
TRIM44 Promotes Rabies Virus Replication by Autophagy-Dependent Mechanism
 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. RABV Infection Upregulates TRIM Proteins in NA Cells
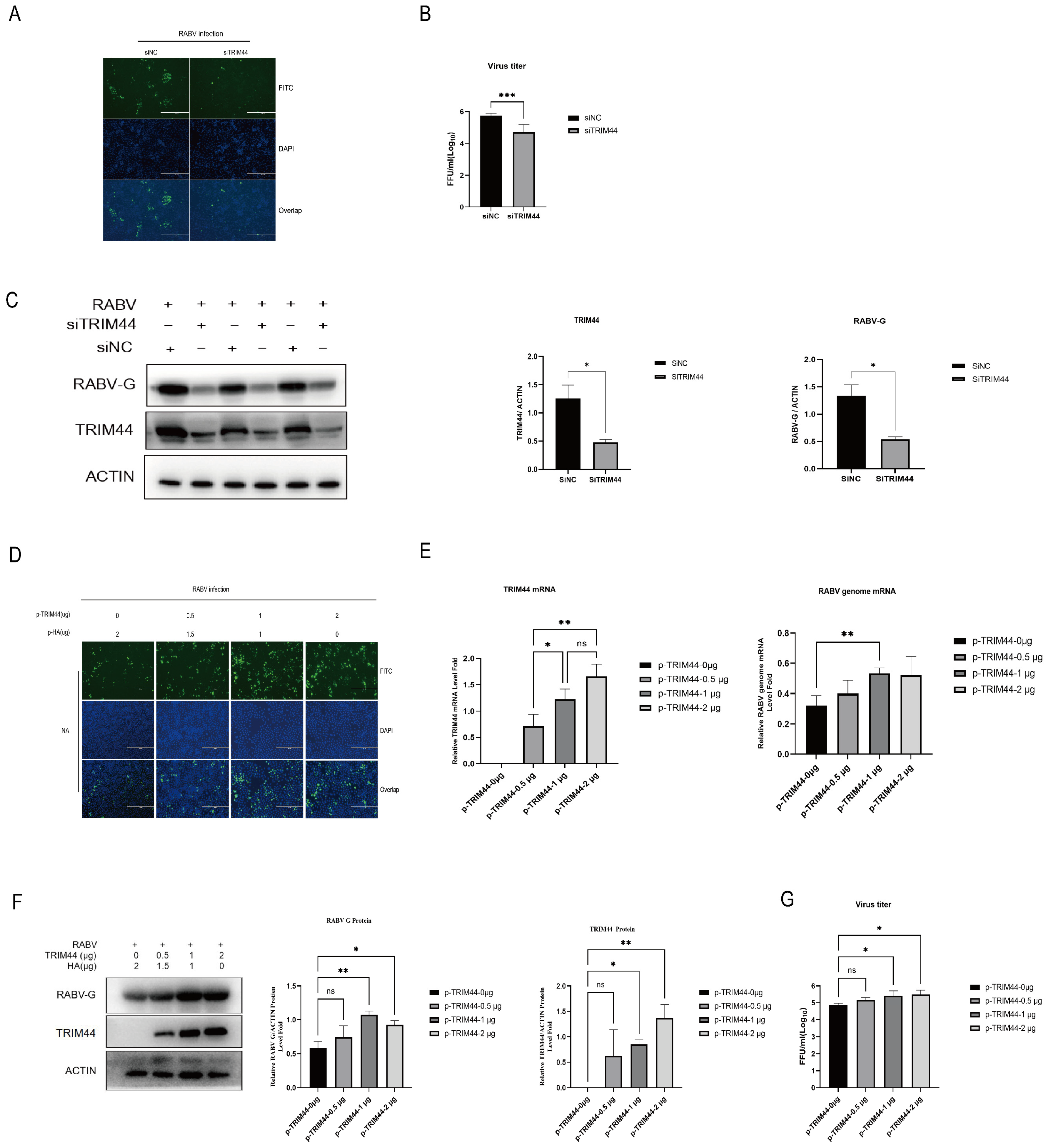
2.2. Silencing of TRIM44 Inhibits RABV Replication
2.3. TRIM44 Is Crucial for Virus Replication
2.4. TRIM44 Promotes RABV Replication through an Autophagy-Dependent Mechanism
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Strains and Cells
4.3. RNA Sequencing (RNA-Seq)
4.4. Plasmids and siRNA
4.5. Virus Infection and Titer Determination
4.6. Real-Time Fluorescence Quantitative PCR (RT-qPCR)
4.7. Western Blotting
4.8. siRNA and Plasmid DNA Transfection
4.9. Laser Confocal Immunofluorescence Microscopy
4.10. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, X.; Wen, Z.; Cao, H.; Luo, J.; Shuai, L.; Wang, C.; Ge, J.; Wang, X.; Bu, Z.; Wang, J. Transferrin Receptor Protein 1 Is an Entry Factor for Rabies Virus. J. Virol. 2023, 97, e0161222. [Google Scholar] [CrossRef] [PubMed]
- Taylor, L.H.; Hampson, K.; Fahrion, A.; Abela-Ridder, B.; Nel, L.H. Difficulties in estimating the human burden of canine rabies. Acta Trop. 2017, 165, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, H.; Mahmood, F.; Fu, Z.F. Rabies virus glycoprotein is an important determinant for the induction of innate immune responses and the pathogenic mechanisms. Vet. Microbiol. 2013, 162, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liao, M.; Yan, Y.; Yang, H.; Wang, H.; Zhou, J. Rabies virus phosphoprotein P5 binding to BECN1 regulates self-replication by BECN1-mediated autophagy signaling pathway. Cell Commun. Signal. 2020, 18, 153. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zhu, S.; Hu, L.; Ye, P.; Wang, Y.; Tian, Q.; Mei, M.; Chen, H.; Guo, X. Wild-type rabies virus induces autophagy in human and mouse neuroblastoma cell lines. Autophagy 2016, 12, 1704–1720. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, Y.; Li, M.; Zhu, J.; Li, X.; Luo, T.R.; Liang, J. Host Desmin Interacts with RABV Matrix Protein and Facilitates Virus Propagation. Viruses 2023, 15, 434. [Google Scholar] [CrossRef] [PubMed]
- Heo, H.; Park, H.; Lee, M.S.; Kim, J.; Kim, J.; Jung, S.Y.; Kim, S.K.; Lee, S.; Chang, J. TRIM22 facilitates autophagosome-lysosome fusion by mediating the association of GABARAPs and PLEKHM1. Autophagy 2023, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, H.; Huang, A.; Zhao, Y.; Xiao, C.; Dong, J.; Liu, X.; Shao, N. Mutual regulation between TRIM21 and TRIM8 via K48-linked ubiquitination. Oncogene 2023, 42, 3708–3718. [Google Scholar] [CrossRef]
- Park, J.S.; Burckhardt, C.J.; Lazcano, R.; Solis, L.M.; Isogai, T.; Li, L.; Chen, C.S.; Gao, B.; Minna, J.D.; Bachoo, R.; et al. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 2020, 578, 621–626. [Google Scholar] [CrossRef]
- Peng, Z.; Zhang, C.; Yin, B.; He, Y.; Li, W.; Wang, J.; Xiao, J.; Peng, K.; Bao, C.; Zhu, R. TRIM21 of Micropterus salmoides exerts antiviral roles against largemouth bass ulcer syndrome virus. Fish Shellfish Immunol. 2023, 142, 109176. [Google Scholar] [CrossRef]
- Niu, Y.; Fu, X.; Lin, Q.; Liang, H.; Luo, X.; Zuo, S.; Liu, L.; Li, N. The composition and antiviral activity of scTRIM59 in Mandarin fish. Fish Shellfish Immunol. 2022, 130, 86–92. [Google Scholar] [CrossRef]
- Sparrer, K.M.J.; Gableske, S.; Zurenski, M.A.; Parker, Z.M.; Full, F.; Baumgart, G.J.; Kato, J.; Pacheco-Rodriguez, G.; Liang, C.; Pornillos, O.; et al. TRIM23 mediates virus-induced autophagy via activation of TBK1. Nat. Microbiol. 2017, 2, 1543–1557. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, M.I.; Xia, H.; Aguilera-Aguirre, L.; Hage, A.; van Tol, S.; Shan, C.; Xie, X.; Sturdevant, G.L.; Robertson, S.J.; McNally, K.L.; et al. Envelope protein ubiquitination drives entry and pathogenesis of Zika virus. Nature 2020, 585, 414–419. [Google Scholar] [CrossRef]
- Fan, W.; Mar, K.B.; Sari, L.; Gaszek, I.K.; Cheng, Q.; Evers, B.M.; Shelton, J.M.; Wight-Carter, M.; Siegwart, D.J.; Lin, M.M.; et al. TRIM7 inhibits enterovirus replication and promotes emergence of a viral variant with increased pathogenicity. Cell 2021, 184, 3410–3425.e17. [Google Scholar] [CrossRef]
- Zhang, B.; Cai, T.; He, H.; Huang, X.; Luo, Y.; Huang, S.; Luo, J.; Guo, X. TRIM25 Suppresses Rabies Virus Fixed HEP-Flury Strain Production by Activating RIG-1-Mediated Type I Interferons. Genes 2023, 14, 1555. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Zhang, B.; Xu, S.; Liu, M.; Wei, Y.; Wang, Q.; Shen, W.; Lei, C.Q.; Zhu, Q. The nucleoprotein of influenza A virus inhibits the innate immune response by inducing mitophagy. Autophagy 2023, 19, 1916–1933. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Sun, C.; Han, Y.; Huang, L.; Sheng, H.; Wang, J.; Zhang, Y.; Lai, J.; Yuan, J.; Chen, X.; et al. Neutrophil autophagy and NETosis in COVID-19: Perspectives. Autophagy 2023, 19, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Liu, J.; Wang, H.; Gu, J.; Deng, T.; Yuan, Z.; Hu, B.; Xu, Y.; Yan, Y.; Zan, J.; Liao, M.; et al. BECN1-dependent CASP2 incomplete autophagy induction by binding to rabies virus phosphoprotein. Autophagy 2017, 13, 739–753. [Google Scholar] [CrossRef]
- Hoenigsperger, H.; Koepke, L.; Acharya, D.; Hunszinger, V.; Freisem, D.; Grenzner, A.; Wiese, S.; Kirchhoff, F.; Gack, M.U.; Sparrer, K.M.J. CSNK2 suppresses autophagy by activating FLN-NHL-containing TRIM proteins. Autophagy 2023, 1–21. [Google Scholar] [CrossRef]
- Yang, B.; Wang, J.; Wang, Y.; Zhou, H.; Wu, X.; Tian, Z.; Sun, B. Novel function of Trim44 promotes an antiviral response by stabilizing VISA. J. Immunol. 2013, 190, 3613–3619. [Google Scholar] [CrossRef]
- Zheng, J.; Zhang, Y.; Zhi, L.; Lv, S.; Xiao, L.; Huang, X.; Huang, Y.; Qin, Q. The novel gene TRIM44L from orange-spotted grouper negatively regulates the interferon response. Fish Shellfish Immunol. 2019, 92, 746–755. [Google Scholar] [CrossRef]
- Lyu, L.; Chen, Z.; McCarty, N. TRIM44 links the UPS to SQSTM1/p62-dependent aggrephagy and removing misfolded proteins. Autophagy 2022, 18, 783–798. [Google Scholar] [CrossRef]
- Munir, M. TRIM proteins: Another class of viral victims. Sci. Signal. 2010, 3, jc2. [Google Scholar] [CrossRef]
- Zhang, B.; Cai, T.; He, H.; Huang, X.; Chen, G.; Lai, Y.; Luo, Y.; Huang, S.; Luo, J.; Guo, X. TRIM21 Promotes Rabies Virus Production by Degrading IRF7 through Ubiquitination. Int. J. Mol. Sci. 2023, 24, 10892. [Google Scholar] [CrossRef]
- Watanabe, M.; Hatakeyama, S. TRIM proteins and diseases. J. Biochem. 2017, 161, 135–144. [Google Scholar] [CrossRef]
- Zhang, M.; Tan, H.; Gong, Y.; Faleti, O.D.; Li, D.; Yang, J.; Huang, J.; Long, J.; Luo, Q.; Wu, G.; et al. TRIM26 restricts Epstein-Barr virus infection in nasopharyngeal epithelial cells through K48-linked ubiquitination of HSP-90beta. FASEB J. 2024, 38, e23345. [Google Scholar] [CrossRef]
- Su, C.M.; Hung, Y.F.; Tang, J.; Han, M.; Everett, R.; Yoo, D. Suppression of TRIM19 by arterivirus nonstructural protein 1 promotes viral replication. Virus Res. 2024, 340, 199302. [Google Scholar] [CrossRef]
- Yuan, Y.; Fang, A.; Wang, Z.; Chen, H.; Fu, Z.F.; Zhou, M.; Zhao, L. The matrix protein of lyssavirus hijacks autophagosome for efficient egress by recruiting nedd4 through its ppxy motif. Autophagy 2024, 1–18. [Google Scholar] [CrossRef]
- Hatakeyama, S. Trim family proteins: Roles in autophagy, immunity, and carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kuang, M.; Lu, Y.; Lin, L.; Liu, X. Characterization and biological function analysis of the trim47 gene from common carp (Cyprinus carpio). Gene 2017, 627, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Yeung, S.H.; Lee, R.H.; Cheng, G.W.; Ma, I.W.; Kofler, J.; Kent, C.; Ma, F.; Herrup, K.; Fornage, M.; Arai, K.; et al. White matter hyperintensity genetic risk factor trim47 regulates autophagy in brain endothelial cells. bioRxiv 2024. [Google Scholar] [CrossRef] [PubMed]
- Seo, G.J.; Kim, C.; Shin, W.J.; Sklan, E.H.; Eoh, H.; Jung, J.U. Trim56-mediated monoubiquitination of cgas for cytosolic dna sensing. Nat. Commun. 2018, 9, 613. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Zhou, X.; Jiao, Q.; Chen, X. The functions of trim56 in antiviral innate immunity and tumorigenesis. Int. J. Mol. Sci. 2023, 24, 5046. [Google Scholar] [CrossRef] [PubMed]
- Boutou, E.; Matsas, R.; Mamalaki, A. Isolation of a mouse brain cDNA expressed in developing neuroblasts and mature neurons. Brain Res. Mol. Brain Res. 2001, 86, 153–167. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, Y.; Liu, X.; Yang, Y.; Yang, X.; Zheng, Z.; Deng, X.; Wu, X.; Guo, X. Characterization of a wild rabies virus isolate of porcine origin in China. Infect. Genet. Evol. 2013, 17, 147–152. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence (5′~3′) |
|---|---|
| siTRIM44 | F: GCAAUGAUAGAGUUGGUGGAATT |
| R: UUCCACCAACUCUAUCAUUGCTT | |
| siTRIM47 | F: CUACAGAAACUCGGCUCAGAATT |
| R: UUCUGAGCCGAGUUUCUGUAGTT | |
| siTRIM56 | F: CGAUAGAACCAAGAUAGGGAATT |
| R: UUCCCUAUCUUGGUUCUAUCGTT | |
| NC | F: UUC UCC GAA CGU GUC ACG UTT |
| R: ACG UGA CAC GUU CGG AGA ATT |
| Gene | Sequence (5′~3′) |
|---|---|
| TRIM44 | F:GCGGACATCCAATCTCACA |
| R:TCGTCACCCTCTGCCTTT | |
| TRIM47 | F:GAGTTTCCAGAACGAGGTGAT |
| R:TGCCGTGCCTTGCTTAG | |
| TRIM56 | F:GTTGACTTGGTGGGTTACAGAGC |
| R:GAGAACAAGGTTGACAGAGGAAGC | |
| GAPDH | F:CGTCCCGTAGACAAAATGGT |
| R:TTGATGGCAACAATCTCCAC | |
| HEP-Flury genome | F:AGAAGAAGCAGACATCGTCAGTTG |
| R:GGAGACCACCTGATTATTGACTTTGA | |
| HEP-G | F:GCCTTGATTGCCCTGATGTTGATAA |
| R:CATTTCTCCCTGTCCCTCCAAGAT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, H.; Cai, T.; Chen, Q.; Chen, Z.; Zhang, B.; Chen, C.; Wang, Y.; Liu, Y.; Wang, Y.; Luo, Y.; et al. TRIM44 Promotes Rabies Virus Replication by Autophagy-Dependent Mechanism. Int. J. Mol. Sci. 2024, 25, 4616. https://doi.org/10.3390/ijms25094616
He H, Cai T, Chen Q, Chen Z, Zhang B, Chen C, Wang Y, Liu Y, Wang Y, Luo Y, et al. TRIM44 Promotes Rabies Virus Replication by Autophagy-Dependent Mechanism. International Journal of Molecular Sciences. 2024; 25(9):4616. https://doi.org/10.3390/ijms25094616
Chicago/Turabian StyleHe, Hongling, Ting Cai, Qiaozhu Chen, Zilian Chen, Boyue Zhang, Changyi Chen, Yueze Wang, Yan Liu, Yueming Wang, Yongwen Luo, and et al. 2024. "TRIM44 Promotes Rabies Virus Replication by Autophagy-Dependent Mechanism" International Journal of Molecular Sciences 25, no. 9: 4616. https://doi.org/10.3390/ijms25094616