
Chronic Traumatic Encephalopathy as the Course of Alzheimer’s Disease
 , , , ,
, , , ,
Abstract
:1. Methodology Based on Preferred Presenting Items for Systematic Reviews and Meta-Analyses (PRISMA)
2. Introduction
3. Alzheimer’s Disease—Basic Facts
4. Chronic Traumatic Encephalopathy
Molecular Mechanisms of Tau Protein in CTE and AD
5. Assembly of Recombinant Tau into Filaments (CTE and AD)
5.1. Tau Protein Structure and Isoforms
5.2. Pathological Tau Phosphorylation
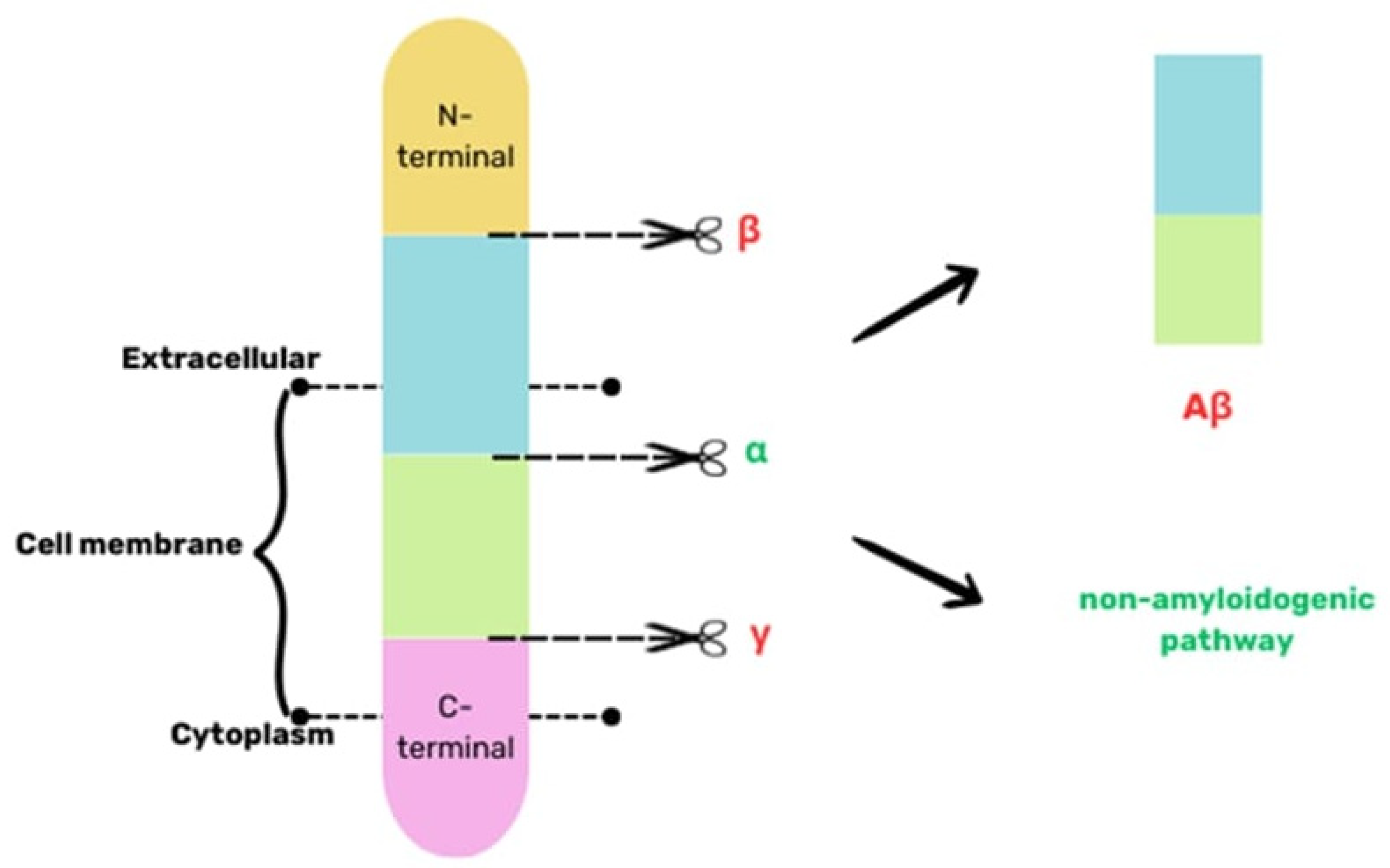
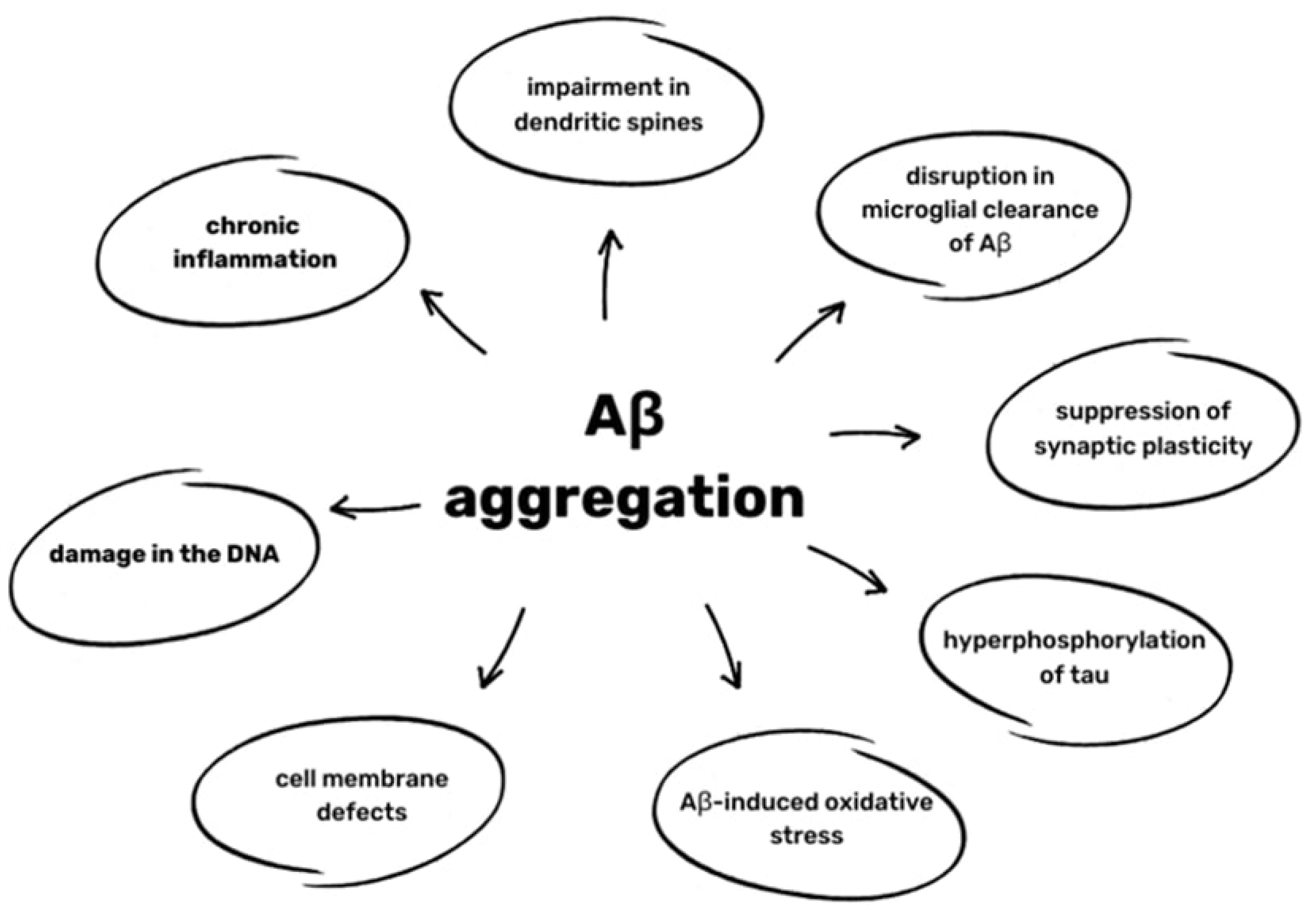
6. Aβ pathology in AD and CTE
7. Endothelial Cell and BBB Damage in CTE and AD
8. Mitochondrial Dysfunction in the Vasculature in CTE and AD
9. Cerebrovascular Inflammation in CTE and AD
10. Treatment Methods of CTE
11. Imagining of CTE
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fesharaki-Zadeh, A. Chronic Traumatic Encephalopathy: A Brief Overview. Front. Neurol. 2019, 10, 713. [Google Scholar] [CrossRef]
- Shively, S.; Scher, A.I.; Perl, D.P.; Diaz-Arrastia, R. Dementia Resulting from Traumatic Brain Injury. Arch. Neurol. 2012, 69, 1245–1251. [Google Scholar] [CrossRef]
- Turner, R.C.; Lucke-Wold, B.P.; Robson, M.J.; Lee, J.M.; Bailes, J.E. Alzheimer’s disease and chronic traumatic encephalopathy: Distinct but possibly overlapping disease entities. Brain Inj. 2016, 30, 1279–1292. [Google Scholar] [CrossRef]
- Fesharaki-Zadeh, A. A Case of Possible Chronic Traumatic Encephalopathy and Alzheimer’s Disease in an Ex-Football Player. Neurologist 2022, 27, 249–252. [Google Scholar] [CrossRef]
- Brett, B.L.; Wilmoth, K.; Cummings, P.; Solomon, G.S.; McCrea, M.A.; Zuckerman, S.L. The Neuropathological and Clinical Diagnostic Criteria of Chronic Traumatic Encephalopathy: A Critical Examination in Relation to Other Neurodegenerative Diseases. J. Alzheimers Dis. 2019, 68, 591–608. [Google Scholar] [CrossRef] [PubMed]
- 2023 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2023, 19, 1598–1695. [CrossRef] [PubMed]
- Apostolova, L.G. Alzheimer Disease. Contin. Lifelong Learn. Neurol. 2016, 22, 419–434. [Google Scholar] [CrossRef]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef]
- Kapasi, A.; Schneider, J.A. Vascular contributions to cognitive impairment, clinical Alzheimer’s disease, and dementia in older persons. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2016, 1862, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, TX, USA, 2013. [Google Scholar]
- World Health Organization (WHO). International Classification of Diseases; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Atri, A. The Alzheimer’s Disease Clinical Spectrum. Med. Clin. N. Am. 2019, 103, 263–293. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Mckee, A.C.; Abdolmohammadi, B.; Stein, T.D. The Neuropathology of Chronic Traumatic Encephalopathy; Elsevier: Amsterdam, The Netherlands, 2018; pp. 297–307. [Google Scholar] [CrossRef]
- Byard, R.; Tiemensma, M.; Buckland, M.E.; Vink, R. Chronic traumatic encephalopathy (CTE)—Features and forensic considerations. Forensic Sci. Med. Pathol. 2023, 19, 620–624. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Mez, J.; Abdolmohammadi, B.; Butler, M.; Huber, B.R.; Uretsky, M.; Babcock, K.; Cherry, J.D.; Alvarez, V.E.; Martin, B.; et al. Neuropathologic and Clinical Findings in Young Contact Sport Athletes Exposed to Repetitive Head Impacts. JAMA Neurol. 2023, 80, 1037. [Google Scholar] [CrossRef] [PubMed]
- Martland, H.S. Punch Drunk. J. Am. Med. Assoc. 1928, 91, 1103. [Google Scholar] [CrossRef]
- Bieniek, K.F.; Cairns, N.J.; Crary, J.F.; Dickson, D.W.; Folkerth, R.D.; Keene, C.D.; Litvan, I.; Perl, D.P.; Stein, T.D.; Vonsattel, J.-P.; et al. The Second NINDS/NIBIB Consensus Meeting to Define Neuropathological Criteria for the Diagnosis of Chronic Traumatic Encephalopathy. J. Neuropathol. Exp. Neurol. 2021, 80, 210–219. [Google Scholar] [CrossRef]
- McKee, A.C.; Cairns, N.J.; Dickson, D.W.; Folkerth, R.D.; Keene, C.D.; Litvan, I.; Perl, D.P.; Stein, T.D.; Vonsattel, J.-P.; Stewart, W.; et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. 2016, 131, 75–86. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Stein, T.D.; Huber, B.R.; Crary, J.F.; Bieniek, K.; Dickson, D.; Alvarez, V.E.; Cherry, J.D.; Farrell, K.; Butler, M.; et al. Chronic traumatic encephalopathy (CTE): Criteria for neuropathological diagnosis and relationship to repetitive head impacts. Acta Neuropathol. 2023, 145, 371–394. [Google Scholar] [CrossRef] [PubMed]
- Casper, S.T. Punch-Drunk Slugnuts: Violence and the Vernacular History of Disease. Isis 2022, 113, 266–288. [Google Scholar] [CrossRef]
- Smolen, P.; Dash, P.K.; Redell, J.B. Traumatic brain injury-associated epigenetic changes and the risk for neurodegenerative diseases. Front. Neurosci. 2023, 17, 1259405. [Google Scholar] [CrossRef]
- Nowinski, C.J.; Bureau, S.C.; Buckland, M.E.; Curtis, M.A.; Daneshvar, D.H.; Faull, R.L.M.; Grinberg, L.T.; Hill-Yardin, E.L.; Murray, H.C.; Pearce, A.J.; et al. Applying the Bradford Hill Criteria for Causation to Repetitive Head Impacts and Chronic Traumatic Encephalopathy. Front. Neurol. 2022, 13, 938163. [Google Scholar] [CrossRef] [PubMed]
- Omalu, B.I.; DeKosky, S.T.; Minster, R.L.; Kamboh, M.I.; Hamilton, R.L.; Wecht, C.H. Chronic Traumatic Encephalopathy in a National Football League Player. Neurosurgery 2005, 57, 128–134. [Google Scholar] [CrossRef]
- Berson, A.; Nativio, R.; Berger, S.L.; Bonini, N.M. Epigenetic Regulation in Neurodegenerative Diseases. Trends Neurosci. 2018, 41, 587–598. [Google Scholar] [CrossRef]
- Randolph, C. Chronic traumatic encephalopathy is not a real disease. Arch. Clin. Neuropsychol. 2018, 33, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Muralidar, S.; Ambi, S.V.; Sekaran, S.; Thirumalai, D.; Palaniappan, B. Role of tau protein in Alzheimer’s disease: The prime pathological player. Int. J. Biol. Macromol. 2020, 163, 1599–1617. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Murzin, A.G.; Falcon, B.; Epstein, A.; Machin, J.; Tempest, P.; Newell, K.L.; Vidal, R.; Garringer, H.J.; Sahara, N.; et al. Cryo-EM structures of tau filaments from Alzheimer’s disease with PET ligand APN-1607. Acta Neuropathol. 2021, 141, 697–708. [Google Scholar] [CrossRef]
- Goedert, M.; Eisenberg, D.S.; Crowther, R.A. Propagation of Tau Aggregates and Neurodegeneration. Annu. Rev. Neurosci. 2017, 40, 189–210. [Google Scholar] [CrossRef]
- Mamun, A.; Uddin, M.; Mathew, B.; Ashraf, G. Toxic tau: Structural origins of tau aggregation in Alzheimer’s disease. Neural Regen. Res. 2020, 15, 1417. [Google Scholar] [CrossRef]
- Boyarko, B.; Hook, V. Human Tau Isoforms and Proteolysis for Production of Toxic Tau Fragments in Neurodegeneration. Front. Neurosci. 2021, 15, 702788. [Google Scholar] [CrossRef]
- Katsumoto, A.; Takeuchi, H.; Tanaka, F. Tau Pathology in Chronic Traumatic Encephalopathy and Alzheimer’s Disease: Similarities and Differences. Front. Neurol. 2019, 10, 980. [Google Scholar] [CrossRef]
- Shahpasand, K.; Uemura, I.; Saito, T.; Asano, T.; Hata, K.; Shibata, K.; Toyoshima, Y.; Hasegawa, M.; Hisanaga, S. Regulation of Mitochondrial Transport and Inter-Microtubule Spacing by Tau Phosphorylation at the Sites Hyperphosphorylated in Alzheimer’s Disease. J. Neurosci. 2012, 32, 2430–2441. [Google Scholar] [CrossRef]
- Moszczynski, A.J.; Strong, W.; Xu, K.; McKee, A.; Brown, A.; Strong, M.J. Pathologic Thr 175 tau phosphorylation in CTE and CTE with ALS. Neurology 2018, 90, e380–e387. [Google Scholar] [CrossRef] [PubMed]
- Kondo, A.; Shahpasand, K.; Mannix, R.; Qiu, J.; Moncaster, J.; Chen, C.-H.; Yao, Y.; Lin, Y.-M.; Driver, J.A.; Sun, Y.; et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 2015, 523, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Lövestam, S.; Koh, F.A.; van Knippenberg, B.; Kotecha, A.; Murzin, A.G.; Goedert, M.; Scheres, S.H. Assembly of recombinant tau into filaments identical to those of Alzheimer’s disease and chronic traumatic encephalopathy. eLife 2022, 11, e76494. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.; Valpuesta, J.M.; Medina, M.; de Garcini, E.M.; Avila, J. Polymerization of τ into Filaments in the Presence of Heparin: The Minimal Sequence Required for τ-τ Interaction. J. Neurochem. 1996, 67, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Kampers, T.; Friedhoff, P.; Biernat, J.; Mandelkow, E.-M.; Mandelkow, E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996, 399, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Chen, W.-D.; Wang, Y.-D. β-Amyloid: The key peptide in the pathogenesis of Alzheimer’s disease. Front. Pharmacol. 2015, 6, 221. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Trejo-Lopez, J.A.; Yachnis, A.T.; Prokop, S. Neuropathology of Alzheimer’s Disease. Neurotherapeutics 2022, 19, 173–185. [Google Scholar] [CrossRef]
- Yamada, M.; Naiki, H. Cerebral Amyloid Angiopathy; Elsevier: Amsterdam, The Netherlands, 2012; pp. 41–78. [Google Scholar] [CrossRef]
- Stein, T.D.; Montenigro, P.H.; Alvarez, V.E.; Xia, W.; Crary, J.F.; Tripodis, Y.; Daneshvar, D.H.; Mez, J.; Solomon, T.; Meng, G.; et al. Beta-amyloid deposition in chronic traumatic encephalopathy. Acta Neuropathol. 2015, 130, 21–34. [Google Scholar] [CrossRef]
- Turk, K.W.; Geada, A.; Alvarez, V.E.; Xia, W.; Cherry, J.D.; Nicks, R.; Meng, G.; Daley, S.; Tripodis, Y.; Huber, B.R.; et al. A comparison between tau and amyloid-β cerebrospinal fluid biomarkers in chronic traumatic encephalopathy and Alzheimer disease. Alzheimers Res. Ther. 2022, 14, 28. [Google Scholar] [CrossRef]
- McKee, A.C.; Stein, T.D.; Kiernan, P.T.; Alvarez, V.E. The Neuropathology of Chronic Traumatic Encephalopathy. Brain Pathol. 2015, 25, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Abrahamson, E.E.; Ikonomovic, M.D. Brain injury-induced dysfunction of the blood brain barrier as a risk for dementia. Exp. Neurol. 2020, 328, 113257. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Xu, Z.; Cao, J.; Fu, Q.; Wu, Y.; Zhang, X.; Long, Y.; Zhang, X.; Yang, Y.; Li, Y.; et al. Elamipretide (SS-31) improves mitochondrial dysfunction, synaptic and memory impairment induced by lipopolysaccharide in mice. J. Neuroinflamm. 2019, 16, 230. [Google Scholar] [CrossRef] [PubMed]
- Archie, S.R.; Al Shoyaib, A.; Cucullo, L. Blood-Brain Barrier Dysfunction in CNS Disorders and Putative Therapeutic Targets: An Overview. Pharmaceutics 2021, 13, 1779. [Google Scholar] [CrossRef]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W. Targeted delivery of protein and gene medicines through the blood–brain barrier. Clin. Pharmacol. Ther. 2015, 97, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Toledo, J.B.; Arnold, S.E.; Raible, K.; Brettschneider, J.; Xie, S.X.; Grossman, M.; Monsell, S.E.; Kukull, W.A.; Trojanowski, J.Q. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain 2013, 136, 2697–2706. [Google Scholar] [CrossRef]
- Zipser, B.D.; Johanson, C.E.; Gonzalez, L.; Berzin, T.M.; Tavares, R.; Hulette, C.M.; Vitek, M.P.; Hovanesian, V.; Stopa, E.G. Microvascular injury and blood–brain barrier leakage in Alzheimer’s disease. Neurobiol. Aging 2007, 28, 977–986. [Google Scholar] [CrossRef]
- Omalu, B.I.; Fitzsimmons, R.P.; Hammers, J.; Bailes, J. Chronic traumatic encephalopathy in a professional American wrestler. J. Forensic Nurs. 2010, 6, 130–136. [Google Scholar] [CrossRef]
- Kuhn, J.; Sharman, T. Cerebral Amyloid Angiopathy. [Updated 2023 Jun 5]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK556105/ (accessed on 18 March 2024).
- Nelson, A.R.; Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Doherty, C.P.; O’Keefe, E.; Wallace, E.; Loftus, T.; Keaney, J.; Kealy, J.; Humphries, M.M.; Molloy, M.G.; Meaney, J.F.; Farrell, M.; et al. Blood–Brain Barrier Dysfunction as a Hallmark Pathology in Chronic Traumatic Encephalopathy. J. Neuropathol. Exp. Neurol. 2016, 75, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Farrell, M.; Aherne, S.; O’Riordan, S.; O’Keeffe, E.; Greene, C.; Campbell, M. Blood-brain barrier dysfunction in a boxer with chronic traumatic encephalopathy and schizophrenia. Clin. Neuropathol. 2019, 38, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Sawa, Y.; Kitagawa-Sakakida, S.; Nishimura, M.; Morishita, R.; Kaneda, Y.; Kohmura, E.; Yoshimine, T.; Matsuda, H. Nuclear factor-κB decoy attenuates neuronal damage after global brain ischemia: A future strategy for brain protection during circulatory arrest. J. Thorac. Cardiovasc. Surg. 2001, 122, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Raval, A.P.; Hirsch, N.; Perez-Pinzon, M.A. Ischemic Preconditioning Mediates Cyclooxygenase-2 Expression Via Nuclear Factor-Kappa B Activation in Mixed Cortical Neuronal Cultures. Transl. Stroke Res. 2010, 1, 40–47. [Google Scholar] [CrossRef]
- Manzanero, S.; Santro, T.; Arumugam, T.V. Neuronal oxidative stress in acute ischemic stroke: Sources and contribution to cell injury. Neurochem. Int. 2013, 62, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Dahm, C.C.; Moore, K.; Murphy, M.P. Persistent S-Nitrosation of Complex I and Other Mitochondrial Membrane Proteins by S-Nitrosothiols but Not Nitric Oxide or Peroxynitrite. J. Biol. Chem. 2006, 281, 10056–10065. [Google Scholar] [CrossRef]
- Singh, R.; Lakhanpal, D.; Kumar, S.; Sharma, S.; Kataria, H.; Kaur, M.; Kaur, G. Late-onset intermittent fasting dietary restriction as a potential intervention to retard age-associated brain function impairments in male rats. Age 2012, 34, 917–933. [Google Scholar] [CrossRef]
- Kosenko, E.; Montoliu, C.; Giordano, G.; Kaminsky, Y.; Venediktova, N.; Buryanov, Y.; Felipo, V. Acute ammonia intoxication induces an NMDA receptor-mediated increase in poly(ADP-ribose) polymerase level and NAD + metabolism in nuclei of rat brain cells. J. Neurochem. 2004, 89, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Soane, L.; Kahraman, S.; Kristian, T.; Fiskum, G. Mechanisms of impaired mitochondrial energy metabolism in acute and chronic neurodegenerative disorders. J. Neurosci. Res. 2007, 85, 3407–3415. [Google Scholar] [CrossRef] [PubMed]
- Bernassola, F.; Catani, M.V.; Corazzari, M.; Rossi, A.; Melino, G. Inactivation of multiple targets by nitric oxide in CD95-triggered apoptosis. J. Cell Biochem. 2001, 82, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Vieira, H.; Kroemer, G. Mitochondria as Targets of Apoptosis Regulation by Nitric Oxide. IUBMB Life 2003, 55, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Kislin, M.; Sword, J.; Fomitcheva, I.V.; Croom, D.; Pryazhnikov, E.; Lihavainen, E.; Toptunov, D.; Rauvala, H.; Ribeiro, A.S.; Khiroug, L.; et al. Reversible Disruption of Neuronal Mitochondria by Ischemic and Traumatic Injury Revealed by Quantitative Two-Photon Imaging in the Neocortex of Anesthetized Mice. J. Neurosci. 2017, 37, 333–348. [Google Scholar] [CrossRef]
- Owens, K.; Park, J.H.; Gourley, S.; Jones, H.; Kristian, T. Mitochondrial dynamics: Cell-type and hippocampal region specific changes following global cerebral ischemia. J. Bioenerg. Biomembr. 2015, 47, 13–31. [Google Scholar] [CrossRef]
- Peng, C.; Rao, W.; Zhang, L.; Wang, K.; Hui, H.; Wang, L.; Su, N.; Luo, P.; Hao, Y.; Tu, Y.; et al. Mitofusin 2 ameliorates hypoxia-induced apoptosis via mitochondrial function and signaling pathways. Int. J. Biochem. Cell Biol. 2015, 69, 29–40. [Google Scholar] [CrossRef]
- Cho, D.-H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-Nitrosylation of Drp1 Mediates β-Amyloid-Related Mitochondrial Fission and Neuronal Injury. Science (1979) 2009, 324, 102–105. [Google Scholar] [CrossRef]
- Lu, M.-H.; Zhao, X.-Y.; Yao, P.-P.; Xu, D.-E.; Ma, Q.-H. The Mitochondrion: A Potential Therapeutic Target for Alzheimer’s Disease. Neurosci. Bull. 2018, 34, 1127–1130. [Google Scholar] [CrossRef]
- Sarkar, S.; Malovic, E.; Harischandra, D.S.; Ngwa, H.A.; Ghosh, A.; Hogan, C.; Rokad, D.; Zenitsky, G.; Jin, H.; Anantharam, V.; et al. Manganese exposure induces neuroinflammation by impairing mitochondrial dynamics in astrocytes. Neurotoxicology 2018, 64, 204–218. [Google Scholar] [CrossRef]
- Guilhaume-Correa, F.; Pickrell, A.M.; VandeVord, P.J. The Imbalance of Astrocytic Mitochondrial Dynamics Following Blast-Induced Traumatic Brain Injury. Biomedicines 2023, 11, 329. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.H.; Chen, X.-H.; Pierce, J.E.S.; Wolf, J.A.; Trojanowski, J.Q.; Graham, D.I.; Mcintosh, T.K. Progressive Atrophy and Neuron Death for One Year Following Brain Trauma in the Rat. J. Neurotrauma 1997, 14, 715–727. [Google Scholar] [CrossRef]
- Kabadi, S.; Faden, A. Neuroprotective Strategies for Traumatic Brain Injury: Improving Clinical Translation. Int. J. Mol. Sci. 2014, 15, 1216–1236. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.G.; Thompson, M.B.; Scheff, S.W. Cyclosporin A Attenuates Acute Mitochondrial Dysfunction Following Traumatic Brain Injury. Exp. Neurol. 1999, 160, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Nagamoto-Combs, K.; McNeal, D.W.; Morecraft, R.J.; Combs, C.K. Prolonged Microgliosis in the Rhesus Monkey Central Nervous System after Traumatic Brain Injury. J. Neurotrauma 2007, 24, 1719–1742. [Google Scholar] [CrossRef]
- Acosta, S.A.; Tajiri, N.; Shinozuka, K.; Ishikawa, H.; Sanberg, P.R.; Sanchez-Ramos, J.; Song, S.; Kaneko, Y.; Borlongan, C.V. Combination Therapy of Human Umbilical Cord Blood Cells and Granulocyte Colony Stimulating Factor Reduces Histopathological and Motor Impairments in an Experimental Model of Chronic Traumatic Brain Injury. PLoS ONE 2014, 9, e90953. [Google Scholar] [CrossRef]
- Giunta, B.; Obregon, D.; Velisetty, R.; Sanberg, P.R.; Borlongan, C.V.; Tan, J. The immunology of traumatic brain injury: A prime target for Alzheimer’s disease prevention. J. Neuroinflamm. 2012, 9, 678. [Google Scholar] [CrossRef]
- Mehta, A.; Prabhakar, M.; Kumar, P.; Deshmukh, R.; Sharma, P.L. Excitotoxicity: Bridge to various triggers in neurodegenerative disorders. Eur. J. Pharmacol. 2013, 698, 6–18. [Google Scholar] [CrossRef]
- Martin, L.J.; Al-Abdulla, N.A.; Brambrink, A.M.; Kirsch, J.R.; Sieber, F.E.; Portera-Cailliau, C. Neurodegeneration in Excitotoxicity, Global Cerebral Ischemia, and Target Deprivation: A Perspective on the Contributions of Apoptosis and Necrosis. Brain Res. Bull. 1998, 46, 281–309. [Google Scholar] [CrossRef]
- Tajiri, N.; Kellogg, S.L.; Shimizu, T.; Arendash, G.W.; Borlongan, C.V. Traumatic Brain Injury Precipitates Cognitive Impairment and Extracellular Aβ Aggregation in Alzheimer’s Disease Transgenic Mice. PLoS ONE 2013, 8, e78851. [Google Scholar] [CrossRef]
- Tajiri, N.; Hernandez, D.; Acosta, S.; Shinozuka, K.; Ishikawa, H.; Ehrhart, J.; Diamandis, T.; Gonzales-Portillo, C.; Borlongan, M.C.; Tan, J.; et al. Suppressed cytokine expression immediatey following traumatic brain injury in neonatal rats indicates an expeditious endogenous anti-inflammatory response. Brain Res. 2014, 1559, 65–71. [Google Scholar] [CrossRef]
- Cherry, J.D.; Stein, T.D.; Tripodis, Y.; Alvarez, V.E.; Huber, B.R.; Au, R.; Kiernan, P.T.; Daneshvar, D.H.; Mez, J.; Solomon, T.M.; et al. CCL11 is increased in the CNS in chronic traumatic encephalopathy but not in Alzheimer’s disease. PLoS ONE 2017, 12, e0185541. [Google Scholar] [CrossRef]
- Hernandez-Ontiveros, D.G.; Tajiri, N.; Acosta, S.; Giunta, B.; Tan, J.; Borlongan, C.V. Microglia Activation as a Biomarker for Traumatic Brain Injury. Front. Neurol. 2013, 4, 30. [Google Scholar] [CrossRef]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Oré, M.-V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Sävman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.-Y.; Bao, X.-Q.; Sun, H.; Zhang, D. Scavenger receptor on astrocytes and its relationship with neuroinflammation. Zhongguo Yi Xue Ke Xue Yuan Xue Bao 2014, 36, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Szmydynger-Chodobska, J.; Gandy, J.R.; Varone, A.; Shan, R.; Chodobski, A. Synergistic Interactions between Cytokines and AVP at the Blood-CSF Barrier Result in Increased Chemokine Production and Augmented Influx of Leukocytes after Brain Injury. PLoS ONE 2013, 8, e79328. [Google Scholar] [CrossRef]
- Kovesdi, E.; Kamnaksh, A.; Wingo, D.; Ahmed, F.; Grunberg, N.E.; Long, J.B.; Kasper, C.E.; Agoston, D.V. Acute Minocycline Treatment Mitigates the Symptoms of Mild Blast-Induced Traumatic Brain Injury. Front. Neurol. 2012, 3, 111. [Google Scholar] [CrossRef]
- Xu, L.; Fagan, S.C.; Waller, J.L.; Edwards, D.; Borlongan, C.V.; Zheng, J.; Hill, W.D.; Feuerstein, G.; Hess, D.C. Low dose intravenous minocycline is neuroprotective after middle cerebral artery occlusion-reperfusion in rats. BMC Neurol. 2004, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Wang, H.; Xu, J.; Li, T.; Zhang, L.; Ding, Y.; Zhu, L.; He, J.; Zhou, M. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: The Nrf2–ARE signaling pathway as a potential mechanism. Free Radic. Biol. Med. 2014, 73, 1–11. [Google Scholar] [CrossRef]
- Uekawa, K.; Hasegawa, Y.; Ma, M.; Nakagawa, T.; Katayama, T.; Sueta, D.; Toyama, K.; Kataoka, K.; Koibuchi, N.; Kawano, T.; et al. Rosuvastatin Ameliorates Early Brain Injury after Subarachnoid Hemorrhage via Suppression of Superoxide Formation and Nuclear Factor-Kappa B Activation in Rats. J. Stroke Cerebrovasc. Dis. 2014, 23, 1429–1439. [Google Scholar] [CrossRef]
- Loane, D.J.; Faden, A.I. Neuroprotection for traumatic brain injury: Translational challenges and emerging therapeutic strategies. Trends Pharmacol. Sci. 2010, 31, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Tajiri, N.; Duncan, K.; Antoine, A.; Pabon, M.; Acosta, S.A.; de la Pena, I.; Hernadez-Ontiveros, D.G.; Shinozuka, K.; Ishikawa, H.; Kaneko, Y.; et al. Stem cell-paved biobridge facilitates neural repair in traumatic brain injury. Front. Syst. Neurosci. 2014, 8, 116. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, I.; Pantalone, A.; Tete, S.; Salini, V.; Borlongan, C.V.; Hess, D.; Stuppia, L. Amniotic Fluid Stem Cells: A Promising Therapeutic Resource for Cell-Based Regenerative Therapy. Curr. Pharm. Des. 2012, 18, 1846–1863. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.-W.; Chang, D.-Y.; Lee, H.-S.; Kim, G.-H.; Park, J.-S.; Ryu, B.-Y.; Joe, E.-H.; Lee, Y.-D.; Kim, S.-S.; Suh-Kim, H. Immune following suppression mesenchymal stem cell transplantation in the ischemic brain is mediated by TGF-β. Neurobiol. Dis. 2013, 58, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Liu, Y.; Yan, K.; Chen, L.; Chen, X.-R.; Li, P.; Chen, F.-F.; Jiang, X.-D. Anti-inflammatory and immunomodulatory mechanisms of mesenchymal stem cell transplantation in experimental traumatic brain injury. J. Neuroinflamm. 2013, 10, 871. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, G.; Munafò, A.; Bellanca, C.M.; La Cognata, V.; Iemmolo, R.; Attaguile, G.A.; Di Mauro, R.; Di Benedetto, G.; Cantarella, G.; Barcellona, M.L.; et al. Stem Cells: Innovative Therapeutic Options for Neurodegenerative Diseases? Cells 2021, 10, 1992. [Google Scholar] [CrossRef] [PubMed]
- Vizoso, F.J.; Eiro, N.; Cid, S.; Schneider, J.; Perez-Fernandez, R. Mesenchymal Stem Cell Secretome: Toward Cell-Free Therapeutic Strategies in Regenerative Medicine. Int. J. Mol. Sci. 2017, 18, 1852. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, M.; Camussi, G.; Valadi, H.; Nazarenko, I.; Ekström, K.; Wang, X.; Principe, S.; Shah, N.; Ashraf, N.M.; Fatima, F.; et al. The emerging role of extracellular vesicles as biomarkers for urogenital cancers. Nat. Rev. Urol. 2014, 11, 688–701. [Google Scholar] [CrossRef]
- Kim, K.-S.; Kim, H.S.; Park, J.-M.; Kim, H.W.; Park, M.-K.; Lee, H.-S.; Lim, D.S.; Lee, T.H.; Chopp, M.; Moon, J. Long-term immunomodulatory effect of amniotic stem cells in an Alzheimer’s disease model. Neurobiol. Aging 2013, 34, 2408–2420. [Google Scholar] [CrossRef]
- Watanabe, J.; Shetty, A.K.; Hattiangady, B.; Kim, D.-K.; Foraker, J.E.; Nishida, H.; Prockop, D.J. Administration of TSG-6 improves memory after traumatic brain injury in mice. Neurobiol. Dis. 2013, 59, 86–99. [Google Scholar] [CrossRef]
- Milner, C.M.; Higman, V.A.; Day, A.J. TSG-6: A pluripotent inflammatory mediator? Biochem. Soc. Trans. 2006, 34, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, D.J.; Mulloy, B.; Forster, M.J.; Blundell, C.D.; Fries, E.; Milner, C.M.; Day, A.J. Characterization of the Interaction between Tumor Necrosis Factor-stimulated Gene-6 and Heparin. J. Biol. Chem. 2005, 280, 27044–27055. [Google Scholar] [CrossRef] [PubMed]
- McManus, M.J.; Murphy, M.P.; Franklin, J.L. The Mitochondria-Targeted Antioxidant MitoQ Prevents Loss of Spatial Memory Retention and Early Neuropathology in a Transgenic Mouse Model of Alzheimer’s Disease. J. Neurosci. 2011, 31, 15703–15715. [Google Scholar] [CrossRef] [PubMed]
- Muthukumaran, K.; Kanwar, A.; Vegh, C.; Marginean, A.; Elliott, A.; Guilbeault, N.; Badour, A.; Sikorska, M.; Cohen, J.; Pandey, S. Ubisol-Q10 (a Nanomicellar Water-Soluble Formulation of CoQ10) Treatment Inhibits Alzheimer-Type Behavioral and Pathological Symptoms in a Double Transgenic Mouse (TgAPEswe, PSEN1dE9) Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 61, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Lautrup, S.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’Connell, J.F.; Baptiste, B.A.; et al. NAD + supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E1876–E1885. [Google Scholar] [CrossRef]
- Fato, R.; Bergamini, C.; Bortolus, M.; Maniero, A.L.; Leoni, S.; Ohnishi, T.; Lenaz, G. Differential effects of mitochondrial Complex I inhibitors on production of reactive oxygen species. Biochim. Biophys. Acta (BBA)-Bioenerg. 2009, 1787, 384–392. [Google Scholar] [CrossRef]
- Polich, G.; Iaccarino, M.A.; Zafonte, R. Psychopharmacology of Traumatic Brain Injury; Elsevier: Amsterdam, The Netherlands, 2019; pp. 253–267. [Google Scholar] [CrossRef]
- Anderson, N.D. State of the science on mild cognitive impairment (MCI). CNS Spectr. 2019, 24, 78–87. [Google Scholar] [CrossRef]
- Cantu, R.; Budson, A. Management of chronic traumatic encephalopathy. Expert Rev. Neurother. 2019, 19, 1015–1023. [Google Scholar] [CrossRef]
- Kakehi, S.; Tompkins, D.M. A Review of Pharmacologic Neurostimulant Use During Rehabilitation and Recovery After Brain Injury. Ann. Pharmacother. 2021, 55, 1254–1266. [Google Scholar] [CrossRef]
- Salter, K.L.; McClure, J.A.; Foley, N.C.; Sequeira, K.; Teasell, R.W. Pharmacotherapy for Depression Posttraumatic Brain Injury: A Meta-analysis. J. Head Trauma Rehabil. 2016, 31, E21–E32. [Google Scholar] [CrossRef]
- Silverberg, N.D.; Panenka, W.J. Antidepressants for depression after concussion and traumatic brain injury are still best practice. BMC Psychiatry 2019, 19, 100. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, E.; Lemelle, T.M.; Samarbafzadeh, E.; Kablinger, A.S. Pharmacological Treatment of Agitation and/or Aggression in Patients with Traumatic Brain Injury: A Systematic Review of Reviews. J. Head Trauma Rehabil. 2021, 36, E262–E283. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C. The Neuropathology of Chronic Traumatic Encephalopathy: The Status of the Literature. Semin. Neurol. 2020, 40, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.H.; Johnson, V.E.; Trojanowski, J.Q.; Stewart, W. Chronic traumatic encephalopathy—Confusion and controversies. Nat. Rev. Neurol. 2019, 15, 179–183. [Google Scholar] [CrossRef] [PubMed]
- VanItallie, T.B. Traumatic brain injury (TBI) in collision sports: Possible mechanisms of transformation into chronic traumatic encephalopathy (CTE). Metabolism 2019, 100, 153943. [Google Scholar] [CrossRef] [PubMed]
- Katz, D.I.; Bernick, C.; Dodick, D.W.; Mez, J.; Mariani, M.L.; Adler, C.H.; Alosco, M.L.; Balcer, L.J.; Banks, S.J.; Barr, W.B.; et al. National Institute of Neurological Disorders and Stroke Consensus Diagnostic Criteria for Traumatic Encephalopathy Syndrome. Neurology 2021, 96, 848–863. [Google Scholar] [CrossRef] [PubMed]
- Holleran, L.; Kim, J.H.; Gangolli, M.; Stein, T.; Alvarez, V.; McKee, A.; Brody, D.L. Axonal disruption in white matter underlying cortical sulcus tau pathology in chronic traumatic encephalopathy. Acta Neuropathol. 2017, 133, 367–380. [Google Scholar] [CrossRef]
- McKee, A.C.; Gavett, B.E.; Stern, R.A.; Nowinski, C.J.; Cantu, R.C.; Kowall, N.W.; Perl, D.P.; Hedley-Whyte, E.T.; Price, B.; Sullivan, C.; et al. TDP-43 Proteinopathy and Motor Neuron Disease in Chronic Traumatic Encephalopathy. J. Neuropathol. Exp. Neurol. 2010, 69, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Charney, M.; Shenton, M.E.; Koerte, I.K. Chronic Traumatic Encephalopathy: Neuroimaging Biomarkers; Elsevier: Amsterdam, The Netherlands, 2018; pp. 309–322. [Google Scholar] [CrossRef]
- Sundman, M.; Doraiswamy, P.M.; Morey, R.A. Neuroimaging assessment of early and late neurobiological sequelae of traumatic brain injury: Implications for CTE. Front. Neurosci. 2015, 9, 334. [Google Scholar] [CrossRef]
- Logothetis, N.K. What we can do and what we cannot do with fMRI. Nature 2008, 453, 869–878. [Google Scholar] [CrossRef]
- McKee, A.C.; Cantu, R.C.; Nowinski, C.J.; Hedley-Whyte, E.T.; Gavett, B.E.; Budson, A.E.; Santini, V.E.; Lee, H.-S.; Kubilus, C.A.; Stern, R.A. Chronic Traumatic Encephalopathy in Athletes: Progressive Tauopathy After Repetitive Head Injury. J. Neuropathol. Exp. Neurol. 2009, 68, 709–735. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Stein, T.D.; Nowinski, C.J.; Stern, R.A.; Daneshvar, D.H.; Alvarez, V.E.; Lee, H.-S.; Hall, G.; Wojtowicz, S.M.; Baugh, C.M.; et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013, 136, 43–64. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Daneshvar, D.H.; Alvarez, V.E.; Stein, T.D. The neuropathology of sport. Acta Neuropathol. 2014, 127, 29–51. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.; Zivanov, J.; Zhang, W.; Murzin, A.G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Newell, K.L.; Ghetti, B.; Goedert, M.; et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019, 568, 420–423. [Google Scholar] [CrossRef]
- Cherry, J.D.; Baucom, Z.H.; Eppich, K.G.; Kirsch, D.; Dixon, E.R.; Tripodis, Y.; Bieniek, K.F.; Farrell, K.; Whitney, K.; Uretsky, M.; et al. Neuroimmune proteins can differentiate between tauopathies. J. Neuroinflamm. 2022, 19, 278. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pszczołowska, M.; Walczak, K.; Miśków, W.; Antosz, K.; Batko, J.; Kurpas, D.; Leszek, J. Chronic Traumatic Encephalopathy as the Course of Alzheimer’s Disease. Int. J. Mol. Sci. 2024, 25, 4639. https://doi.org/10.3390/ijms25094639
Pszczołowska M, Walczak K, Miśków W, Antosz K, Batko J, Kurpas D, Leszek J. Chronic Traumatic Encephalopathy as the Course of Alzheimer’s Disease. International Journal of Molecular Sciences. 2024; 25(9):4639. https://doi.org/10.3390/ijms25094639
Chicago/Turabian StylePszczołowska, Magdalena, Kamil Walczak, Weronika Miśków, Katarzyna Antosz, Joanna Batko, Donata Kurpas, and Jerzy Leszek. 2024. "Chronic Traumatic Encephalopathy as the Course of Alzheimer’s Disease" International Journal of Molecular Sciences 25, no. 9: 4639. https://doi.org/10.3390/ijms25094639