A DFT Study on Deactivation of Triplet Excited State Riboflavin by Polyphenols

Abstract
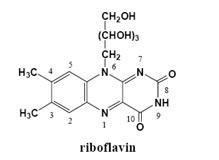
:
1. Introduction
2. Theoretical Methods
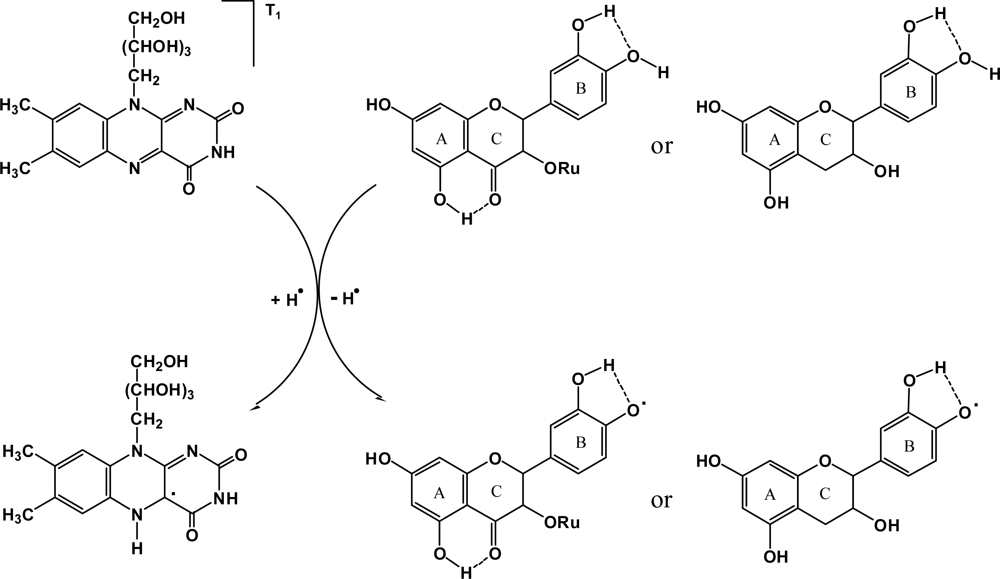
3. Results and Discussion
4. Conclusions
Acknowledgments
References and Notes
- Baier, J; Maisch, T; Maier, M; Engel, E; Landthaler, M; Bäumler, W. Singlet oxygen generation by UVA light exposure of endogenous photosensitizers. Biophys. J. 2006, 91, 1452–1459. [Google Scholar]
- Wondrak, GT; Jacobson, MK; Jacobson, EL. Endogenous UVA-photosensitizers: Mediators of skin photodamage and novel targets for skin photoprotection. Photochem. Photobiol. Sci 2006, 5, 215–237. [Google Scholar]
- Grzelak, A; Rychlik, B; Bartosz, G. Light-dependent generation of reactive oxygen species in cell culture media. Free Radic. Biol. Med 2001, 30, 1418–1425. [Google Scholar]
- Shen, L; Ji, HF; Zhang, HY. Computational note on the photosensitization mechanisms of riboflavin. J. Mol. Struct. (Theochem) 2007, 821, 171–172. [Google Scholar]
- Ito, K; Inoue, S; Yamamoto, K; Kawanishi, S. 8-Hydroxydeoxyguanosine formation at the 5′ site of 5′-GG-3′ sequences in double-stranded DNA by UV radiation with riboflavin. J. Biol. Chem. 1993, 268, 13221–13227. [Google Scholar]
- Joshi, PC. Comparison of the DNA-damaging property of photosensitised riboflavin via singlet oxygen (1O2) and superoxide radical O2-. mechanisms. Toxicol. Lett 1985, 26, 211–217. [Google Scholar]
- Becker, EM; Cardoso, DR; Skibsted, LH. Deactivation of riboflavin triplet-excited state by phenolic antioxidants: mechanism behind protective effects in photooxidation of milk-based beverages. Eur. Food. Res. Technol. 2005, 221, 382–386. [Google Scholar]
- Zhang, HY. Structure-activity relationships and rational design strategies for radical-scavenging antioxidants. Curr. Computer-Aided Drug Des. 2005, 1, 257–273. [Google Scholar]
- Shen, L; Ji, HF; Zhang, HY. A theoretical elucidation on the solvent-dependent photosensitive behaviors of C60. Photochem. Photobiol 2006, 82, 798–800. [Google Scholar]
- Shen, L; Ji, HF; Zhang, HY. A TD-DFT study on triplet excited-state properties of curcumin and its implications in elucidating the photosensitizing mechanisms of the pigment. Chem. Phys. Lett. 2005, 409, 300–303. [Google Scholar]
- Shen, L; Ji, HF; Zhang, HY. Hypericin anion is crucial to elucidating the pigment’s photosensitive features. Bioorg. Med. Chem. Lett 2006, 16, 1414–1417. [Google Scholar]
- Shen, L; Ji, HF. How α-tocopherol quenches triplet state riboflavin? Insights from theory. J. Photochem. Photobiol. A: Chem. 2008, 199, 119–121. [Google Scholar]
- Shen, L; Ji, HF. A theoretical study on the quenching mechanisms of triplet state riboflavin by tryptophan and tyrosine. J. Photochem. Photobiol. B: Biol. 2008, 92, 10–12. [Google Scholar]
- Hohenberg, P; Kohn, W. Inhomogeneous electron gas. Phys. Rev 1964, 136, B864–B871. [Google Scholar]
- Kohn, W; Sham, LJ. Self-consistent equations including exchange and correlation effects. Phys. Rev 1965, 140, A1133–A1138. [Google Scholar]
- Lee, C; Yang, W; Parr, RG. Development of the Colle-Salvetti correlation energy formula into a functional of the electron density. Phys. Rev. B. 1988, 37, 785–789. [Google Scholar]
- Becke, AD. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar]
- Stephens, PJ; Devlin, FJ; Chabalowski, CF; Frisch, MJ. Ab Initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar]
- Stratmann, RE; Scuseria, GE; Frisch, MJ. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 1998, 109, 8218–8224. [Google Scholar]
- Bauernschmitt, R; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett 1996, 256, 454–464. [Google Scholar]
- Casida, ME; Jamorski, C; Casida, KC; Salahub, DR. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem. Phys. 1998, 108, 4439–4449. [Google Scholar]
- Miertus, S; Scrocco, E; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar]
- Miertus, S; Tomasi, J. Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem. Phys 1982, 65, 239–241. [Google Scholar]
- Cossi, M; Barone, V; Cammi, J. Ab initio study of solvated molecules: a new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar]
- Frisch, MJ; Trucks, GW; Schlegel, HB; Scuseria, GE; Robb, MA; Cheeseman, JR; Montgomery, JA; Vreven, T; Kudin, KN; Burant, JC; et al. Gaussian 03 2003.
- Pannala, SA; Chan, TS; O′Brien, PJ; Rice-Evans, CA. Flavonoid B-ring chemistry and antioxidant activity: fast reaction kinetics. Biochem. Biophys. Res. Commun. 2001, 282, 1161–1168. [Google Scholar]
- Cardoso, DR; Olsen, K; Skibsted, LH. Mechanism of deactivation of triplet-excited riboflavin by ascorbate, carotenoids, and tocopherols in homogeneous and heterogeneous aqueous food model systems. J. Agric. Food Chem. 2007, 55, 6285–6291. [Google Scholar]



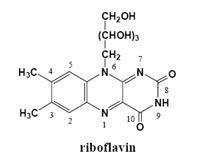{kind=link}
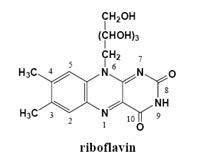{kind=link}
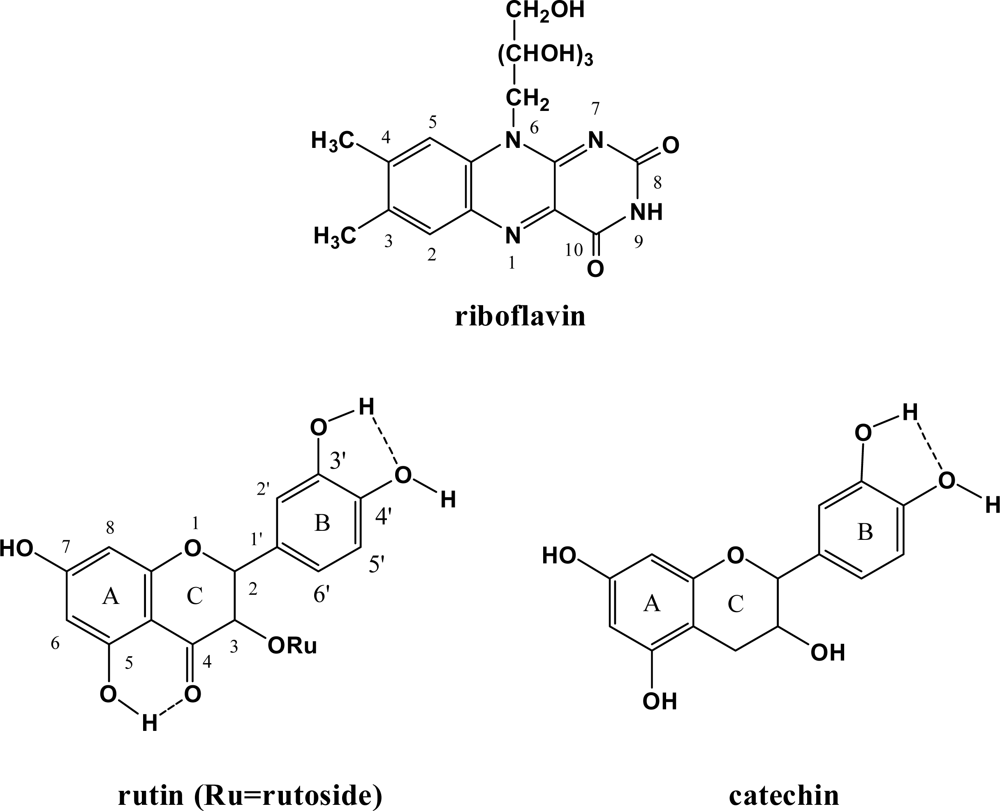{kind=link}
{kind=link}
| Compounds | Solvents | ET1 | VIPS0 | VEAS0 | VEAT1a |
|---|---|---|---|---|---|
| Rutin | benzene | 3.12 | 6.76 | ||
| water | 3.13 | 5.95 | |||
| Catechin | benzene | 3.62 | 6.49 | ||
| water | 3.63 | 5.82 | |||
| Riboflavinb | benzene | 2.10 | –2.52 | –4.62 | |
| water | 2.09 | –3.32 | –5.41 |
| Compounds | Solvents | O-H BDE | HAAT1a |
|---|---|---|---|
| Rutin | benzene | 78.18 | |
| water | 79.97 | ||
| Catechin | benzene | 78.97 | |
| water | 80.73 | ||
| Riboflavin | benzene | −97.24 | |
| water | −106.19 |
© 2008 by MDPI This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ji, H.-F.; Shen, L. A DFT Study on Deactivation of Triplet Excited State Riboflavin by Polyphenols. Int. J. Mol. Sci. 2008, 9, 1908-1914. https://doi.org/10.3390/ijms9101908
Ji H-F, Shen L. A DFT Study on Deactivation of Triplet Excited State Riboflavin by Polyphenols. International Journal of Molecular Sciences. 2008; 9(10):1908-1914. https://doi.org/10.3390/ijms9101908
Chicago/Turabian StyleJi, Hong-Fang, and Liang Shen. 2008. "A DFT Study on Deactivation of Triplet Excited State Riboflavin by Polyphenols" International Journal of Molecular Sciences 9, no. 10: 1908-1914. https://doi.org/10.3390/ijms9101908
APA StyleJi, H.-F., & Shen, L. (2008). A DFT Study on Deactivation of Triplet Excited State Riboflavin by Polyphenols. International Journal of Molecular Sciences, 9(10), 1908-1914. https://doi.org/10.3390/ijms9101908