

Is the Existence of Two Lineages for Hamadryas glauconome (Lepidoptera: Nymphalidae) True? Molecular and Ecological Evidence
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ecological Analyses
2.1.1. Databases
2.1.2. Accessible Area
2.1.3. Model Calibration
2.1.4. Ecological Niche Modeling
2.1.5. Identity Test
2.1.6. Fundamental Niche Overlap
2.2. Molecular Analysis
2.2.1. Sequences
2.2.2. Phylogenetic Inferences
2.2.3. Generalized Mixed Yule Coalescent (GMYC) Analysis
3. Results
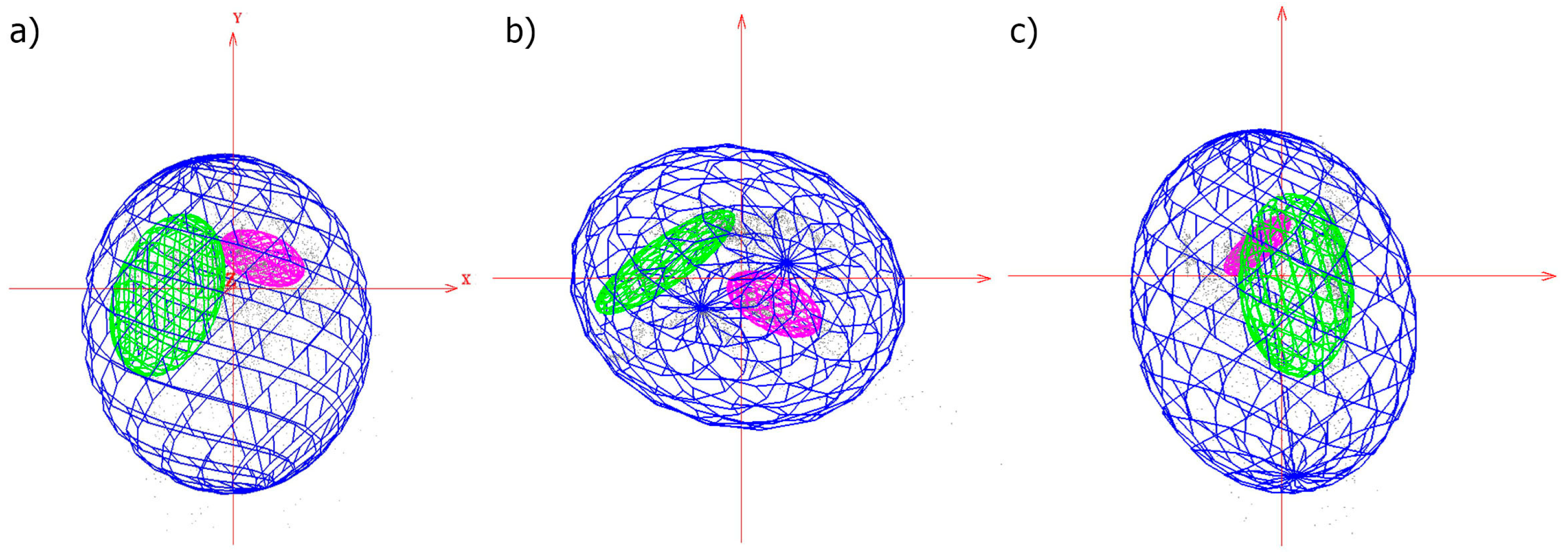
3.1. Ecological Analyses
3.2. Molecular Analyses
3.2.1. Phylogenetic Inferences
3.2.2. Generalized Mixed Yule Coalescent (GMYC) Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jenkins, D.W. Neotropical Nymphalidae I. Revision of Hamadryas. Bull. Allyn. Mus. 1983, 81, 1–146. [Google Scholar]
- Fruhstorfer, H. Ageronia. In Die Gross-Schmetterlinge der Erde: Eine Systematische Bearbeitung der bis Jetzt Bekannten Gross-Schmetterlinge; Seitz, A., Ed.; Alfred Kernen: Stuttgart, Germany, 1916; Volume Bd.5, pp. 537–542. [Google Scholar]
- Rzedowski, J. Capítulo 6. Provincias Florísticas de México. In Vegetación de México; Limusa: Mexico City, Mexico, 1978. [Google Scholar]
- Morrone, J.J. Hacia una síntesis biogeográfica de México. Rev. Mex. Biodivers. 2005, 76, 207–252. [Google Scholar] [CrossRef]
- Luis Martínez, A.; Llorente-Bosuquets, J.; Vargas-Fernández, I. Nymphalidae de México I (Danainae, Apaturinae, Biblidinae y Heliconiinae): Distribución Geográfica e Ilustración; Prensas de Ciencias: Mexico City, Mexico, 2003; ISBN 9789703206933. [Google Scholar]
- Meerman, J.; Boomsma, T. Occasional Papers of the Belize Natural History Society. J. Belizean Na. Hist. 1993, 2, 37–46. [Google Scholar]
- Prado, B.R.; Pozo, C.; Valdez-Moreno, M.; Hebert, P.D.N. Beyond the Colours: Discovering Hidden Diversity in the Nymphalidae of the Yucatan Peninsula in Mexico through DNA Barcoding. PLoS ONE 2011, 6, e27776. [Google Scholar] [CrossRef] [PubMed]
- Salinas-Gutiérrez, J.L.; Méndez, C.; Barrios, M.; Pozo, C.; Llorente-Bousquets, J. Hacia Una Síntesis de Los Papilionoidea (Insecta: Lepidoptera) de Guatemala Con Una Reseña Histórica. Caldasia 2009, 31, 407–440. [Google Scholar]
- Garzón-Orduña, I.J.; Marini-Filho, O.; Johnson, S.G.; Penz, C.M. Phylogenetic Relationships of Hamadryas (Nymphalidae: Biblidinae) Based on the Combined Analysis of Morphological and Molecular Data. Cladistics 2013, 29, 629–642. [Google Scholar] [CrossRef]
- Garzón-Orduña, I.J.; Brower, A.V.Z.; Kamilari, M.; Iribar, A.; Murienne, J. Cracking the Code: Examination of Species Delimitations among Hamadryas Butterflies with DNA Barcodes Suggests Caribbean Cracker Is Hamadryas Februa Hübner (Nymphalidae: Biblidinae). J. Lepid. Soc. 2018, 72, 53–73. [Google Scholar] [CrossRef]
- Nieves-Uribe, S.; Flores-Gallardo, A.; Llorente-Bousquets, J.; Luis-Martínez, A.; Pozo, C. Use of Exochorion Characters for the Systematics of Hamadryas Hübner and Ectima Doubleday (Nymphalidae: Biblidinae: Ageroniini). Zootaxa 2019, 4619, 77–108. [Google Scholar] [CrossRef]
- Esquivel-Ruíz, E.S. Reevaluación Del Status Taxonómico de Hamadryas Julitta Respecto a Hamadryas Glauconome Por Medio de Análisis Cuantitativos de Caracteres Morfológicos; Maestría, UNAM: Ciudad de México, Mexico, 2022. [Google Scholar]
- De Queiroz, K. Species Concepts and Species Delimitation. Syst. Biol. 2007, 56, 879–886. [Google Scholar] [CrossRef]
- Raxworthy, C.J.; Ingram, C.M.; Rabibisoa, N.; Pearson, R.G. Applications of Ecological Niche Modeling for Species Delimitation: A Review and Empirical Evaluation Using Day Geckos (Phelsuma) from Madagascar. Syst. Biol. 2007, 56, 907–923. [Google Scholar] [CrossRef]
- Rissler, L.J.; Apodaca, J.J. Adding More Ecology into Species Delimitation: Ecological Niche Models and Phylogeography Help Define Cryptic Species in the Black Salamander (Aneides Flavipunctatus). Syst. Biol. 2007, 56, 924–942. [Google Scholar] [CrossRef] [PubMed]
- Vaissi, S.; Rezaei, S. Niche Divergence at Intraspecific Level in the Hyrcanian Wood Frog, Rana Pseudodalmatina: A Phylogenetic, Climatic, and Environmental Survey. Front. Ecol. Evol. 2022, 10, 774481. [Google Scholar] [CrossRef]
- Imada, Y.; Kawakita, A.; Kato, M. Allopatric Distribution and Diversification without Niche Shift in a Bryophyte-Feeding Basal Moth Lineage (Lepidoptera: Micropterigidae). Proc. R. Soc. B Biol. Sci. 2011, 278, 3026–3033. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Kaliszewska, Z.A.; Heath, A.; Pierce, N.E. Recent Diversification of Chrysoritis Butterflies in the South African Cape (Lepidoptera: Lycaenidae). Mol. Phylogenet. Evol. 2020, 148, 106817. [Google Scholar] [CrossRef]
- Moraes, S.D.E.S.; Gueratto, P.E.; Dos Santos, J.P.; Santos, M.H.; Freitas, A.V.L.; Duarte, M. Niche Modelling and Comparative Morphology Untangle Taxonomy of the Dysschema Eurocilia Clade (Lepidoptera: Erebidae) and Reveal a Relictual Pleistocene Arc Distribution. Syst. Biodivers. 2022, 20, 1–39. [Google Scholar] [CrossRef]
- Campbell, E.O.; MacDonald, Z.G.; Gage, E.V.; Gage, R.V.; Sperling, F.A.H. Genomics and Ecological Modelling Clarify Species Integrity in a Confusing Group of Butterflies. Mol. Ecol. 2022, 31, 2400–2417. [Google Scholar] [CrossRef] [PubMed]
- Andersson, L. The Driving Force: Species Concepts and Ecology. Taxon 1990, 39, 375–382. [Google Scholar] [CrossRef]
- Van Valen, L. Ecological species, multispecies, and oaks. Taxon 1976, 25, 233–239. [Google Scholar] [CrossRef]
- Hutchinson, G.E. Concluding Remarks. Cold Spring Harb. Symp. Quant. Biol. 1957, 22, 415–427. [Google Scholar] [CrossRef]
- Maguire, B. Niche Response Structure and the Analytical Potentials of Its Relationship to the Habitat. Am. Nat. 1973, 107, 213–246. [Google Scholar] [CrossRef]
- Soberón, J.; Peterson, A. Interpretation of Models of Fundamental Ecological Niches and Species’ Distributional Areas. Biodivers. Inform. 2005, 2, 1–10. [Google Scholar] [CrossRef]
- Barve, N.; Barve, V.; Jiménez-Valverde, A.; Lira-Noriega, A.; Maher, S.P.; Peterson, A.T.; Soberón, J.; Villalobos, F. The Crucial Role of the Accessible Area in Ecological Niche Modeling and Species Distribution Modeling. Ecol. Model. 2011, 222, 1810–1819. [Google Scholar] [CrossRef]
- Soberón, J.M. Niche and Area of Distribution Modeling: A Population Ecology Perspective. Ecography 2010, 33, 159–167. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; deWaard, J.R. Biological Identifications through DNA Barcodes. Proc. R. Soc. Lond. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.N.; Ratnasingham, S.; de Waard, J.R. Barcoding Animal Life: Cytochrome c Oxidase Subunit 1 Divergences among Closely Related Species. Proc. R. Soc. Lond. B Biol. Sci. 2003, 270, S96–S99. [Google Scholar] [CrossRef]
- Dumas, P.; Barbut, J.; Le Ru, B.; Silvain, J.-F.; Clamens, A.-L.; d’Alençon, E.; Kergoat, G.J. Phylogenetic Molecular Species Delimitations Unravel Potential New Species in the Pest Genus Spodoptera Guenée, 1852 (Lepidoptera, Noctuidae). PLoS ONE 2015, 10, e0122407. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Barraclough, T.G. Delimiting Species Using Single-Locus Data and the Generalized Mixed Yule Coalescent Approach: A Revised Method and Evaluation on Simulated Data Sets. Syst. Biol. 2013, 62, 707–724. [Google Scholar] [CrossRef]
- Pons, J.; Barraclough, T.G.; Gomez-Zurita, J.; Cardoso, A.; Duran, D.P.; Hazell, S.; Kamoun, S.; Sumlin, W.D.; Vogler, A.P. Sequence-Based Species Delimitation for the DNA Taxonomy of Undescribed Insects. Syst. Biol. 2006, 55, 595–609. [Google Scholar] [CrossRef]
- Miller, J.; Matthews, D.; Warren, A.; Solis, M.; Harvey, D.; Gentili-Poole, P.; Lehman, R.; Emmel, T.; Covell, C. An Annotated List of the Lepidoptera of Honduras. Insecta Mundi. 2012, 205, 1–72. Available online: https://digitalcommons.unl.edu/cgi/viewcontent.cgi?article=1724&context=insectamundi (accessed on 31 October 2021).
- Calderón-Vásquez, M.A. Evaluación de Cuatro Años de Monitoreo de Mariposas En La Finca Agroecológica de Zamorano, Honduras; Escuela Agrícola Panamericana: Zamorano, Honduras, 2020. [Google Scholar]
- Medina, M.; Orozco, J.; Hernández, J. Diversidad de Especies de Lepidoptera: Nymphalidae, Papilionidae y Pieridae, En “El Cacao”, Telica, León, Nicaragua, En 2018. Revista Nicaragüense de Entomología. 2020, 211, 3–64. [Google Scholar]
- Gallardo, R.J.; Diaz, O. Guide to the Butterflies of Honduras; Robert Gallardo (privately published): Santa Bárbara, Honduras, 2022. [Google Scholar]
- GBIF Occurrence Download. Available online: https://www.gbif.org/occurrence/download/0008894-231002084531237 (accessed on 9 October 2023).
- Rios, N.E.; Bart, H.L. GEOLocate 2010 (Version 3.22). Tulane University Museum of Natural History: Belle Chasse, LA, USA. Available online: https://www.geo-locate.org/standalone/default.html (accessed on 31 October 2021).
- QGIS.org QGIS Geographic Information System. Available online: https://www.qgis.org/ (accessed on 31 October 2021).
- Google Earth. Available online: https://earth.google.com/ (accessed on 4 February 2022).
- Hijmans, R.J.; Cameron, S.E.; Parra, J.L.; Jones, P.G.; Jarvis, A. Very High Resolution Interpolated Climate Surfaces for Global Land Areas. Int. J. Climatol. 2005, 25, 1965–1978. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2023. [Google Scholar]
- Di Cola, V.; Broennimann, O.; Petitpierre, B.; Breiner, F.T.; D’Amen, M.; Randin, C.; Engler, R.; Pottier, J.; Pio, D.; Dubuis, A.; et al. Ecospat: An R Package to Support Spatial Analyses and Modeling of Species Niches and Distributions. Ecography 2017, 40, 774–787. [Google Scholar] [CrossRef]
- Lourenço, G.M.; Dáttilo, W.; Ribeiro, S.P.; Freitas, A.V.L. Biological Aspects and Movements of Neotropical Fruit-Feeding Butterflies. Neotrop. Entomol. 2022, 51, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Machado-Stredel, F.; Cobos, M.E.; Peterson, A.T. A Simulation-Based Method for Selecting Calibration Areas for Ecological Niche Models and Species Distribution Models. Front. Biogeogr. 2021, 13, e48814. [Google Scholar] [CrossRef]
- Hijmans, R.J.; van Etten, J. Raster: Geographic Analysis and Modeling with Raster Data. R Package Version 2.0-12 2012. Available online: https://rspatial.org/raster (accessed on 31 October 2021).
- Hijmans, R.J.; Phillips, S.; Leathwick, J.; Elith, J. Dismo: Species Distribution Modeling. R Package Version 1.0-12 2015. Available online: https://rspatial.org/raster/sdm/ (accessed on 31 October 2021).
- Lares, B. Lares: Analytics & Machine Learning Sidekick. R Package Version 5.2.4.9000 2023. Available online: https://github.com/laresbernardo/lares (accessed on 31 October 2021).
- Phillips, S.J.; Dudík, M. Modeling of Species Distributions with Maxent: New Extensions and a Comprehensive Evaluation. Ecography 2008, 31, 161–175. [Google Scholar] [CrossRef]
- Warren, D.L.; Beaumont, L.J.; Dinnage, R.; Baumgartner, J.B. New Methods for Measuring ENM Breadth and Overlap in Environmental Space. Ecography 2019, 42, 444–446. [Google Scholar] [CrossRef]
- Warren, D.L.; Glor, R.E.; Turelli, M. Environmental Niche Equivalency Versus Conservatism: Quantitative Approaches to Niche Evolution. Evolution 2008, 62, 2868–2883. [Google Scholar] [CrossRef]
- Qiao, H.; Peterson, A.T.; Campbell, L.P.; Soberón, J.; Ji, L.; Escobar, L.E. NicheA: Creating Virtual Species and Ecological Niches in Multivariate Environmental Scenarios. Ecography 2016, 39, 805–813. [Google Scholar] [CrossRef]
- Ratnasingham, S.; Hebert, P.D.N. Bold: The Barcode of Life Data System (Http://Www.Barcodinglife.Org). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef]
- Ivanova, N.V.; DeWaard, J.R.; Hebert, P.D.N. An Inexpensive, Automation-Friendly Protocol for Recovering High-Quality DNA. Mol. Ecol. Notes 2006, 6, 998–1002. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A Simple Method for Estimating Evolutionary Rates of Base Substitutions through Comparative Studies of Nucleotide Sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.; Joyce, P.; Posada, D.; Crandall, K.A. JModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 36, 716–723. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree v1.3.1. Institute of Evolutionary Biology, University of Edinburgh, Edinburgh. 2010. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 31 October 2021).
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian Evolutionary Analysis by Sampling Trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Hillis, D.M. Success of Phylogenetic Methods in the Four-Taxon Case. Syst. Biol. 1993, 42, 247–264. [Google Scholar] [CrossRef]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An Advanced Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Drummond, A. Improving the Performance of Bayesian Phylogenetic Inference under Relaxed Clock Models. BMC Evol. Biol. 2020, 20, 54. [Google Scholar] [CrossRef] [PubMed]
- Ezard, T.; Fujisawa, T.; Barraclough, T.G. Splits: Species’ Limits by Threshold Statistics. R Package Version 2009. Available online: https://r-forge.r-project.org/projects/splits/ (accessed on 31 October 2021).
- Pozo, C.; Luis-Martínez, A.; Llorente-Bousquets, J.; Salas-Suárez, N.; Maya-Martínez, A.; Vargas-Fernández, I.; Warren, A.D. Seasonality and Phenology of the Butterflies (Lepidoptera: Papilionoidea and Hesperioidea) of Mexico’s Calakmul Region. Fla. Entomol. 2008, 91, 407–422. [Google Scholar] [CrossRef]
- Scoble, M.J. The Lepidoptera: Form, Function and Diversity; Natural History Museum Publications; Natural History Museum: New York, NY, USA, 1995; ISBN 9780198549529. [Google Scholar]
- Warren, A.D.; Davis, K.J.; Stangeland, E.M.; Pelham, J.P.; Willmott, K.; Grishin, N.V. Illustrated Lists of American Butterflies. Available online: https://www.butterfliesofamerica.com (accessed on 27 November 2023).
- Jasso-Martínez, J.M.; Machkour-M’Rabet, S.; Vila, R.; Rodríguez-Arnaiz, R.; Castañeda-Sortibrán, A.N. Molecular Evidence of Hybridization in Sympatric Populations of the Enantia Jethys Complex (Lepidoptera: Pieridae). PLoS ONE 2018, 13, e0197116. [Google Scholar] [CrossRef] [PubMed]
- Graham, C.H.; Ron, S.R.; Santos, J.C.; Schneider, C.J.; Moritz, C. Integrating phylogenetics and environmental niche models to explore speciation mechanisms in dendrobatid frogs. Evolution 2004, 58, 1781–1793. [Google Scholar] [CrossRef] [PubMed]
- Pitteloud, C.; Arrigo, N.; Suchan, T.; Mastretta-Yanes, A.; Vila, R.; Dincă, V.; Hernández-Roldán, J.; Brockmann, E.; Chittaro, Y.; Kleckova, I.; et al. Climatic Niche Evolution Is Faster in Sympatric than Allopatric Lineages of the Butterfly Genus Pyrgus. Proc. R. Soc. B Biol. Sci. 2017, 284, 20170208. [Google Scholar] [CrossRef] [PubMed]
- Maya-Martínez, A.; Schmitter-Soto, J.J.; Pozo, C. Panbiogeography of the Yucatan Peninsula Based on Charaxinae (Lepidoptera: Nymphalidae). Fla. Entomol. 2011, 94, 527–533. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Penton, E.H.; Burns, J.M.; Janzen, D.H.; Hallwachs, W. Ten Species in One: DNA Barcoding Reveals Cryptic Species in the Neotropical Skipper Butterfly Astraptes Fulgerator. Proc. Natl. Acad. Sci. USA 2004, 101, 14812–14817. [Google Scholar] [CrossRef]
- DeSalle, R.; Goldstein, P. Review and Interpretation of Trends in DNA Barcoding. Front. Ecol. Evol. 2019, 7, 302. [Google Scholar] [CrossRef]
- Lassance, J.-M.; Svensson, G.P.; Kozlov, M.V.; Francke, W.; Löfstedt, C. Pheromones and Barcoding Delimit Boundaries between Cryptic Species in the Primitive Moth Genus Eriocrania (Lepidoptera: Eriocraniidae). J. Chem. Ecol. 2019, 45, 429–439. [Google Scholar] [CrossRef]
- Smadja, C.; Butlin, R.K. On the Scent of Speciation: The Chemosensory System and Its Role in Premating Isolation. Heredity (Edinb) 2009, 102, 77–97. [Google Scholar] [CrossRef] [PubMed]
- González-Rojas, M.F.; Darragh, K.; Robles, J.; Linares, M.; Schulz, S.; McMillan, W.O.; Jiggins, C.D.; Pardo-Diaz, C.; Salazar, C. Chemical Signals Act as the Main Reproductive Barrier between Sister and Mimetic Heliconius Butterflies. Proc. R. Soc. B Biol. Sci. 2020, 287, 20200587. [Google Scholar] [CrossRef] [PubMed]
- Vane-Wright, R.I.; Boppré, M.; Butlin, R.K.; Guilford, T.; Krebs, J.R. Visual and Chemical Signalling in Butterflies: Functional and Phylogenetic Perspectives. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1997, 340, 197–205. [Google Scholar] [CrossRef]
- Janzen, D.H.; Hallwachs, W. Dynamic Database for an Inventory of the Macrocaterpillar Fauna, and Its Food Plants and Parasitoids, of Area de Conservacion Guanacaste (ACG), Northwestern Costa Rica (Nn-SRNP-Nnnnn Voucher Codes). Available online: http://janzen.sas.upenn.edu (accessed on 30 September 2020).
- D’Amen, M.; Zimmermann, N.E.; Pearman, P.B. Conservation of Phylogeographic Lineages under Climate Change. Glob. Ecol. Biogeogr 2013, 22, 93–104. [Google Scholar] [CrossRef]
- Islebe, G.A.; Schmook, B.; Calmé, S.; León-Cortés, J.L. Introduction: Biodiversity and Conservation of the Yucatán Peninsula, Mexico. In Biodiversity and Conservation of the Yucatán Peninsula; Islebe, G., Calmé, S., León-Cortés, J., Schmook, B., Eds.; Springer: Cham, Switzerland, 2015. [Google Scholar]
- Pfeiler, E. Butterfly Biodiversity in a Threatened Coastal Desert Ecosystem of Northwestern Mexico, with a Focus on the Life History and Ecology of Potentially Endangered Species. J. Lepid. Soc. 2016, 70, 47–60. [Google Scholar] [CrossRef]
- Garza-García, J. Environmental NGOs and the Political Ecology of Biodiversity Conservation in Mexican Forests. Ph.D. Thesis, Macquarie University, Sidney, Australia, 2023. [Google Scholar]
- Rissler, L.J.; Hijmans, R.J.; Graham, C.H.; Moritz, C.; Wake, D.B. Phylogeographic Lineages and Species Comparisons in Conservation Analyses: A Case Study of California Herpetofauna. Am. Nat. 2006, 167, 655–666. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Variable | Permutation Importance | Percent Contribution |
|---|---|---|
| Precipitation of Driest Month (bio14) * | 15 | 3.6 |
| Mean Temperature of Wettest Quarter (bio08) * | 13.8 | 9.2 |
| Min Temperature of Coldest Month (bio06) * | 10.8 | 12.2 |
| Isothermality (bio03) * | 9.3 | 1.2 |
| Precipitation of Coldest Quarter (bio19) * | 8.3 | 16.3 |
| Temperature Seasonality (bio04) | 7.7 | 5.9 |
| Mean Diurnal Range (bio02) | 7.3 | 3.5 |
| Max Temperature of Warmest Month (bio05) | 6.9 | 6.2 |
| Mean Temperature of Warmest Quarter (bio10) | 4 | 6 |
| Precipitation of Warmest Quarter (bio18) | 3.7 | 7.6 |
| Precipitation of Wettest Month (bio13) | 3.5 | 0.4 |
| Mean Temperature of Driest Quarter (bio09) | 3.3 | 3.3 |
| Annual Precipitation (bio12) | 2.4 | 11.1 |
| Precipitation of Wettest Quarter (bio16) | 2.4 | 3.3 |
| Precipitation of Driest Quarter (bio17) | 1 | 0.5 |
| Annual Mean Temperature (bio01) | 0.2 | 6.3 |
| Mean Temperature of Coldest Quarter (bio11) | 0.2 | 1.6 |
| Precipitation Seasonality (bio15) | 0.2 | 0.5 |
| Temperature Annual Range (bio07) | 0.1 | 1.4 |
| Variable | H. g. glauconome | H. g. grisea | H. julitta | |||
|---|---|---|---|---|---|---|
| Percent Contribution | Permutation Importance | Percent Contribution | Permutation Importance | Percent Contribution | Permutation Importance | |
| bio03 | 12.9 | 10.8 | 0.1 | 2 | 18.9 | 25.4 |
| bio06 | 19.5 | 27 | 56.1 | 41.3 | 13.4 | 7.3 |
| bio08 | 8.3 | 9.5 | 9.8 | 3.3 | 14 | 16.7 |
| bio14 | 19.4 | 16.5 | 28.2 | 48.4 * | 43.6 | 30.3 * |
| bio19 | 39.9 | 36.2 * | 5.8 | 5 | 10.1 | 20.3 |
| Schoender’s D Estimation | ||
|---|---|---|
| Species | Empirical Value | Critical Value |
| H. g. glauconome-H. julitta | 0.258 | 0.837 |
| H. g. glauconome-H. g. grisea | 0.613 | 0.724 |
| H. g. grisea-H. julitta | 0.279 | 0.646 |
| BOLD Process ID | GenBank Accession Number | Species | BOLD Process ID | GenBank Accession Number | Species | ||
|---|---|---|---|---|---|---|---|
| 1 | BRPC085-23 | OR891501 | H. g. glauconome | 23 | BRPC057-23 | OR891504 | H. g. glauconome |
| 2 | BRPC082-23 | OR891505 | H. g. glauconome | 24 | BRPC008-23 | OR891516 | H. g. grisea |
| 3 | BRPC081-23 | OR891513 | H. g. glauconome | 25 | BRPC007-23 | OR891500 | H. g. grisea |
| 4 | BRPC080-23 | OR891499 | H. g. glauconome | 26 | BRPC005-23 | OR891510 | H. g. grisea |
| 5 | BRPC079-23 | OR891491 | H. g. glauconome | 27 | BRPC003-23 | OR891493 | H. g. grisea |
| 6 | BRPC092-23 | OR891506 | H. g. glauconome | 28 | BRPC001-23 | OR891509 | H. g. grisea |
| 7 | BRPC073-23 | OR891498 | H. g. glauconome | 29 | LPMX210-07 | JN201289 | H. julitta |
| 8 | BRPC072-23 | OR891503 | H. g. glauconome | 30 | LYPAP807-09 | GU659503 | H. julitta |
| 9 | BRPC069-23 | OR891511 | H. g. glauconome | 31 | LYPAP769-09 | GU659537 | H. julitta |
| 10 | BRPC093-23 | OR891490 | H. g. glauconome | 32 | LPYPC059-08 | JN201290 | H. julitta |
| 11 | BRPC060-23 | OR891508 | H. g. glauconome | 33 | LYPAP810-09 | GU659498 | H. julitta |
| 12 | BRPC058-23 | OR891494 | H. g. glauconome | 34 | LYPAP809-09 | GU659497 | H. julitta |
| 13 | BRPC056-23 | OR891507 | H. g. glauconome | 35 | LYPAP808-09 | GU659496 | H. julitta |
| 14 | BRPC055-23 | OR891495 | H. g. glauconome | 36 | LYPAP805-09 | GU659502 | H. julitta |
| 15 | BRPC053-23 | OR891502 | H. g. glauconome | 37 | LYPAP804-09 | GU659501 | H. julitta |
| 16 | BRPC017-23 | OR891512 | H. g. glauconome | 38 | LYPAP803-09 | GU659500 | H. julitta |
| 17 | BRPC040-23 | OR891514 | H. g. glauconome | 39 | LYPAP802-09 | GU659507 | H. julitta |
| 18 | BRPC035-23 | OR891496 | H. g. glauconome | 40 | LYPAP801-09 | GU659506 | H. julitta |
| 19 | BRPC031-23 | OR891497 | H. g. glauconome | 41 | LYPAP800-09 | GU659505 | H. julitta |
| 20 | BRPC030-23 | OR891492 | H. g. glauconome | 42 | LYPAP799-09 | GU659504 | H. julitta |
| 21 | BRPC029-23 | OR891515 | H. g. glauconome | 43 | LYPAP798-09 | GU659511 | H. julitta |
| 22 | BRPC059-23 | OR891489 | H. g. glauconome | 44 | LYPAP797-09 | GU659510 | H. julitta |
| Species | H. g. grisea | H. julitta |
|---|---|---|
| H. g. glauconome | 0.0153 | 0.0251 |
| H. g. grisea | - | 0.0260 |
| GMYC Entity Assignment of H. glauconome Sequences | ||
|---|---|---|
| GMYC Species | Number of Sequences | Species |
| 1 | 5 | H. g. grisea |
| 2 | 22 | H. g. glauconome |
| 3 | 1 | H. g. glauconome * |
| GMYC entity assignment of sequences, including H. julitta | ||
| 1 | 5 | H. g. grisea |
| 2 | 23 | H. g. glauconome |
| 3 | 16 | H. julitta |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prado-Cuellar, B.R.; Lara-Pérez, L.A.; Trujano-Ortega, M.; Machkour-M’Rabet, S.; Pozo, C. Is the Existence of Two Lineages for Hamadryas glauconome (Lepidoptera: Nymphalidae) True? Molecular and Ecological Evidence. Diversity 2023, 15, 1196. https://doi.org/10.3390/d15121196
Prado-Cuellar BR, Lara-Pérez LA, Trujano-Ortega M, Machkour-M’Rabet S, Pozo C. Is the Existence of Two Lineages for Hamadryas glauconome (Lepidoptera: Nymphalidae) True? Molecular and Ecological Evidence. Diversity. 2023; 15(12):1196. https://doi.org/10.3390/d15121196
Chicago/Turabian StylePrado-Cuellar, Blanca R., Luis A. Lara-Pérez, Marysol Trujano-Ortega, Salima Machkour-M’Rabet, and Carmen Pozo. 2023. "Is the Existence of Two Lineages for Hamadryas glauconome (Lepidoptera: Nymphalidae) True? Molecular and Ecological Evidence" Diversity 15, no. 12: 1196. https://doi.org/10.3390/d15121196