabetes 2017, 66, 1103–1110. [Google Scholar] [CrossRef]
- Elena, J.W.; Steplowski, E.; Yu, K.; Hartge, P.; Tobias, G.S.; Brotzman, M.J.; Chanock, S.J.; Stolzenberg-Solomon, R.Z.; Arslan, A.A.; Bueno-De-Mesquita, H.B.; et al. Diabetes and risk of pancreatic cancer: A pooled analysis from the pancreatic cancer cohort consortium. Cancer Causes Control 2013, 24, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Wang, B.; Zhang, X.; Hao, L.; Hu, X.; Li, Z.; Sun, S. Long-Term Diabetes Mellitus Is Associated with an Increased Risk of Pancreatic Cancer: A Meta-Analysis. PLoS ONE 2015, 10, e0134321. [Google Scholar] [CrossRef]
- Lu, Y.; Gentiluomo, M.; Lorenzo-Bermejo, J.; Morelli, L.; Obazee, O.; Campa, D.; Canzian, F. Mendelian randomisation study of the effects of known and putative risk factors on pancreatic cancer. J. Med. Genet. 2020, 57, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Kar, S.; Carter, P.; Vithayathil, M.; Mason, A.M.; Burgess, S.; Larsson, S.C. Is Type 2 Diabetes Causally Associated With Cancer Risk? Evidence From a Two-Sample Mendelian Randomization Study. Diabetes 2020, 69, 1588–1596. [Google Scholar] [CrossRef] [PubMed]
- Carreras-Torres, R.; Johansson, M.; Gaborieau, V.; Haycock, P.C.; Wade, K.H.; Relton, C.L.; Martin, R.M.; Davey Smith, G.; Brennan, P. The Role of Obesity, Type 2 Diabetes, and Metabolic Factors in Pancreatic Cancer: A Mendelian Randomization Study. J. Natl. Cancer Inst. 2017, 109, djx012. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yuan, S.; Fu, T.; Ruan, X.; Qiao, J.; Wang, X.; Li, X.; Gill, D.; Burgess, S.; Giovannucci, E.L.; et al. Gastrointestinal Consequences of Type 2 Diabetes Mellitus and Impaired Glycemic Homeostasis: A Mendelian Randomization Study. Diabetes Care 2023, 46, 828–835. [Google Scholar] [CrossRef]
- Lee, K.; Lim, C.Y. Mendelian Randomization Analysis in Observational Epidemiology. J. Lipid Atheroscler. 2019, 8, 67–77. [Google Scholar] [CrossRef]
- Sanderson, E.; Glymour, M.M.; Holmes, M.V.; Kang, H.; Morrison, J.; Munafò, M.R.; Palmer, T.; Schooling, C.M.; Wallace, C.; Zhao, Q.; et al. Mendelian randomization. Nat. Rev. Methods Primers 2022, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Foley, C.N.; Allara, E.; Staley, J.R.; Howson, J.M.M. A robust and efficient method for Mendelian randomization with hundreds of genetic variants. Nat. Commun. 2020, 11, 376. [Google Scholar] [CrossRef] [PubMed]
- Richmond, R.C.; Davey Smith, G. Mendelian Randomization: Concepts and Scope. Cold Spring Harb. Perspect. Med. 2022, 12, a040501. [Google Scholar] [CrossRef] [PubMed]
- Hemani, G.; Zheng, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 2018, 7, e34408. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, F.P.; Davies, N.M.; Hemani, G.; Davey Smith, G. Two-sample Mendelian randomization: Avoiding the downsides of a powerful, widely applicable but potentially fallible technique. Int. J. Epidemiol. 2016, 45, 1717–1726. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, A.; Taliun, D.; Thurner, M.; Robertson, N.R.; Torres, J.M.; Rayner, N.W.; Payne, A.J.; Steinthorsdottir, V.; Scott, R.A.; Grarup, N.; et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 2018, 50, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Vujkovic, M.; Keaton, J.M.; Lynch, J.A.; Miller, D.R.; Zhou, J.; Tcheandjieu, C.; Huffman, J.E.; Assimes, T.L.; Lorenz, K.; Zhu, X.; et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat. Genet. 2020, 52, 680–691. [Google Scholar] [CrossRef]
- Morris, A.P.; Voight, B.F.; Teslovich, T.M.; Ferreira, T.; Segrè, A.V.; Steinthorsdottir, V.; Strawbridge, R.J.; Khan, H.; Grallert, H.; Mahajan, A.; et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 2012, 44, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, K.J.; Ferreira, T.; Lee, Y.; Raimondo, A.; Mägi, R.; Reschen, M.E.; Mahajan, A.; Locke, A.; Rayner, N.W.; Robertson, N.; et al. Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat. Genet. 2015, 47, 1415–1425. [Google Scholar] [CrossRef]
- Xue, A.; Wu, Y.; Zhu, Z.; Zhang, F.; Kemper, K.E.; Zheng, Z.; Yengo, L.; Lloyd-Jones, L.R.; Sidorenko, J.; Wu, Y.; et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat. Commun. 2018, 9, 2941. [Google Scholar] [CrossRef]
- Pearson-Stuttard, J.; Papadimitriou, N.; Markozannes, G.; Cividini, S.; Kakourou, A.; Gill, D.; Rizos, E.C.; Monori, G.; Ward, H.A.; Kyrgiou, M.; et al. Type 2 Diabetes and Cancer: An Umbrella Review of Observational and Mendelian Randomization Studies. Cancer Epidemiol. Biomark. Prev. 2021, 30, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Vittinghoff, E.; Bertenthal, D.; Corley, D.; Shen, H.; Walter, L.C.; McQuaid, K. New-onset diabetes and pancreatic cancer. Clin. Gastroenterol. Hepatol. 2006, 4, 1366–1372, quiz 1301. [Google Scholar] [CrossRef] [PubMed]
- Toledo, F.G.S.; Chari, S.; Yadav, D. Understanding the Contribution of Insulin Resistance to the Risk of Pancreatic Cancer. Am. J. Gastroenterol. 2021, 116, 669–670. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Herrington, M.; Larsson, J.; Permert, J. The relationship between diabetes and pancreatic cancer. Mol. Cancer 2003, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Guo, Y.; Du, J.; Gu, J.; Kong, L.; Tao, B.; Li, J.; Fu, D. The Intricate Crosstalk Between Insulin and Pancreatic Ductal Adenocarcinoma: A Review From Clinical to Molecular. Front. Cell Dev. Biol. 2022, 10, 844028. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Shen, X.; Lei, J.; Xu, Q.; Yu, Y.; Li, R.; Wu, E.; Ma, Q. Hyperglycemia, a neglected factor during cancer progression. Biomed. Res. Int. 2014, 2014, 461917. [Google Scholar] [CrossRef] [PubMed]
- Giri, B.; Dey, S.; Das, T.; Sarkar, M.; Banerjee, J.; Dash, S.K. Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction, infection, cancer progression and other pathophysiological consequences: An update on glucose toxicity. Biomed. Pharmacother. 2018, 107, 306–328. [Google Scholar] [CrossRef] [PubMed]
- Ramteke, P.; Deb, A.; Shepal, V.; Bhat, M.K. Hyperglycemia Associated Metabolic and Molecular Alterations in Cancer Risk, Progression, Treatment, and Mortality. Cancers 2019, 11, 1402. [Google Scholar] [CrossRef]
- Sato, K.; Hikita, H.; Myojin, Y.; Fukumoto, K.; Murai, K.; Sakane, S.; Tamura, T.; Yamai, T.; Nozaki, Y.; Yoshioka, T.; et al. Hyperglycemia enhances pancreatic cancer progression accompanied by elevations in phosphorylated STAT3 and MYC levels. PLoS ONE 2020, 15, e0235573. [Google Scholar] [CrossRef]
- Shadhu, K.; Xi, C. Inflammation and pancreatic cancer: An updated review. Saudi J. Gastroenterol. 2019, 25, 3–13. [Google Scholar] [CrossRef]
- Padoan, A.; Plebani, M.; Basso, D. Inflammation and Pancreatic Cancer: Focus on Metabolism, Cytokines, and Immunity. Int. J. Mol. Sci. 2019, 20, 676. [Google Scholar] [CrossRef]
- Sammallahti, H.; Kokkola, A.; Rezasoltani, S.; Ghanbari, R.; Asadzadeh Aghdaei, H.; Knuutila, S.; Puolakkainen, P.; Sarhadi, V.K. Microbiota Alterations and Their Association with Oncogenomic Changes in Pancreatic Cancer Patients. Int. J. Mol. Sci. 2021, 22, 12978. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Jin, M.; Liu, Y.; Jin, L. Gut Microbiota: Its Potential Roles in Pancreatic Cancer. Front. Cell. Infect. Microbiol. 2020, 10, 572492. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.; Huang, Z.; Shen, X.; Lin, T.; Zhang, Y.; Feng, X.; Mao, Q.; Liang, Y. Microbiota Regulates Pancreatic Cancer Carcinogenesis through Altered Immune Response. Microorganisms 2023, 11, 1240. [Google Scholar] [CrossRef]
- Wei, M.Y.; Shi, S.; Liang, C.; Meng, Q.C.; Hua, J.; Zhang, Y.Y.; Liu, J.; Zhang, B.; Xu, J.; Yu, X.J. The microbiota and microbiome in pancreatic cancer: More influential than expected. Mol. Cancer 2019, 18, 97. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, J.; Zhu, Y. Potential Roles of the Gut Microbiota in Pancreatic Carcinogenesis and Therapeutics. Front. Cell. Infect. Microbiol. 2022, 12, 872019. [Google Scholar] [CrossRef] [PubMed]
- Parida, S.; Siddharth, S.; Sharma, D. Adiponectin, Obesity, and Cancer: Clash of the Bigwigs in Health and Disease. Int. J. Mol. Sci. 2019, 20, 2519. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, H.; Ding, Y.; Wan, M.; Xu, M. The Role of Adipokines in Pancreatic Cancer. Front. Oncol. 2022, 12, 926230. [Google Scholar] [CrossRef] [PubMed]
- Brocco, D.; Florio, R.; De Lellis, L.; Veschi, S.; Grassadonia, A.; Tinari, N.; Cama, A. The Role of Dysfunctional Adipose Tissue in Pancreatic Cancer: A Molecular Perspective. Cancers 2020, 12, 1849. [Google Scholar] [CrossRef]
- Chang, H.-H.; Eibl, G. Obesity-Induced Adipose Tissue Inflammation as a Strong Promotional Factor for Pancreatic Ductal Adenocarcinoma. Cells 2019, 8, 673. [Google Scholar] [CrossRef]
- Kim, J.; Oh, C.M.; Kim, H. The Interplay of Adipokines and Pancreatic Beta Cells in Metabolic Regulation and Diabetes. Biomedicines 2023, 11, 2589. [Google Scholar] [CrossRef]
- Hart, P.A.; Bellin, M.D.; Andersen, D.K.; Bradley, D.; Cruz-Monserrate, Z.; Forsmark, C.E.; Goodarzi, M.O.; Habtezion, A.; Korc, M.; Kudva, Y.C.; et al. Type 3c (pancreatogenic) diabetes mellitus secondary to chronic pancreatitis and pancreatic cancer. Lancet Gastroenterol. Hepatol. 2016, 1, 226–237. [Google Scholar] [CrossRef]
- Ruze, R.; Liu, T.; Zou, X.; Song, J.; Chen, Y.; Xu, R.; Yin, X.; Xu, Q. Obesity and type 2 diabetes mellitus: Connections in epidemiology, pathogenesis, and treatments. Front. Endocrinol. 2023, 14, 1161521. [Google Scholar] [CrossRef]
- Maggio, C.A.; Pi-Sunyer, F.X. Obesity and type 2 diabetes. Endocrinol. Metab. Clin. North. Am. 2003, 32, 805–822. [Google Scholar] [CrossRef] [PubMed]
- Frayling, T.M.; Timpson, N.J.; Weedon, M.N.; Zeggini, E.; Freathy, R.M.; Lindgren, C.M.; Perry, J.R.; Elliott, K.S.; Lango, H.; Rayner, N.W.; et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 2007, 316, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, D.A.; Harbord, R.M.; Sterne, J.A.; Timpson, N.; Davey Smith, G. Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Stat. Med. 2008, 27, 1133–1163. [Google Scholar] [CrossRef] [PubMed]
- Garfield, V.; Farmaki, A.E.; Fatemifar, G.; Eastwood, S.V.; Mathur, R.; Rentsch, C.T.; Denaxas, S.; Bhaskaran, K.; Smeeth, L.; Chaturvedi, N. Relationship Between Glycemia and Cognitive Function, Structural Brain Outcomes, and Dementia: A Mendelian Randomization Study in the UK Biobank. Diabetes 2021, 70, 2313–2321. [Google Scholar] [CrossRef]
- Burgess, S.; Davey Smith, G.; Davies, N.M.; Dudbridge, F.; Gill, D.; Glymour, M.M.; Hartwig, F.P.; Kutalik, Z.; Holmes, M.V.; Minelli, C.; et al. Guidelines for performing Mendelian randomization investigations: Update for summer 2023. Wellcome Open Res. 2019, 4, 186. [Google Scholar] [CrossRef]
- Kurki, M.I.; Karjalainen, J.; Palta, P.; Sipilä, T.P.; Kristiansson, K.; Donner, K.M.; Reeve, M.P.; Laivuori, H.; Aavikko, M.; Kaunisto, M.A.; et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 2023, 613, 508–518. [Google Scholar] [CrossRef] [PubMed]
- FinnGen R10 + UKBB. Available online: https://public-metaresults-fg-ukbb.finngen.fi/about (accessed on 29 January 2024).
- Garfield, V.; Salzmann, A.; Burgess, S.; Chaturvedi, N. A Guide for Selection of Genetic Instruments in Mendelian Randomization Studies of Type 2 Diabetes and HbA1c: Toward an Integrated Approach. Diabetes 2023, 72, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Andrews, I.; Stock, J.H.; Sun, L. Weak Instruments in Instrumental Variables Regression: Theory and Practice. Annu. Rev. Econ. 2019, 11, 727–753. [Google Scholar] [CrossRef]
- Staiger, D.; Stock, J.H. Instrumental Variables Regression with Weak Instruments. Econometrica 1997, 65, 557–586. [Google Scholar] [CrossRef]
- You, R.; Chen, L.; Xu, L.; Zhang, D.; Li, H.; Shi, X.; Zheng, Y.; Chen, L. High Level of Uromodulin Increases the Risk of Hypertension: A Mendelian Randomization Study. Front. Cardiovasc. Med. 2021, 8, 736001. [Google Scholar] [CrossRef]
- Burgess, S.; Thompson, S.G. Avoiding bias from weak instruments in Mendelian randomization studies. Int. J. Epidemiol. 2011, 40, 755–764. [Google Scholar] [CrossRef]
- Brion, M.J.; Shakhbazov, K.; Visscher, P.M. Calculating statistical power in Mendelian randomization studies. Int. J. Epidemiol. 2013, 42, 1497–1501. [Google Scholar] [CrossRef]
- mRnd: Power Calculations for Mendelian Randomization. Available online: https://shiny.cnsgenomics.com/mRnd/ (accessed on 30 January 2024).
- Burgess, S.; Butterworth, A.; Thompson, S.G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 2013, 37, 658–665. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Haycock, P.C.; Burgess, S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar] [CrossRef]
- Hartwig, F.P.; Davey Smith, G.; Bowden, J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 2017, 46, 1985–1998. [Google Scholar] [CrossRef]
- Verbanck, M.; Chen, C.Y.; Neale, B.; Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018, 50, 693–698. [Google Scholar] [CrossRef]
- Hemani, G.; Tilling, K.; Davey Smith, G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 2017, 13, e1007081. [Google Scholar] [CrossRef]
- Skrivankova, V.W.; Richmond, R.C.; Woolf, B.A.R.; Davies, N.M.; Swanson, S.A.; VanderWeele, T.J.; Timpson, N.J.; Higgins, J.P.T.; Dimou, N.; Langenberg, C.; et al. Strengthening the reporting of observational studies in epidemiology using mendelian randomisation (STROBE-MR): Explanation and elaboration. BMJ 2021, 375, n2233. [Google Scholar] [CrossRef]
- Sanderson, E. Multivariable Mendelian Randomization and Mediation. Cold Spring Harb. Perspect. Med. 2021, 11, a038984. [Google Scholar] [CrossRef]
- Kamat, M.A.; Blackshaw, J.A.; Young, R.; Surendran, P.; Burgess, S.; Danesh, J.; Butterworth, A.S.; Staley, J.R. PhenoScanner V2: An expanded tool for searching human genotype-phenotype associations. Bioinformatics 2019, 35, 4851–4853. [Google Scholar] [CrossRef]
- PhenoScanner V2. Available online: http://www.phenoscanner.medschl.cam.ac.uk/ (accessed on 5 February 2024).
- R Software. Available online: https://www.r-project.org/ (accessed on 30 January 2024).
- MR-PRESSO R Package. Available online: https://github.com/rondolab/MR-PRESSO (accessed on 30 January 2024).
- TwoSampleMR: Mendelian Randomization with GWAS Summary Data. Available online: https://mrcieu.github.io/TwoSampleMR/index.html (accessed on 30 January 2024).
- Skrivankova, V.W.; Richmond, R.C.; Woolf, B.A.R.; Yarmolinsky, J.; Davies, N.M.; Swanson, S.A.; VanderWeele, T.J.; Higgins, J.P.T.; Timpson, N.J.; Dimou, N.; et al. Strengthening the Reporting of Observational Studies in Epidemiology using Mendelian Randomization (STROBE-MR) Statement. JAMA 2021, 326, 1614–1621. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |
| Multivariable MR | |||||
|---|---|---|---|---|---|
| Exposure | nsnp | OR | p-Value | 95% Lower CI | 95% Upper CI |
| BMI | 43 | 2.753 | 0.395 | 0.267 | 28.365 |
| T2DM | 43 | 1.485 | <0.001 | 1.228 | 1.796 |
| Waist circumference | 43 | 0.190 | 0.266 | 0.010 | 3.545 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ke, T.-M.; Lophatananon, A.; Muir, K.R. Strengthening the Evidence for a Causal Link between Type 2 Diabetes Mellitus and Pancreatic Cancer: Insights from Two-Sample and Multivariable Mendelian Randomization. Int. J. Mol. Sci. 2024, 25, 4615. https://doi.org/10.3390/ijms25094615
Ke T-M, Lophatananon A, Muir KR. Strengthening the Evidence for a Causal Link between Type 2 Diabetes Mellitus and Pancreatic Cancer: Insights from Two-Sample and Multivariable Mendelian Randomization. International Journal of Molecular Sciences. 2024; 25(9):4615. https://doi.org/10.3390/ijms25094615
Chicago/Turabian StyleKe, Te-Min, Artitaya Lophatananon, and Kenneth R. Muir. 2024. "Strengthening the Evidence for a Causal Link between Type 2 Diabetes Mellitus and Pancreatic Cancer: Insights from Two-Sample and Multivariable Mendelian Randomization" International Journal of Molecular Sciences 25, no. 9: 4615. https://doi.org/10.3390/ijms25094615