

Survival and Genome Evolution Signatures of Klebsiella pneumoniae Isolates Originated in Seven Species of Aquatic Animals

1
Key Laboratory of Quality and Safety Risk Assessment for Aquatic Products on Storage and Preservation (Shanghai), Ministry of Agriculture and Rural Affairs of the People’s Republic of China, Shanghai 201306, China
2
College of Food Science and Technology, Shanghai Ocean University, Shanghai 201306, China
3
Shanghai-MOST Key Laboratory of Health and Disease Genomics, Institute for Genome and Bioinformatics, Shanghai Institute for Biomedical and Pharmaceutical Technologies, Shanghai 200032, China
*
Author to whom correspondence should be addressed.
Diversity 2023, 15(4), 527; https://doi.org/10.3390/d15040527
Submission received: 25 January 2023
/
Revised: 31 March 2023
/
Accepted: 2 April 2023
/
Published: 6 April 2023
(This article belongs to the Special Issue Generation of Genome-Wide Genetic Data and Evolutionary Analyses)
Abstract
:Klebsiella pneumoniae can cause life-threatening pneumonia in humans. The bacterium is also the causative agent of nosocomial infection diseases. In our recent research, we reported, for the first time, the presence of K. pneumoniae in fourteen species of aquatic animals sampled in Shanghai, China. Here, we further investigated the bacterial survival and genome evolution traits. The results revealed that K. pneumoniae isolates (n = 7), recovered from 7 species of commonly consumed aquatic animals, had multiple antibiotic and heavy metal resistance profiles. The isolates were capable of growing vigorously at pH 4.5−7.5 and 0.5−1.0% NaCl in TSB medium at 37 °C. Draft genome sequences of the K. pneumoniae isolates were determined (5,256,522−5,857,823 bp, 56.35–57.81% GC contents), which carried many mobile genetic elements, including genomic islands (n = 87), prophages (n = 14), integrons (n = 4), and insertion sequences (n = 22), indicating possible active horizontal gene transfer during the genome evolution. Meanwhile, numerous strain-specific (n = 199−605) genes, antibiotic resistance (n = 20−35, e.g., β-lactamase) genes, and virulence (n = 43−59, e.g., enterobactin)-related genes, were also identified, demonstrating considerable genome variation in the K. pneumoniae isolates. Overall, the results of this study fill prior gaps in understanding the K. pneumoniae genomes derived from aquatic animals.
1. Introduction
Klebsiella pneumoniae was first described by Carl Friedlander in 1882, which was isolated from the lungs of patients who died from pneumonia [1]. Since then, the Gram-negative bacterium has also been reported to cause bloodstream infections (BSIs), meningitis, osteomyelitis, thrombophlebitis, urinary tract infections (UTIs), and invasive pyogenic liver abscess syndrome [2,3,4]. To date, more than 79 capsular (K antigen) serotypes have been reported in K. pneumoniae isolates, of which K1, K2, K5, K20, K54, and K57 serotypes are strongly associated with the bacterial pathogenesis [5,6].
Antibiotics are widely used in clinics for prevention and therapy of bacterial infections [3]. Nevertheless, the overuse and/or misuse of antibiotics have accelerated the spread of antibiotic-resistant pathogens, particularly in developing countries [2,7,8]. For instance, in China, more than 40% (267/666) of clinical K. pneumoniae isolates, collected from 30 medical centers across the country, were identified as carbapenem-resistant K. pneumoniae (CRKP) in 2017 [7]. The mortality of patients with CRKP infection (main BSIs) was up to 70% [8]. The rising incidence of multiple drug resistant (MDR) K. pneumoniae creates serious threats to public health.
K. pneumoniae are found growing in hospital wastewaters, urban rivers, and even in shrimp farms [9,10,11,12]. Intensive farming in aquaculture drives indiscriminate use of antibiotics, which results in antibiotic residues in aquatic products and MDR pathogens [13]. For example, recently, Luo et al. [14] detected chloramphenicol (CHL) residue in shrimp, shellfish, and fish samples (n = 291) sampled in Shenzhen, China. They found that positive detection rates of CHL were 13.6% (3/22), 37.2% (64/172), and 16.5% (16/97) in shrimp, shellfish, and fish, respectively [14]. Xu et al. [15] recently reported K. pneumoniae isolates (n = 94) present in 14 species of aquatic animals sampled in Shanghai, China, which showed higher resistance rates to sulfamethoxazole–trimethoprim (SXT, 52.1%) and CHL (31.9%). On the other hand, heavy metal concentrations in marine environments, rivers, and soils have been increasing because of industrialization and environmental pollution, which has given rise to heavy-metal-tolerant bacteria [16]. Moreover, heavy metals could trigger the proliferation of antibiotic resistance by increasing mobile genetic element (MGE) abundance or by influencing bacterial communities [17]. It has been reported that the majority of the K. pneumoniae isolates were also tolerant to heavy metals e.g., Cr3+ (96.8%), Pb2+ (89.4%), and Hg2+ (81.9%) [15]. The co-selection between antibiotics and heavy metals, leading to MDR K. pneumoniae, threatens population health [18,19].
Aquatic environments, considered as a pool of antibiotic resistance genes (ARGs), contain diverse microbial communities, where the dissemination of ARGs could partially be attributed to horizontal gene transfer (HGT) mediated via MGEs in bacterial genomes [20,21]. To date, complete genome sequences of over 1757 K. pneumoniae isolates are available in the GenBank database (https://www.ncbi.nlm.nih.gov/, accessed on 14 March 2023). Of these, the majority of the K. pneumoniae strains (n = 1659) were isolated from humans, followed by animals (n = 87), water environments (n = 4), and others (n = 7). To the best of our knowledge, current literature on the genomes of K. pneumoniae isolates originating in aquatic animals is rare.
In our recent research, we reported, for the first time, the presence of K. pneumoniae in 14 species of aquatic animals [15]. Based on the finding, in this study, we further investigated the survival and genome evolution traits of the K. pneumoniae isolates recovered from seven species of commonly consumed aquatic animals. The major objectives of this study were (1) to examine phenotypes, genotypes, and growth traits of the K. pneumoniae isolates; (2) to determine draft genome sequences of the K. pneumoniae isolates using the Illumina Hiseq × Ten sequencing technique and to identify MGEs, such as genomic islands (GIs), prophages, integrons (INs), and insertion sequences (ISs) in the K. pneumoniae genomes; (3) to identify virulence- and resistance-related genes in the K. pneumoniae genomes; and (4) to analyze phylogenetic relatedness of the K. pneumoniae isolates. The results of this study will enrich genome data and fill prior gaps in understanding K. pneumoniae genomes derived from aquatic animals.
2. Materials and Methods
2.1. K. pneumoniae Isolates and Cultural Conditions
K. pneumoniae strains (Table S1) were isolated from three species of shellfish: Maoctra veneriformis, Cipangopaludina cahayensis and Tegillarca granosa; two species of crustaceans: Eriocheir sinensis and Procambarus clarkii; and two species of fish: Epinephelus fuscoguttatus and Misgurnus anguillicaudatus, which were sampled in Shanghai, China in July−September of 2018−2019 [15]. The K. pneumoniae isolates were identified by biochemical and molecular biological methods [15] and stored at −80 °C in a freezer in our laboratory at Shanghai Ocean University, in Shanghai, China. The isolates were routinely incubated in Tryptic Soy Broth (TSB) medium (pH 7.2, 0.5% NaCl) (Beijing Land Bridge Technology, Beijing, China) at 37 °C aerobically, with shaking at 175 rpm [15]. K. pneumoniae ATCC13883 was used as a positive control strain.
2.2. Antibiotic Susceptibility and Heavy Metal Tolerance Assays
Antibiotic susceptibility of the K. pneumoniae isolates were tested according to the disc diffusion method approved by the Clinical and Laboratory Standards Institute in the United State (CLSI, M100-S28, 2018). The Mueller–Hinton (MH) medium and antibiotic discs were purchased from OXOID, Basingstoke, UK, as described in our recent reports [15,22]. Heavy metal tolerance of the K. pneumoniae isolates was performed according to the broth dilution testing (microdilution, CLSI) [15,22]. The HgCl2, NiCl2, CrCl3, CdCl2, PbCl2, CuCl2, ZnCl2, and MnCl2 (Analytical Reagent, Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) were applied in the range from 3200 to 3.125 μg/mL. The results were interpreted by minimal inhibitory concentrations (MICs) that completely inhibited the growth of the bacteria. Escherichia coli ATCC25922 and K12 strains (Institute of Industrial Microbiology, Shanghai, China) were used as quality control strains [15,22].
2.3. Growth Curve Assay
The TSB was adjusted to different pH (3.5, 4.5, 5.5, 6.5, and 7.5) and NaCl concentrations (0.5%, 1%, 2%, 3%, and 4%) as described in our previous studies [23,24]. Growth curves of the K. pneumoniae isolates under different pH (3.5–7.5) and NaCl (0.5–4%) conditions were individually determined at 37 °C for 25 h using Multimode Microplate Reader (BioTek Instruments, Winooski, VT, USA).
2.4. Polymerase Chain Reaction (PCR) Assay
The 16S rRNA gene, virulence-associated genes (aerobactin, magA, tarT, wcaG, iroN, rmpA, entB, fimH, mrkD, and ybtA), and capsule serotypes (K1, K2, K5, K20, K54, and K57) were detected using the PCR assay as described in our recent report [15]. The primers (Table S2) were synthesized by the Sangon (Shanghai, China).
2.5. Genome Sequencing, Assembly, and Annotation
The K. pneumoniae isolates were individually incubated in the TSB (pH 7.2, 0.5% NaCl) to logarithmic growth stage (LGS). Bacterial cells were harvested by centrifugation, and genomic DNA was extracted using TIANamp Bacteria DNA Kit (Tiangen Biochemical Technology Co., Ltd., Beijing, China) according to the manufacture’s instruction. Three separately produced DNA samples were used for each of the K. pneumoniae isolates. DNA samples were analyzed, as described in our previous report [21,24], and only high quality samples were subjected to the genome sequencing.
Whole-genome sequencing of the K. pneumoniae isolates was conducted by Shanghai Majorbio Bio-Pharm Technology Co., Ltd., Shanghai, China, using Illumina Hiseq × Ten (Illumina, San Diego, CA, USA) platform [21]. High-quality sequence assembly was performed using SOAPdenovo (version 2.04) software [25]. Coding sequences (CDSs), rRNA genes, tRNA genes, and Clusters of Orthologous Groups (COG) of proteins were predicted using the same software Glimmer (version 3.02) [26], Barrnap tool (https://github.com/tseemann/barrnap, accessed on 31 July 2022), tRNAscan-SE (version 2.0) [27], and Basic Local Alignment Search Tool (BLAST, http://www.ncbi.nlm.nih.gov/BLAST, accessed on 31 July 2022) with default parameters as described in our recent report [18]. The virulence factor database (http://www.mgc.ac.cn/VFs, accessed on 31 July 2022) and the ARGs database (http://arpcard.Mcmaster.ca, accessed on 31 July 2022) were used to detect virulence- and antibiotic resistance-related genes, respectively [18].
2.6. Comparative Genome Analysis
GIs, prophages, INs, ISs, and CRISPR-Cas repeats in the K. pneumoniae genomes were predicted using the same software IslandViewer (version 1.2) [28], Phage_Finder [29], Integron_Finder (version 2.0) [30], ISEScan (version 1.7.2.1) [31], as well as Mined software (version 3) and CRISPRtyper [32] with default parameters as described in our recent report [21].
Core genes were the set of genes encoding orthologous proteins in all the genomes tested, while pan–genes were the set of all genes present in all the tested genomes. Only proteins with ≥60% amino acid similarity and ≥80% sequence coverage were designated as direct relatives, while those with ≤30% or no hits were assigned as strain-specific genes at E ≤ 1 × 10−5 [21].
A phylogenetic tree was constructed on the basis of amino acid data sets of single-copy orthologs that were present in all the analyzed genomes of 72 K. pneumoniae isolates, of which complete genome sequences of 65 K. pneumoniae isolates were downloaded from the GenBank database (Table S3). The maximum likelihood method was used to build a tree by RAxML (version 8) software [33], with 1000 bootstrap replications and a cut-off threshold of ≥50% bootstrap values.
2.7. Statistical Analysis
The SPSS statistical analysis software (version 17.0, SPSS Inc., Chicago, IL, USA) was used to analyze the data. All tests were conducted in triplicate.
3. Results
3.1. Phenotypes and Genotypes of the K. pneumoniae Isolates
K. pneumoniae 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, and 8-2-10-5 strains (Table S1) were isolated from 7 species of aquatic animals, including M. anguillicaudatus, M. veneriformis, E. sinensis, C. cahayensis, P. clarkii, T. granosa, and E. fuscoguttatus, respectively [15]. The isolates were confirmed by 16S rRNA gene sequencing and analysis, and the obtained 16S rDNA sequences were deposited in the National Center for Biotechnology Information (NCBI) database under the accession numbers shown in Table S1.
The K. pneumoniae isolates had different antibiotic resistance profiles (Table S1). For example, K. pneumoniae 7-5-4 from M. anguillicaudatus displayed resistance to CHL, ciprofloxacin (CIP), kanamycin (KAN), norfloxacin (NOR), SXT, and tetracycline (TET), while K. pneumoniae 7-10-14 from M. veneriformis was resistant to ampicillin (AMP), CHL, gentamicin (GEN), KAN, SXT, and TET. Conversely, K. pneumoniae 7-13-2, 7-17-8, and 8-1-12-1 isolates were solely resistant to AMP.
Meanwhile, the K. pneumoniae isolates tolerated 3−7 heavy metals, showing different tolerance patterns. For example, K. pneumoniae 8-1-12-1 from P. clarkii was tolerant to Zn2+/Pb2+/Mn2+/Hg2+/Cu2+/Cr3+/Cd2+, while K. pneumoniae 8-2-10-5 from E. fuscoguttatus was tolerant to Zn2+/Pb2+/Hg2+/Cu2+/Cr3+/Cd2+. Conversely, K. pneumoniae 7-17-8 from C. cahayensis tolerated the minimum number of heavy metals (Zn2+/Cu2+/Cr3+) tested.
In addition, all the K. pneumoniae isolates tested negative for the virulence-related genes, including the aerobactin, magA, tarT, wcaG, iroN, and rmpA. However, some virulence-related genes tested positive in the K. pneumoniae isolates. For instance, K. pneumoniae 7-10-14 carried the entB gene; K. pneumoniae 7-5-4, 7-13-2, 8-2-5-4, and 8-2-10-5 isolates tested positive for the entB/fimH/mrkD genes; and K. pneumoniae 7-17-8, and 8-1-12-1 isolates had the entB/fimH/mrkD/ybtA gene profile.
3.2. Survival of the K. pneumoniae Isolates at Different pH and NaCl Conditions
The pH of the human stomach normally ranges pH 1−3, but can rise above 6.0 after food consumption [23]. Therefore, we examined survival of the K. pneumoniae isolates at different pH conditions (pH 3.5–7.5) when incubated in the TSB (0.5% NaCl) at 37 °C. As shown in Figure 1A–G, the growth of all K. pneumoniae isolates was severely inhibited at the acidic pH 3.5. However, notably, the acidic pH 4.5 condition strongly promoted the bacterial growth, and all the isolates were capable of growing vigorously at pH 4.5−7.5. The highest biomass was observed at pH 7.5, with the maximum OD600 at stationary growth phase (SGP) in the range of 1.37−1.51 (Figure 1).
Given that the K. pneumoniae isolates were recovered from different aquatic animals in water environments, we further determined growth curves of the K. pneumoniae isolates at different salinity concentrations (0.5−4% NaCl) when incubated in the TSB (pH 7.5) at 37 °C. As shown in Figure 2A−G, the growth of all K. pneumoniae isolates was severely inhibited at 4% NaCl. The biomass of the K. pneumoniae isolates gradually increased with the decreased NaCl concentrations (3–0.5%), and the highest biomass was observed at 0.5% NaCl, showing the maximum OD600 values ranging from 1.26 to 1.44 at SGP.
For example, K. pneumoniae 7-5-4 from M. anguillicaudatus appeared the most susceptible to the higher salinity concentrations, as this isolate was strongly repressed at 3% NaCl as well. Conversely, this condition stimulated the growth of the other 6 isolates, but showed a longer retarded growth phase (RGP) (11−19 h) with lower biomass (OD600 = 0.68−0.97). The decreased NaCl concentration (2%) promoted the growth of K. pneumoniae 7-5-4, although it had an RGP for 8 h. K. pneumoniae 7-5-4 was able to grow vigorously at 1.0−0.5%, the same case as the other isolates.
Taken together, the results demonstrated that the K. pneumoniae isolates of aquatic animal origins were able to grow vigorously at pH 4.5−7.5, 0.5−1.0% NaCl in the TSB at 37 °C. K. pneumoniae 7-5-4 was the most susceptible to higher salinity concentrations (4−3 NaCl) among the isolates tested in this study.
3.3. Genome Features of the K. pneumoniae Isolates of Aquatic Animal Origins
Based on the obtained results, we further determined draft genome sequences of the 7 K. pneumoniae isolates using the Illumina Hiseq × Ten sequencing platform, which generated approximately 76,382−107,851 clean single reads. The final assembly yielded 45−113 scaffolds with sequencing depth (on average) of 188.3–fold to 271.9−fold. The obtained genome sizes ranged from 5,256,522 to 5,857,823 bp with GC contents of 56.35–57.81% (Table 1, Figure S2). A total of 4885–5558 protein-coding genes were predicted, of which approximately 4639–4986 genes were classified into 22 functional catalogs in the COG database. Remarkably, the K. pneumoniae genomes carried many MGEs, including GIs (n = 87), prophages (n = 14), INs (n = 4), and ISs (n = 22), suggesting the HGT during the K. pneumoniae genome evolution via these MGEs.
The draft genomes of the K. pneumoniae 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4 and 8-2-10-5 isolates were deposited in the GenBank database under the accession numbers JALJQY000000000, JALJQX000000000, JALJQW000000000, JALJQV000000000, JALJQT000000000, JALJQQ000000000, and JALJQR00000000, respectively.
3.4. MGEs in the K. pneumoniae Genomes of Aquatic Animal Origins
3.4.1. GIs
GIs can carry large foreign DNA fragments (~200 Kb) and endorse the host’s diverse biological functions [21]. In this study, remarkably, a total of 87 GIs were identified in the 7 K. pneumoniae genomes (Table S4), each of which contained 8−17 GIs, ranging from 3228 to 44,595 bp and encoding 5−47 genes (Figure 3). Interestingly, various functions carried by the GIs were found, e.g., virulence, resistance, substrate hydrolysis, transporting and utilization, restriction and modification, as well as phage and stress regulation.
The K. pneumoniae 7-5-4 genome contained the maximum number of the GIs (n = 17, GIs 1−17), whereas K. pneumoniae 7-10-14 had the fewest GIs (n = 8, GIs 1−8). Of note, virulence-related genes were found in 10 GIs in the 7 K. pneumoniae genomes (Table S5). For example, the hcp gene was present in the GI 12, GI 1, GI 3, GI 16, and GI 3 in K. pneumoniae 7-5-4, 7-10-14, 7-17-8, 8-1-12-1, and 8-2-5-4 genomes, respectively. It is a core gene of the T6SS in Acinetobacter baumannii, causing respiratory tract infection [34]. Antibiotic resistance and heavy-metal-tolerance-related genes were identified in some GIs, e.g., the GI 3 (17,350 bp) and GI 8 (6626 bp) in the K. pneumoniae 7-13-2 genome, respectively. Interestingly, the conjugative transfer-related genes were found in the GI 11 in K. pneumoniae 7-17-8. Additionally, there were some identified GIs carrying phage regulator genes in the K. pneumoniae genomes. For example, the GI 5 in K. pneumoniae 7-5-4, and the GI 11 in K. pneumoniae 8-1-12-1 contained the gene (Kp 7-5-4_1988; Kp 8-1-12-1_3134) encoding a lysB family phage lysis regulatory protein.
3.4.2. Prophages
Prophages are viruses that infect bacteria. They can transfer important biological characteristics to their bacterial hosts [35]. In this study, a total of 14 prophage gene clusters were identified in the 7 K. pneumoniae genomes (Table S6), each of which carried 1−3 prophages, ranging from 21,338 to 108,967 bp and encoding 34−115 genes (Figure 4).
The K. pneumoniae 8-1-12-1 genome contained the maximum number of prophage gene clusters (n = 3), which had sequence similarity to Ralstonia_phage_RSA1 (38,760 bp, NCBI accession number: NC_009382), Enterobacteria_phage_186 (30,624 bp, NCBI accession number: NC_001317), and Klebsiella_phage_phiKO2 (51,601 bp, NCBI accession number: NC_005857). Conversely, K. pneumoniae 8-2-5-4 carried only 1 prophage gene cluster similar to Enterobacteria_phage_HK022 (15,456 bp, NCBI accession number: NC_002161).
The identified 14 prophages in the 7 K. pneumoniae genomes were derived from 4 different genera, including Enterobacteria, Klebsiella, Pseudomonas, and Ralstonia, indicating extensive phage transmission across the genera boundaries. Moreover, the Enterobacteria_phage_186 homologue was present in the K. pneumoniae 7-5-4 and 8-1-12-1 genomes, but in different lengths, encoding different numbers of genes. The same was the case for the Pseudomonas_phage_D3 homologue, which was present in the K. pneumoniae 7-5-4 and 7-10-14 genomes. Likewise, the Klebsiella_phage_phiKO2 homologue was found in the K. pneumonia 7-17-8, 8-1-12-1 and 8-2-10-5 genomes carrying a similar set of phage structure genes but different accessory genes. These results also provided evidence of extensive genome rearrangement during the K. pneumoniae genome evolution.
3.4.3. INs
INs are considered as determinants in acquisition and evolution of virulence and antibiotic resistance [36]. They are classified into type I, type II, type III, and super integrons based on integrase genes (intI1, intI2, intI3, and intI4) [21]. In this study, INs were identified in the K. pneumoniae 7-5-4 (n = 1), 7-10-14 (n = 2), and 8-2-10-5 (n = 1) genomes, but absent from K. pneumoniae 7-13-2, 7-17-8, 8-1-12-1, and 8-2-5-4 genomes (Figure 5, Table S7).
The K. pneumoniae 7-5-4 genome contained only 1 complete IN (2737 bp), encoding a NAD (+)-rifampin ADP-ribosyltransferase Arr-2 (Kp 7_5_4_5567), an ANT(3″)-Ia family aminoglycoside nucleotidyltransferase AadA3 (Kp 7_5_4_5566), an Orf3/QacEdelta1 fusion protein (Kp 7_5_4_5565), and an intl1 (Kp 7_5_4_5568). The type 1 INs are strongly associated with the dissemination of antibiotic resistance in bacteria [37].
The K. pneumoniae 7-10-14 genome contained 2 incomplete INs (INs 1−2) with gene cassettes. The IN 1 encoded an aac(6′)-Ib family aminoglycoside 6′-N-acetyltransferase (Kp 7_10_14_5082), a NAD(+)-rifampin ADP-ribosyltransferase (Kp 7_10_14_5083), and a dihydrofolate reductase type 15 (Kp 7_10_14_5084), while the IN 2 encoded a quaternary ammonium compound efflux SMR (Kp 7_10_14_5103) and an aminoglycoside resistance protein (Kp 7_10_14_5104).
The K. pneumoniae 8-2-10-5 genome contained 1 incomplete IN, encoding a glycoside hydrolase family 31 protein (Kp 8_2_10_5_0283) and an ABC-F family ATPase (Kp 8_2_10_5_0284).
3.4.4. ISs
ISs are short discrete DNA fragments that can move themselves to a new position in DNA almost randomly in a single cell [38]. In this study, all the 7 K. pneumoniae genomes contained ISs (n = 2 to 6), ranging from 741 to 1588 bp (Table S8).
For instance, the K. pneumoniae 7-13-2 genome contained the maximum numbers of ISs (n = 6, IS001−IS006). The IS001 (807 bp) coded for a tyrosine-type recombinase/integrase (Kp 7_13_2_0367); IS002 (1265 bp) for a transposase IS116/IS110/IS902 family (Kp 7_13_2_4341); IS003 (1161 bp) for transposases (Kp 7_13_2_4675, Kp 7_13_2_4676); IS004 (1173 bp) for a IS5 family transposase (Kp 7_13_2_4820); IS005 (1257 bp) for a IS3 family transposase (Kp 7_13_2_5141, Kp 7_13_2_5142); and IS006 (819 bp) for a IS6-like element IS26 family transposase (Kp 7_13_2_5153).
3.5. CRISPR-Cas Repeats
The CRISPR-Cas systems provide adaptive immunity to prokaryotes from invasion by foreign nucleic acids in their hosts [39]. The systems are associated with drug resistance in K. pneumoniae [40,41]. In this study, a number of CRISPR-Cas repeats (n = 54) were identified in the 7 K. pneumoniae genomes, each of which contained 2−14 such gene clusters, ranging from 75 to 2649 bp. However, none of these systems contained the Cas protein, suggesting partial or inactive CRISPR-Cas systems in the K. pneumoniae isolates (Figure 6).
For instance, the K. pneumoniae 7-5-4 genome contained the maximum number of the CRISPR-Cas repeats (n = 14, CRISPRs 1−14), ranging from 77 to 2649 bp. The CRISPR 4 was the longest in size (2649 bp), with the maximum number of repetitive sequences (n = 44), whereas the CRISPR 3 was the shortest (77 bp), with the fewest repeats (n = 2). Conversely, the K. pneumoniae 7-17-8 genome had the fewest CRISPR-Cas repeats (n = 2, CRISPRs 1−2). The CRISPR 1 (201 bp) had 4 repeats, while the CRISPR 2 (188 bp) had 3 repeats.
3.6. Putative Virulence-Associated Genes in the K. pneumoniae Genomes
Many putative virulence-related genes (n = 43−59) were identified in the 7 K. pneumoniae genomes (Table 2). K. pneumoniae 8-1-12-1 form P. clarkii contained the maximum number of such genes (n = 59), whereas K. pneumoniae 7-10-14 from M. veneriformis carried relatively fewer (n = 43).
For instance, the entABCDEFS and fepABCDG gene clusters were identified in all the K. pneumoniae genomes, both of which are related to enterobactin in K. pneumoniae. The former is responsible for enterobactin biosynthesis, while the latter mediates enterobactin transport. Specifically, the fepA gene encodes a receptor for enterobactin uptake [3]. Of note, the iucABCD gene cluster was found in the K. pneumoniae 8-2-5-4 genome, which is involved in the biosynthesis of aerobactin, commonly detected in hypervirulent K. pneumoniae [42].
The other virulence-related genes involved in the bacterial persistence in the host were also identified in the seven K. pneumoniae genomes. For example, the fimH gene encoding a fimbrial protein was found in the K. pneumoniae 7-5-4, 7-17-8, 8-1-12-1, 8-2-5-4, and 8-2-10-5 genomes, which can bind to highly mannosylated UPIa to ensure stable adhesion of bacteria to tissues in the host [43].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 2.
The putative virulence-related genes identified in the K. pneumoniae genomes.
| Virulence-Related Gene | K. pneumoniae Genome | Reference |
|---|---|---|
| ecpABCDER | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [44] |
| entABCDEFS | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [45] |
| fepABCDG | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [46] |
| fes | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [46] |
| gnd | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [47] |
| kdsA | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [48] |
| rcsAB | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [45] |
| tuf | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [49] |
| wza | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [50] |
| galU | 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [51] |
| hcp | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4 | [52] |
| impBCGHJKL | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4 | [52,53] |
| vasDG | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4 | [53] |
| yhjH | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [54] |
| fimABCDEFGI | 7-5-4, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [55] |
| fimH | 7-5-4, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [43] |
| manBC | 7-5-4, 7-10-14, 7-17-8, 8-1-12-1, 8-2-5-4 | [56] |
| vgrG | 7-5-4, 7-10-14, 7-13-2, 8-1-12-1, 8-2-5-4 | [57] |
| iroE | 7-13-2, 7-17-8, 8-2-5-4, 8-2-10-5 | [46] |
| glf | 7-10-14, 7-13-2, 8-2-5-4 | [58] |
| vasJ | 7-10-14, 8-2-5-4 | [59] |
| fyuA | 7-17-8, 8-1-12-1 | [60] |
| irp12345 | 7-17-8, 8-1-12-1 | [55,61] |
| mbtI | 7-17-8, 8-1-12-1 | [62] |
| ybtAX | 7-17-8, 8-1-12-1 | [63,64] |
| allABCDRS | 8-2-10-5 | [65] |
| iucABCD | 8-2-5-4 | [45] |
3.7. Antibiotic and Heavy Metal Resistance-Associated Genes in the K. pneumoniae Genomes
Antimicrobial resistance-related genes (n = 20−35) were also identified in the 7 K. pneumoniae genomes (Table 3). K. pneumoniae 7-5-4 from M. anguillicaudatus contained the maximum number of such genes (n = 35), whereas K. pneumoniae 7-13-2, 7-17-8, and 8-1-12-1 isolates had the least (n = 20).
All the K. pneumoniae genomes contained the genes for MDR, e.g., a multidrug efflux SMR transporter subunit (KpnE), a spermidine export protein mdtJ (KpnF), a multidrug efflux MFS transporter periplasmic adaptor subunit emrA (KpnG), a multidrug resistance protein (KpnH), a multidrug efflux MFS transporter (mdtG), a multidrug efflux RND transporter permease subunit (acrB), a multidrug efflux RND transporter periplasmic adaptor subunit (oqxA), a multidrug efflux RND transporter permease subunit (oqxB), and a multidrug ABC transporter permease/ATP-binding protein (yojI).
The qnrs1 gene, encoding a quinolone resistance pentapeptide repeat protein, was present in the K. pneumoniae 7-5-4 and 8-2-10-5 genomes, while the floR, mphA, and sul1 genes were found in the K. pneumoniae 7-5-4, 7-10-14, and 8-2-10-5 genomes, which encoded a chloramphenicol/florfenicol efflux MFS transporter (floR), a mph(A) family macrolide 2-phosphotransferase (mphA), and a sulfonamide-resistant dihydropteroate synthase (sul1), respectively. Additionally, the genes involved in the resistance to β-lactam antibiotics (penicillins, cephalosporins, carbapenems, and monobactams) and aminoglycosides were also found in some of the K. pneumoniae genomes (Table 3). These results provided genome-wide evidence for antibiotic resistance phenotypes of the seven K. pneumoniae isolates.
Several genes involved in heavy metal tolerance were identified in the K. pneumoniae genomes as well (Table 3). For example, the copA gene, which plays a key role in the export of excess copper [66], was present in the seven K. pneumoniae genomes. Moreover, the cusARS genes, which are involved in the heavy metal efflux RND transporter [66,67], were also found in the seven K. pneumoniae genomes. Additionally, the K. pneumoniae 7-10-14 genome carried the zntA gene as well, which encodes a Zn/Cd/Hg/Pb-transporting ATPase, while K. pneumoniae 7-13-2 had the arsABCR genes, which are essential for heavy metal As resistance [68].
Table 3.
The antibiotic and heavy metal resistance-related genes identified in the K. pneumoniae genomes.
Table 3.
The antibiotic and heavy metal resistance-related genes identified in the K. pneumoniae genomes.
| Antibiotic/Heavy Metal | Gene | K. pneumoniae Genome | Reference |
|---|---|---|---|
| Cephalosporin | acrB | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [69] |
| Fluoroquinolone | hns | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [70] |
| emrR | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [71] | |
| marA | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [72] | |
| ramA | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [72] | |
| crp | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [73] | |
| qnrS1 | 7-5-4, 8-2-10-5 | [74] | |
| qnrB2 | 7-10-14, 8-2-10-5 | [75] | |
| Tetracycline | oqxAB | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [44] |
| tet(A) | 7-5-4, 7-10-14, 8-2-10-5, 8-2-5-4 | [44] | |
| Aminoglycoside | KpnEFGH | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [76,77] |
| ompK37 | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [78] | |
| aph(3′)-Ia | 7-5-4, 7-10-14, 8-2-10-5 | [79] | |
| aph(3″)-Ib | 7-5-4, 7-10-14, 8-2-10-5 | [79] | |
| aph(6)-Id | 7-5-4, 7-10-14, 8-2-10-5 | [79] | |
| aac(3)-IId | 7-10-14, 8-2-10-5 | [79] | |
| aac(6′)-Ib-cr | 7-10-14, 8-2-10-5 | [80] | |
| aadA16 | 7-10-14, 8-2-10-5 | [79] | |
| aadA8 | 7-5-4 | [81] | |
| Diaminopyrimidine | dfrA27 | 7-10-14, 8-2-10-5 | [82] |
| Beta-lactam | SHV-11 | 7-5-4, 7-17-8 | [83] |
| SHV-1 | 7-10-14, 7-13-2 | [84] | |
| SHV-38 | 8-1-12-1, 8-2-5-4 | [85] | |
| TEM-1 | 7-5-4 | [85] | |
| OKP-B-7 | 8-2-10-5 | [86] | |
| TEM-116 | 8-2-5-4 | [85] | |
| Macrolide | mphA | 7-5-4, 7-10-14, 8-2-10-5 | [44] |
| ermB | 7-5-4 | [44] | |
| mphE | 7-5-4 | [87] | |
| msrE | 7-5-4 | [87] | |
| Nitroimidazole | msbA | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [88] |
| Phenicol | floR | 7-5-4, 7-10-14, 8-2-10-5 | [89] |
| Peptide | pmrF | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [90] |
| ugd | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [91] | |
| yojI | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [92] | |
| Sulfonamide | sul1 | 7-5-4, 7-10-14, 8-2-10-5 | [44] |
| sul2 | 7-5-4, 8-2-10-5 | [44] | |
| Rifamycin | arr-2 | 7-5-4 | [80] |
| arr-3 | 7-10-14 | [46] | |
| Fosfomycin | FosA5 | 7-5-4, 7-10-14 | [80] |
| FosA6 | 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [80] | |
| mdtG | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [93] | |
| Heavy metal | cusARS | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [66,67] |
| Heavy metal | copA | 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, 8-2-10-5 | [66] |
| Heavy metal | zntA | 7-10-14 | [94] |
| Heavy metal | arsABCR | 7-13-2 | [68] |
3.8. Strain-Specific Genes of the K. pneumoniae Isolates of Aquatic Animal Origins
Comparative genomic analyses revealed approximately 4111 core genes shared by the K. pneumoniae genomes, which accounted for 65.7% of pan genes (n = 6255). Meanwhile, many strain-specific genes (n = 199−605) were found in the K. pneumoniae isolates (Figure 7). Interestingly, K. pneumoniae 7-5-4 from M. anguillicaudatus contained the highest number of strain-specific genes (n = 605), whereas K. pneumoniae 7-10-14 from T. veneriformis had the fewest (n = 199). Remarkably, higher percentages of the strain-specific genes (30.2−54.4%) encoded unknown proteins. These results also provided the evidence of the considerable genome variation in the K. pneumoniae isolates of aquatic animal origins.
3.9. Phylogenetic Relatedness of the K. pneumoniae Isolates of Aquatic Animal Origins
To address phylogenetic relatedness of the K. pneumoniae isolates of aquatic animal origins, we constructed a phylogenetic tree on the basis of 72 K. pneumoniae genomes. Of these, complete genomes of 65 K. pneumoniae strains were derived from the GenBank database, the majority of which (n = 57) were isolated from human samples, followed by animals (n = 5, bovine, cat, chicken, dog, pig, and rabbit), and water environment (n = 3) in 1999−2021. The 7 K. pneumoniae isolates of aquatic animal origins tested negative for the toxic K1, K2, K5, K20, K54, and K57 serotypes, except K. pneumoniae 8-2-5-4 of serotype K2 [15]. The phylogenetic tree analysis revealed seven distinct clusters, designated as Clusters A−F (Figure 8).
K. pneumoniae 8-1-12-1 from P. clarkii fell into Cluster B, together with K. pneumoniae JX-CR-hvKP (GenBank accession no. NZ_CP064208), which was isolated from human blood in 2019 in China.
Both K. pneumoniae 7-10-14 from M. Veneriformis and K. pneumoniae 7-17-8 from C. cahayensis were classified into Cluster F, together with K. pneumoniae 49,210 (GenBank accession no. NZ_CP089024) isolated from humans in 2016 in China.
Both K. pneumoniae 7-5-4 from M. anguillicaudatus and K. pneumoniae 8-2-10-5 from E. fuscoguttatus were grouped into Cluster E, together with K. pneumoniae SWHE3 (GenBank accession no. NZ_CP055061) isolated from humans in 2018 in China, and K. pneumoniae SB617 (GenBank accession no. NZ_CP084825), which was isolated from water in 2000 in the Netherlands.
K. pneumoniae 7-13-2 from E. sinensis was classified into a single Cluster A, phylogenetically distant from all the other genomes tested, suggesting its unique genome trait.
K. pneumoniae 8-2-5-4 of serotype K2 from T. granosa was classified into Cluster D, showing the closest phylogenetic distance with K. pneumoniae BcKp067 (GenBank accession no. NZ_CP084829), which was isolated from the water environment in 1999 in the Netherlands.
In addition, 8 K. pneumoniae isolates belonging to the capsule serotypes K1, K2, K5, K54 and K57 were classified into Clusters D−F.
Taken together, these results demonstrated the genome diversity of the K. pneumoniae isolates of the clinical and environmental origins.
3.10. Sequence Types (ST) of the K. pneumoniae Isolates of Aquatic Animal Origins
Based on the 7 conserved core genes (gapA, infB, mdh, pgi, phoE, rpoB, and tonB) in K. pneumoniae [15], the multilocus sequence typing (MLST) analysis against the MLST database (https://cge.food.dtu.dk/services/MLST/ (accessed on 23 November 2022)) revealed that K. pneumoniae 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, and 8-2-5-4 isolates belonged to the ST-273, ST-1310, ST-101, ST-353, ST-6289, and ST-2026, respectively, whereas the K. pneumoniae 8-2-10-5 isolate was not classified into any known STs.
4. Discussion
The inappropriate use of antibiotics may result in increasing levels of bacterial resistance [8,95]. For example, Fatima et al. [96] examined the resistance of K. pneumoniae isolates from urine (n = 72) and sputum (n = 35) isolated in Balochistan to 17 antimicrobial agents. They found that the majority of the K. pneumoniae isolates were resistant to GEN (76.2%), followed by SXT (66.7%), and NOR (42.9%) [96]. Marques et al. [97] determined the resistance of K. pneumoniae isolates from companion animals (n = 27) and humans (n = 77), isolated in Lisbon during 2002 to 2015, to 29 antimicrobial agents. Their results showed that most K. pneumoniae isolates were resistant to AMP (95.2%), followed by CHL (31.7%), KAN (27.9%), TET (28.8%), and CIP (25%) [97]. In this study, the K. pneumoniae isolates had different antibiotic resistance profiles. For example, K. pneumoniae 7-5-4 from M. anguillicaudatus and K. pneumoniae 7-10-14 from M. veneriformis displayed resistance to CHL/CIP/KAN/NOR/SXT/TET and AMP/CHL/GEN/KAN/SXT/TET, respectively. K. pneumoniae 8-2-10-5 from E. fuscoguttatus was resistant to CHL/CIP/KAN/SXT/TET, while K. pneumoniae 8-2-5-4 from T. granosa was resistant to AMP/CHL/TET, suggesting different antibiotic exposure levels or pollution sources of aquaculture environments.
Previous studies have reported heavy metal residues in various aquatic environments and aquatic products sampled around the world, especially in developing countries [98,99]. For example, Varol and Sunbul et al. [98] examined the residues of five heavy metals in biota samples, including one species of mussel, crayfish, and farmed fish, respectively, and six species of wild fish, collected from the Euphrates River in Turkey. The highest concentrations of As, Cd, and Pb were detected in mussels, while the highest concentrations of Cu and Zn were detected in crayfish [98]. Recently, Ni et al. [99] reported Cu, Hg, Pb, and Cd residues in 41 species of aquatic animals, sampled in Shanghai, China in July−September of 2018−2019, with positive sample rates of 100%, 100%, 77.4%, and 34.0%, respectively, but none of which exceeded their maximum residue limits [99]. In this study, the K. pneumoniae isolates had different heavy metal tolerance patterns. For example, K. pneumoniae 8-1-12-1 from P. clarkii was tolerant to Cu2+/Cd2+/Cr3+/Hg2+/Mn2+/Pb2+/Zn2+, while K. pneumoniae 8-2-10-5 from E. fuscoguttatus was tolerant to Zn2+/Pb2+/Hg2+/Cu2+/Cr3+/Cd2+. These results suggested a potential risk of consuming these aquatic animals.
The human acidic stomach environment challenges survival and infection of K. pneumoniae. Therefore, we examined survival of the K. pneumoniae isolates at pH 3.5–7.5 when incubated in the TSB (0.5% NaCl) at 37 °C. Unexpectedly, all the isolates were capable of growing vigorously at pH 4.5–7.5, although the highest biomass was observed at pH 7.5. These results provided evidence of the acidic tolerance of the K. pneumoniae isolates of animal origins, which may have been attributed to the bacterial survival across the acidic stomach boundary in the host.
The K. pneumoniae isolates were recovered from different aquatic animals in water environments. M. anguillicaudatus, E. sinensis, C. cahayensis, and P. clarkii were derived from freshwater, while M. Veneriformis, T. granosa, and E. fuscoguttatus from seawater. Therefore, we determined growth curves of the K. pneumoniae isolates at different salinity concentrations (0.5−4% NaCl) when incubated in the TSB (pH 7.5) at 37 °C. Our results indicated that all the K. pneumoniae isolates were able to grow vigorously at 0.5−1.0% NaCl in the TSB (pH 7.5) at 37 °C. Of these, K. pneumoniae 7-5-4 from M. anguillicaudatus was the most susceptible to higher salinity concentrations (4−3% NaCl), consistent with its freshwater culture environment.
Draft genomes of the seven K. pneumoniae isolates were determined using the Illumina Hiseq × Ten sequencing platform. The typical Poisson distribution, with a clear single peak at the only 17–mers frequency, was observed in the sequencing data, indicating less repetitive DNA in the K. pneumoniae genomes (Figure S1, Table S9). The assembled genomes were 5,256,522−5,857,823 bp with GC contents of 56.35−57.81%, similar to the other K. pneumoniae genomes [42,100]. For example, Yu et al. [100] used MiSeq short-read sequencing and Oxford nanopore long-read sequencing to determine whole genomes of 7 CRKP strains, and obtained genome sizes ranging from 5.4 to 5.8 Mb with an average GC content of 57.2% [100]. Du et al. [42] sequenced and de novo assembled the genomes of 6 hvKP strains, which ranged 5.34−5.58 Mb and GC percentages ranging from 57.22 to 57.46% [42]. The genome sizes of 1757 K. pneumoniae strains, which were available in the GenBank database (accessed on 14 March, 2023), ranged from 4.76 Mb to 6.37 Mb, most of which contained 3107 to 5973 predicted genes. In this study, interestingly, a total of 4885–5558 protein-coding genes were predicted, of which 1188−1363 protein-coding genes encoded unknown proteins. Moreover, about 7.1−41.0% of the strain-specific (n = 199−605) genes encoded unknown proteins as well. These results highlighted specific genome traits of the K. pneumoniae isolates of aquatic animal origins, which may result from the high numbers of identified MGEs.
Remarkably, the K. pneumoniae genomes carried many MGEs, including GIs (n = 87), prophages (n = 14), INs (n = 4), and ISs (n = 22). The identified MGEs carrying a large number of genes may constitute an important driving force in K. pneumoniae genome evolution and speciation. For instance, the identified 87 GIs endowed the bacterium with a variety of biological functions for fitness into niches, such as virulence, resistance, substrate hydrolysis, transporting and utilization, and restriction and modification, as well as phage and stress regulation.
There are approximately 1031 bacteriophages on earth, which play a critical role in virulence and evolution of bacterial genomes [101]. Bleriot et al. [102] reported 40 prophages (11.454−84.199 kb) in 16 clinical CRKP strains, 27 of which belonged to the family Myoviridae, 10 to Siphoviridae, and 3 to Podoviridae [102]. In this study, we found 14 prophages (12,633−109,928 bp) in the 7 K. pneumoniae genomes, which were derived from different genera, including Enterobacteria, Klebsiella, Pseudomonas, and Ralstonia. The results in this study, coupled with previous report [102], indicated extensive phage transmission between Klebsiella and the other bacterial genera. In this study, the identified prophage homologues, e.g., Enterobacteria_phage_186, Klebsiella_phage_phiKO2, and Pseudomonas_phage_D3, were present in different K. pneumoniae genomes, but in different lengths encoding different numbers of genes, which provided evidence of extensive genome rearrangement during the K. pneumoniae evolution.
It has been reported that the Type 1 IN is the most prevalent and common in clinical bacteria [103]. For example, Firoozeh et al. [104] isolated K. pneumoniae strains (n = 181) from clinical specimens and found 82.9% (n = 150) of the isolates with MDR phenotypes. Of the MDR isolates, 100% (n = 150) and 36.7% (n = 55) carried intI1 and intI2 genes, respectively, but none had the intI3 gene by PCR amplification [104]. In this study, 4 INs (1229−4332 bp) were identified in the K. pneumoniae 7-5-4, 7-10-14, and 8-2-10-5 genomes. Of these, K. pneumoniae 7-5-4 carried the Type 1 IN. Moreover, this IN contained ARGs, encoding a NAD(+)-rifampin ADP-ribosyltransferase arr-2 (Kp 7_5_4_5567) and an ANT(3″)-Ia family aminoglycoside nucleotidyltransferase aadA3 (Kp 7_5_4_5566), suggesting possible transmission of ARGs mediated by the IN.
ISs consist of two inverted repeat sequences and one or two genes encoding transposases [105]. In this study, all the 7 K. pneumoniae genomes contained ISs (n = 2 to 6), ranging from 741 to 1588 bp. They belonged to the IS3 family, IS5 family, IS6 family, IS91 family, and IS110 family.
The CRISPR-Cas systems defend the prokaryotes from invasion by MGEs [106]. In this study, 54 CRISPR-Cas gene clusters (75−2649 bp) were identified in the 7 K. pneumoniae genomes. However, all the predicted clusters lacked the Cas protein, which plays an essential role in the function of the CRISPR-Cas systems [107]. These results provided indirect evidence for inactive CRISPR-Cas repeats and possible active HGT in the 7 K. pneumoniae isolates.
Many virulence-related genes have been identified in K. pneumoniae isolates [108,109]. For example, Remya et al. [108] detected 9 virulence genes in K. pneumoniae isolates (n = 370) by PCR amplification, including the magA, allS, kfu, K2A, rmpA, entB, ybtS, fimH, and uge genes. They found that 93.2% (345/370) of the isolates carried multiple virulence genes, 4.0% (15/370) carried 1, and 2.7% (10/370) had none [108]. Kuş et al. [109] detected 16 virulence genes in K. pneumoniae strains (n = 53) isolated from nosocomial infections in Turkey by PCR amplification, including the fimH-1, mrkD, kpn, iutA, ycfM, entB, irp-1, irp-2, ybtS, fyuA, iroN, rmpA, magA, traT, hlyA, and cnf-1 genes. Their results showed that the entB gene was the most predominant (96.2%), followed by the ycfM (86.8%), mrkD (83.0%), fimH-1 (64.2%), fyuA (54.7%), and kpn (49.1%). The detection rates of the ybtS, irp-1, irp-2, traT, and iutA genes ranged from 41.5 to 5.7%, whereas the other genes (iroN, rmpA, magA, hlyA, and cnf-1) tested negative in the isolates [109]. In this study, based on the obtained genome sequences, we also identified many virulence-related genes (n = 43−59) in the 7 K. pneumoniae isolates of aquatic animal origins, e.g., ecpABCDE, entABCDEF, fimABCDEFGHI, iucABCD, fepABCDG, fyuA, vgrG, galU, gnd, vgrG, fimH, entB, mrkd, ybta, and T6SS-associated genes, which were involved in adhesion, antiphagocytosis, secretion system, and gene regulation of K. pneumoniae. Of note, K. pneumoniae 8-1-12-1 from P. clarkii contained the maximum number of the virulence-associated genes (n = 59), whereas K. pneumoniae 7-10-14 from M. veneriformis had relatively fewer (n = 43). These virulence-related genes may be candidate targets for the development of new diagnostics, vaccines, and treatments to control K. pneumoniae infection.
The emergence and spread of MDR pathogens poses a serious threat to public safety [110]. For example, Marques et al. [97] reported 15 antibiotic resistance-related genes in K. pneumoniae isolates, causing UTIs from companion animals (n = 27) and humans (n = 77), e.g., qnrB, qnrS, sul1, sul2, sul3, dfrA12, dfrIa, tet(A), tet(B), and floR [97]. In this study, many antibiotic resistance-related genes (n = 20−35) were identified in the 7 K. pneumoniae genomes, e.g., tetA, acrB, hns, oqxA, aac (6′)-Ib-cr, ermB, msbA, floR, pmrF, sul1, arr-2, and fosA5, which are involved in the resistance to cephalosporin, fluoroquinolone, tetracycline, aminoglycoside, macrolide, phenicol, sulfonamide, rifamycin, and fosfomycin. Moreover, several genes in heavy metal tolerance were also identified in the seven K. pneumoniae genomes, such as the cusASR, copA, zntA, and arsABCR genes. These results provided genome-wide evidence for the resistance phenotypes of the K. pneumoniae isolates of aquatic animal origins.
5. Conclusions
The K. pneumoniae 7-10-14, 7-17-8, 8-2-5-4, 7-13-2, 8-1-12-1, 8-2-10-5, and 7-5-4 strains of aquatic animal origins had multiple antibiotic resistance and heavy metal tolerance profiles, and were capable of growing vigorously at pH 4.5−7.5 and 0.5−1.0% NaCl in the TSB medium at 37 °C.
Remarkably, the K. pneumoniae genomes carried many MGEs, including GIs (n = 87), prophages (n = 14), INs (n = 4), and ISs (n = 22), as well as partial or inactive CRISPR-Cas systems (n = 54), indicating possible active HGT during the K. pneumoniae genome evolution. Many antibiotic resistance (n = 20−35) and virulence (n = 43−59)-related genes were found in the K. pneumoniae genomes. K. pneumoniae 7-5-4 from M. anguillicaudatus contained the maximum number of ARGs (n = 35), while K. pneumoniae 8-1-12-1 from P. clarkii carried the most virulence-related genes (n = 59). Additionally, numerous strain-specific (n = 199−605) genes were present in the K. pneumoniae isolates, approximately 30.2−54.4% of which encoded unknown proteins. These results, coupled with the phylogenetic tree analysis, demonstrated considerable genome variation and high genome plasticity of the K. pneumoniae isolates.
Overall, the results of this study enrich genome data and fill prior gaps in understanding the K. pneumoniae genomes derived from aquatic animals.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/d15040527/s1, Table S1: The genotypes and phenotypes of the K. pneumoniae isolates used in this study; Table S2: The oligonucleotide primers used in this study; Table S3: The sixty-five K. pneumoniae strains with complete genomes used in the phylogenetic tree; Table S4: The identified GIs in the K. pneumoniae genomes; Table S5: Various functions of the identified GIs in the K. pneumoniae genomes; Table S6: The identified prophages in the K. pneumoniae genomes; Table S7: The identified INs in the K. pneumoniae genomes; Table S8: The identified ISs in the K. pneumoniae genomes; Table S9: The identified repeats at the end of scaffolds of the K. pneumoniae genomes; Figure S1: The k-mer analysis for K. pneumoniae subread data based on the number of unique 17-mers; (A−G): K. pneumoniae 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, and 8-2-10-5 genomes, respectively; Figure S2: Genome circle maps of the seven K. pneumoniae isolates. Circles from the inwards to outside represented GC content (outward parts mean higher than average, while inward parts mean lower than average); GC-skew (purple values are higher than zero, while green values are lower than zero); the reference genome of K. pneumoniae ATCC43816 (GenBank accession no. NZ_CP064352); K. pneumoniae 8-2-5-4, 8-2-10-5, 8-1-12-1, 7-17-8, 7-13-2, 7-10-14, and 7-5-4 genomes, respectively; and CDSs on the negative and positive chains (inward and outward parts), respectively. References [111,112,113] are cited in the supplementary materials.
Author Contributions
H.G.: investigation, data curation, and writing—original draft preparation; L.X.: supervision and discussion; L.C.: funding acquisition, conceptualization, and writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Shanghai Municipal Science and Technology Commission, grant number 17050502200, and the National Natural Science Foundation of China, grant number 31671946.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Draft genome sequences of the seven K. pneumoniae isolates have been deposited in GenBank database under the accession numbers: JALJQY000000000, JALJQX000000000, JALJQW000000000, JALJQV000000000, JALJQT000000000, JALJQQ000000000, and JALJQR000000000.
Acknowledgments
The authors are grateful to Dingxiang Xu at Shanghai Ocean University for help with the genome sequence analysis in this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Martin, R.M.; Bachman, M.A. Colonization, infection, and the accessory genome of Klebsiella pneumoniae. Front. Cell Infect. Microbiol. 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girometti, N.; Lewis, R.E.; Giannella, M.; Ambretti, S.; Bartoletti, M.; Tedeschi, S.; Tumietto, F.; Cristini, F.; Trapani, F.; Gaibani, P.; et al. Klebsiella pneumoniae bloodstream infection: Epidemiology and impact of inappropriate empirical therapy. Medicine 2014, 93, 298–309. [Google Scholar] [CrossRef]
- Paczosa, M.K.; Mecsas, J. Klebsiella pneumoniae: Going on the offense with a strong defense. Microbiol. Mol. Biol. Rev. 2016, 80, 629–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arato, V.; Raso, M.M.; Gasperini, G.; Berlanda Scorza, F.; Micoli, F. Prophylaxis and treatment against Klebsiella pneumoniae: Current insights on this emerging anti-microbial resistant global threat. Int. J. Mol. Sci. 2021, 22, 4042. [Google Scholar] [CrossRef]
- Turton, J.F.; Perry, C.; Elgohari, S.; Hampton, C.V. PCR characterization and typing of Klebsiella pneumoniae using capsular type-specific, variable number tandem repeat and virulence gene targets. J. Med. Microbiol. 2010, 59, 541–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Zhao, Y.; Liu, C.; Chen, Z.; Zhou, D. Molecular pathogenesis of Klebsiella pneumoniae. Future Microbiol. 2014, 9, 1071–1081. [Google Scholar] [CrossRef]
- Qin, X.; Wu, S.; Hao, M.; Zhu, J.; Ding, B.; Yang, Y. The colonization of carbapenem-resistant Klebsiella pneumoniae: Epidemiology, resistance mechanisms, and risk factors in patients admitted to intensive care units in China. J. Infect. Dis. 2020, 221, S206–S214. [Google Scholar] [CrossRef]
- Liu, P.; Li, X.; Luo, M.; Xu, X.; Su, K.; Chen, S. Risk factors for carbapenem-resistant Klebsiella pneumoniae infection: A meta-analysis. Microb. Drug Resist. 2018, 24, 190–198. [Google Scholar] [CrossRef]
- Galler, H.; Feierl, G.; Petternel, C.; Reinthaler, F.F.; Haas, D.; Grisold, A.J. KPC-2 and OXA-48 carbapenemase-harbouring Enterobacteriaceae detected in an Austrian wastewater treatment plant. Clin. Microbiol. Infect. 2014, 20, O132–O134. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, S.; Moura, R.A.; Silva, K.C.; Pavez, M.; McCulloch, J.A.; Dropa, M. Isolation of KPC-2-producing Klebsiella pneumoniae strains belonging to the high-risk multiresistant clonal complex 11 (ST437 and ST340) in urban rivers. J. Antimicrob. Chemother. 2014, 69, 849–852. [Google Scholar] [CrossRef] [Green Version]
- Jelić, M.; Hrenović, J.; Dekić, S.; Goić-Barišić, I.; Tambić Andrašević, A. First evidence of KPC-producing ST258 Klebsiella pneumoniae in river water. J. Hosp. Infect. 2019, 103, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, S.; Rosado, D.; Moreno-Andrés, J.; Cartuche, L.; Cruz, D.; Acevedo-Merino, A. Inactivation of a wild isolated Klebsiella pneumoniae by photo-chemical processes: UV-C, UV-C/H2O2 and UV-C/H2O2/Fe3+. Catal. Today. 2018, 313, 94–99. [Google Scholar] [CrossRef]
- Chen, J.; Sun, R.; Pan, C.; Sun, Y.; Mai, B.; Li, Q.X. Antibiotics and food safety in aquaculture. J. Agric. Food Chem. 2020, 68, 11908–11919. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Lu, S.; Huang, C.; Wang, F.; Ren, Y.; Cao, H. A survey of chloramphenicol residues in aquatic products of Shenzhen, South China. Food Addit. Contam. Part A 2021, 38, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ni, L.; Guan, H.; Chen, D.; Qin, S.; Chen, L. First report of potentially pathogenic Klebsiella pneumoniae from serotype K2 in Mollusk Tegillarca granosa and genetic diversity of Klebsiella pneumoniae in 14 species of edible aquatic animals. Foods 2022, 11, 4058. [Google Scholar] [CrossRef]
- Pal, A.; Bhattacharjee, S.; Saha, J.; Sarkar, M.; Mandal, P. Bacterial survival strategies and responses under heavy metal stress: A comprehensive overview. Crit. Rev. Microbiol. 2022, 48, 327–355. [Google Scholar] [CrossRef]
- Jiang, H.; Yu, T.; Yang, Y.; Yu, S.; Wu, J.; Lin, R.; Li, Y.; Fang, J.; Zhu, C. Co-occurrence of antibiotic and heavy metal resistance and sequence type diversity of Vibrio parahaemolyticus isolated from Penaeus vannamei at freshwater farms, feawater farms, and markets in Zhejiang Province, China. Front. Microbiol. 2020, 11, 1294. [Google Scholar] [CrossRef]
- Balali-Mood, M.; Naseri, K.; Tahergorabi, Z.; Khazdair, M.R.; Sadeghi, M. Toxic mechanisms of five heavy metals: Mercury, lead, chromium, cadmium, and arsenic. Front. Pharmacol. 2021, 12, 643972. [Google Scholar] [CrossRef]
- Dickinson, A.W.; Power, A.; Hansen, M.G.; Brandt, K.K.; Piliposian, G.; Appleby, P. Heavy metal pollution and co-selection for antibiotic resistance: A microbial palaeontology approach. Environ. Int. 2019, 132, 105117. [Google Scholar] [CrossRef]
- Kunhikannan, S.; Thomas, C.J.; Franks, A.E.; Mahadevaiah, S.; Kumar, S.; Petrovski, S. Environmental hotspots for antibiotic resistance genes. Microbiologyopen 2021, 10, e1197. [Google Scholar] [CrossRef]
- Xu, D.; Peng, X.; Xie, L.; Chen, L. Survival and genome diversity of Vibrio parahaemolyticus isolated from edible aquatic animals. Diversity 2022, 14, 350. [Google Scholar] [CrossRef]
- Chen, D.; Li, X.; Ni, L.; Xu, D.; Xu, Y.; Ding, Y.; Xie, L.; Chen, L. First experimental evidence for the presence of potentially toxic Vibrio cholerae in snails, and virulence, cross-resistance and genetic diversity of the bacterium in 36 species of aquatic food animals. Antibiotics 2021, 10, 412. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, T.; Peng, X.; Chen, L. Insights into Vibrio parahaemolyticus CHN25 response to artificial gastric fluid stress by transcriptomic analysis. Int. J. Mol. Sci. 2014, 15, 22539–22562. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, Y.; Yu, P.; Ren, S.; Zhu, Z.; Jin, Y.; Yan, J.; Peng, X.; Chen, L. Prophage-related gene VpaChn25_0724 contributes to cell membrane integrity and growth of Vibrio parahaemolyticus CHN25. Front. Cell. Infect. Microbiol. 2020, 10, 595709. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [Green Version]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Fouts, D.E. Phage_Finder: Automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 2006, 34, 5839–5851. [Google Scholar] [CrossRef]
- Bland, C.; Ramsey, T.L.; Sabree, F.; Lowe, M.; Brown, K.; Kyrpides, N.C.; Hugenholtz, P. CRISPR recognition tool (CRT): A tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform. 2007, 8, 209. [Google Scholar] [CrossRef] [Green Version]
- Cury, J.; Jové, T.; Touchon, M.; Néron, B.; Rocha, E.P. Identification and analysis of integrons and cassette arrays in bacterial genomes. Nucleic Acids Res. 2016, 44, 4539–4550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siguier, P.; Gourbeyre, E.; Chandler, M. Bacterial insertion sequences: Their genomic impact and diversity. FEMS Microbiol. Rev. 2014, 38, 865–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.Y.; Liu, C.X.; Liu, P.; Wu, Z.Y.; Zhang, Y.D.; Xiong, X.S. Regulation of gene expression of hcp, a core gene of the type VI secretion system in Acinetobacter baumannii causing respiratory tract infection. J. Med. Microbiol. 2018, 67, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wei, Y.; Ji, X. Research progress of prophages. Yi Chuan 2021, 43, 240–248. [Google Scholar] [CrossRef]
- Li, Y.; Yang, L.; Fu, J.; Yan, M.; Chen, D.; Zhang, L. Genotyping and high flux sequencing of the bacterial pathogenic elements-integrons. Microb. Pathog. 2018, 116, 22–25. [Google Scholar] [CrossRef]
- Huyan, J.; Tian, Z.; Zhang, Y.; Zhang, H.; Shi, Y.; Gillings, M.R. Dynamics of class 1 integrons in aerobic biofilm reactors spiked with antibiotics. Environ. Int. 2020, 140, 105816. [Google Scholar] [CrossRef]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 2018, 31, e00088-17. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Jiang, S.; Wang, Y.; Tian, X.; Zhao, P.; Xu, J. CRISPR-Cas adaptive immune systems in Sulfolobales: Genetic studies and molecular mechanisms. Sci. China Life Sci. 2021, 64, 678–696. [Google Scholar] [CrossRef]
- Wang, G.; Song, G.; Xu, Y. Association of CRISPR/Cas system with the drug resistance in Klebsiella pneumoniae. Infect. Drug Resist. 2020, 13, 1929–1935. [Google Scholar] [CrossRef]
- Kamruzzaman, M.; Iredell, J.R. CRISPR-Cas system in antibiotic resistance plasmids in Klebsiella pneumoniae. Front. Microbiol. 2020, 10, 2934. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Zhang, J.; Liu, P.; Li, X.; Su, K.; Yuan, L. Genome sequencing and comparative genome analysis of 6 hypervirulent Klebsiella pneumoniae strains isolated in China. Arch. Microbiol. 2021, 203, 3125–3133. [Google Scholar] [CrossRef] [PubMed]
- Sarshar, M.; Behzadi, P.; Ambrosi, C.; Zagaglia, C.; Palamara, A.T.; Scribano, D. FimH and anti-adhesive therapeutics: A disarming strategy against uropathogens. Antibiotics 2020, 9, 397. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Kim, Y.; Han, D.; Hur, H.G. Emergence of high level carbapenem and extensively drug resistant Escherichia coli ST746 producing NDM-5 in influent of wastewater treatment plant, seoul, South Korea. Front. Microbiol. 2021, 12, 645411. [Google Scholar] [CrossRef]
- Yuan, L.; Li, X.; Du, L.; Su, K.; Zhang, J.; Liu, P. RcsAB and fur coregulate the iron-acquisition system via entC in Klebsiella pneumoniae NTUH-K2044 in response to iron availability. Front. Cell Infect. Microbiol. 2020, 10, 282. [Google Scholar] [CrossRef]
- Huang, L.; Fu, L.; Hu, X.; Liang, X.; Gong, G.; Xie, C. Co-occurrence of Klebsiella variicola and Klebsiella pneumoniae both carrying bla (KPC) from a respiratory intensive care unit patient. Infect. Drug Resist. 2021, 14, 4503–4510. [Google Scholar] [CrossRef]
- Cookson, A.L.; Biggs, P.J.; Marshall, J.C.; Reynolds, A.; Collis, R.M.; French, N.P. Culture independent analysis using gnd as a target gene to assess Escherichia coli diversity and community structure. Sci. Rep. 2017, 7, 841. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.B.; Wu, S.L.; Zhu, D.P.; Wu, H.; Jiang, T.; Qian, Y.H. Expression of the 2-dehydro-3-deoxyphosphooc-tonate aldolase (KdsA) gene in mulberry leaves (Morus alba L.) is down-regulated under high salt and drought stress. Genet. Mol. Res. 2015, 14, 11955–11964. [Google Scholar] [CrossRef]
- Harvey, K.L.; Jarocki, V.M.; Charles, I.G.; Djordjevic, S.P. The diverse functional roles of elongation factor Tu (EF-Tu) in microbial pathogenesis. Front. Microbiol. 2019, 10, 2351. [Google Scholar] [CrossRef]
- Niu, T.; Guo, L.; Luo, Q.; Zhou, K.; Yu, W.; Chen, Y. Wza gene knockout decreases Acinetobacter baumannii virulence and affects wzy-dependent capsular polysaccharide synthesis. Virulence 2020, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- De Luca, E.; Alvarez-Narvaez, S.; Maboni, G.; Baptista, R.P.; Nemeth, N.M.; Niedringhaus, K.D. Comparative genomics analyses support the reclassification of bisgaard taxon 40 as Mergibacter gen. nov., with Mergibacter septicus sp. nov. as type species: Novel insights into the phylogeny and virulence factors of a pasteurellaceae family member associated with mortality events in seabirds. Front. Microbiol. 2021, 12, 667356. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhao, L.; Huang, L.; Qin, Y.; Zhang, J.; Zhang, J. Integration of RNA-seq and RNAi provides a novel insight into the immune responses of Epinephelus coioides to the impB gene of Pseudomonas plecoglossicida. Fish Shellfish. Immunol. 2020, 105, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Pira, H.; Risdian, C.; Musken, M.; Schupp, P.J.; Wink, J. Photobacterium arenosum WH24, isolated from the gill of pacific oyster Crassostrea gigas from the north sea of germany: Co-cultivation and prediction of virulence. Curr. Microbiol. 2022, 79, 219. [Google Scholar] [CrossRef] [PubMed]
- Claret, L.; Miquel, S.; Vieille, N.; Ryjenkov, D.A.; Gomelsky, M.; Darfeuille-Michaud, A. The flagellar sigma factor fliA regulates adhesion and invasion of crohn disease-associated Escherichia coli via a cyclic dimeric GMP-dependent pathway. J. Biol. Chem. 2007, 282, 33275–33283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araújo, B.F.; Ferreira, M.L.; Campos, P.A.; Royer, S.; Gonçalves, I.R.; da Fonseca Batistão, D.W. Hypervirulence and biofilm production in KPC-2-producing Klebsiella pneumoniae CG258 isolated in Brazil. J. Med. Microbiol. 2018, 67, 523–528. [Google Scholar] [CrossRef]
- Jensen, S.O.; Reeves, P.R. Molecular evolution of the GDP-mannose pathway genes (manB and manC) in Salmonella enterica. Microbiology-SGM 2001, 147 Pt 3, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhou, Z.; He, F.; Ruan, Z.; Jiang, Y.; Hua, X. The role of the type VI secretion system vgrG gene in the virulence and antimicrobial resistance of Acinetobacter baumannii ATCC 19606. PLoS ONE 2018, 13, e0192288. [Google Scholar] [CrossRef] [Green Version]
- Barnell, W.O.; Liu, J.; Hesman, T.L.; O’Neill, M.C.; Conway, T. The zymomonas mobilis glf, zwf, edd, and glk genes form an operon: Localization of the promoter and identification of a conserved sequence in the regulatory region. J. Bacteriol. 1992, 174, 2816–2823. [Google Scholar] [CrossRef] [Green Version]
- Planamente, S.; Salih, O.; Manoli, E.; Albesa-Jové, D.; Freemont, P.S.; Filloux, A. TssA forms a gp6-like ring attached to the type VI secretion sheath. EMBO J. 2016, 35, 1613–1627. [Google Scholar] [CrossRef]
- Chen, Y.; Song, K.; Chen, X.; Li, Y.; Lv, R.; Zhang, Q. Attenuation of Yersinia pestis fyuA mutants caused by iron uptake inhibition and decreased survivability in macrophages. Front. Cell. Infect. Microbiol. 2022, 12, 874773. [Google Scholar] [CrossRef]
- Magistro, G.; Magistro, C.; Stief, C.G.; Schubert, S. The high-pathogenicity island (HPI) promotes flagellum-mediated motility in extraintestinal pathogenic. Escherichia coli. PLoS ONE 2017, 12, e0183950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meneghetti, F.; Villa, S.; Gelain, A.; Barlocco, D.; Chiarelli, L.R.; Pasca, M.R. Iron acquisition pathways as targets for antitubercular drugs. Curr. Med. Chem. 2016, 23, 4009–4026. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Wang, Q.; Perlaza-Jimenez, L.; Zheng, X.; Zhao, Y.; Dhanasekaran, V. First description of antimicrobial resistance in carbapenem-susceptible Klebsiella pneumoniae after imipenem treatment, driven by outer membrane remodeling. BMC Microbiol. 2020, 20, 218. [Google Scholar] [CrossRef] [PubMed]
- Bobrov, A.G.; Kirillina, O.; Fosso, M.Y.; Fetherston, J.D.; Miller, M.C.; VanCleave, T.T. Zinc transporters YbtX and ZnuABC are required for the virulence of Yersinia pestis in bubonic and pneumonic plague in mice. Metallomics. 2017, 9, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Arena, F.; Henrici De Angelis, L.; Pieralli, F.; Di Pilato, V.; Giani, T.; Torricelli, F. Draft genome sequence of the first hypermucoviscous Klebsiella quasipneumoniae subsp. quasipneumoniae isolate from a bloodstream infection. Genome Announc. 2015, 3, e00952-15. [Google Scholar] [CrossRef] [Green Version]
- Besaury, L.; Bodilis, J.; Delgas, F.; Andrade, S.; De la Iglesia, R.; Ouddane, B. Abundance and diversity of copper resistance genes cusA and copA in microbial communities in relation to the impact of copper on Chilean marine sediments. Mar. Pollut. Bull. 2013, 67, 16–25. [Google Scholar] [CrossRef]
- Shafer, C.M.; Tseng, A.; Allard, P.; McEvoy, M.M. Strength of Cu-efflux response in Escherichia coli coordinates metal resistance in Caenorhabditis elegans and contributes to the severity of environmental toxicity. J. Biol. Chem. 2021, 297, 101060. [Google Scholar] [CrossRef]
- Zhen, Z.; Yan, C.; Zhao, Y. Influence of epiphytic bacteria on arsenic metabolism in Hydrilla verticillata. Environ. Pollut. 2020, 261, 114232. [Google Scholar] [CrossRef]
- Bratu, S.; Landman, D.; George, A.; Salvani, J.; Quale, J. Correlation of the expression of acrB and the regulatory genes marA, soxS and ramA with antimicrobial resistance in clinical isolates of Klebsiella pneumoniae endemic to New York City. J. Antimicrob. Chemother. 2009, 64, 278–283. [Google Scholar] [CrossRef] [Green Version]
- Hurtado-Escobar, G.A.; Grepinet, O.; Raymond, P.; Abed, N.; Velge, P.; Virlogeux-Payant, I. H-NS is the major repressor of Salmonella Typhimurium Pef fimbriae expression. Virulence 2019, 10, 849–867. [Google Scholar] [CrossRef] [Green Version]
- Barroso, K.C.M.; Previato-Mello, M.; Batista, B.B.; Batista, J.H.; da Silva Neto, J.F. EmrR-dependent upregulation of the efflux pump emrCAB contributes to antibiotic resistance in Chromobacterium violaceum. Front. Microbiol. 2018, 9, 2756. [Google Scholar] [CrossRef] [PubMed]
- Holden, E.R.; Webber, M.A. MarA, ramA, and soxS as mediators of the stress response: Survival at a cost. Front. Microbiol. 2020, 11, 828. [Google Scholar] [CrossRef] [PubMed]
- Aiba, H. Autoregulation of the Escherichia coli crp gene: CRP is a transcriptional repressor for its own gene. Cell 1983, 32, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Monárrez, R.; Wang, Y.; Fu, Y.; Liao, C.H.; Okumura, R.; Braun, M.R. Genes and proteins involved in qnrS1 induction. Antimicrob. Agents Chemother. 2018, 62, e00806-18. [Google Scholar] [CrossRef] [Green Version]
- Furlan, J.P.R.; Lopes, R.; Gonzalez, I.H.L.; Ramos, P.L.; von Zeska Kress, M.R.; Stehling, E.G. Hypermucoviscous/hypervirulent and extensively drug-resistant QnrB2-, QnrS1-, and CTX-M-3-coproducing Klebsiella pneumoniae ST2121 isolated from an infected elephant (Loxodonta africana). Vet. Microbiol. 2020, 251, 108909. [Google Scholar] [CrossRef]
- Srinivasan, V.B.; Rajamohan, G. KpnEF, a new member of the Klebsiella pneumoniae cell envelope stress response regulon, is an SMR-type efflux pump involved in broad-spectrum antimicrobial resistance. Antimicrob. Agents Chemother. 2013, 57, 4449–4462. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, V.B.; Singh, B.B.; Priyadarshi, N.; Chauhan, N.K.; Rajamohan, G. Role of novel multidrug efflux pump involved in drug resistance in Klebsiella pneumoniae. PLoS ONE 2014, 9, e96288. [Google Scholar] [CrossRef] [Green Version]
- Doménech-Sánchez, A.; Hernández-Allés, S.; Martínez-Martínez, L.; Benedí, V.J.; Albertí, S. Identification and characterization of a new porin gene of Klebsiella pneumoniae: Its role in beta-lactam antibiotic resistance. J. Bacteriol. 1999, 181, 2726–2732. [Google Scholar] [CrossRef] [Green Version]
- Galetti, R.; Filho, R.; Ferreira, J.C.; Varani, A.M.; Sazinas, P.; Jelsbak, L. The plasmidome of multidrug-resistant emergent Salmonella serovars isolated from poultry. Infect. Genet. Evol. 2021, 89, 104716. [Google Scholar] [CrossRef]
- Naha, S.; Sands, K.; Mukherjee, S.; Roy, C.; Rameez, M.J.; Saha, B. KPC-2-producing Klebsiella pneumoniae ST147 in a neonatal unit: Clonal isolates with differences in colistin susceptibility attributed to acrAB-TolC pump. Int. J. Antimicrob. Agents. 2020, 55, 105903. [Google Scholar] [CrossRef]
- Ceccarelli, D.; Salvia, A.M.; Sami, J.; Cappuccinelli, P.; Colombo, M.M. New cluster of plasmid-located class 1 integrons in Vibrio cholerae O1 and a dfrA15 cassette-containing integron in Vibrio parahaemolyticus isolated in Angola. Antimicrob. Agents. Chemother. 2006, 50, 2493–2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.F.; Chen, G.S.; Kong, Q.X.; Gao, L.P.; Chen, X.; Ye, Y. Increase in the prevalence of resistance determinants to trimethoprim/sulfamethoxazole in clinical Stenotrophomonas maltophilia isolates in China. PLoS ONE 2016, 11, e0157693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miryala, S.K.; Anbarasu, A.; Ramaiah, S. Role of SHV-11, a class A beta-lactamase, gene in multidrug resistance among Klebsiella pneumoniae strains and understanding Its mechanism by gene Network analysis. Microb. Drug. Resist. 2020, 26, 900–908. [Google Scholar] [CrossRef]
- Dadashpour, R.; Moghaddam, M.J.M.; Salehi, Z. Prevalence of non-extended spectrum beta-lactamases SHV-1 and TEM-1 or -2 types in multidrug-resistant Enterobacteriaceae in northern Iran. Biol. Futur. 2020, 71, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.H.; Siddiqui, M.T.; Sultan, I.; Haq, Q.M.R. Prevalence and diversity of blaTEM, blaSHV and blaCTX-M variants among multidrug resistant Klebsiella spp. from an urban riverine environment in India. Int. J. Environ. Health Res. 2019, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Fevre, C.; Passet, V.; Weill, F.X.; Grimont, P.A.; Brisse, S. Variants of the Klebsiella pneumoniae OKP chromosomal beta-lactamase are divided into two main groups, OKP-A and OKP-B. Antimicrob. Agents Chemother. 2005, 49, 5149–5152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Lu, W.; Zhou, D.; Zheng, G.; Liu, H.; Qian, C. Characterization of two macrolide resistance-related genes in multidrug-resistant Pseudomonas aeruginosa isolates. Pol. J. Microbiol. 2020, 69, 349–356. [Google Scholar] [CrossRef]
- Chiu, H.C.; Lin, T.L.; Yang, J.C.; Wang, J.T. Synergistic effect of imp/ostA and msbA in hydrophobic drug resistance of Helicobacter pylori. BMC Microbiol. 2009, 9, 136. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Zhang, J.; Xu, L.; Liu, Y.; Li, P.; Zhu, T. Spread of the florfenicol resistance floR gene among clinical Klebsiella pneumoniae isolates in China. Antimicrob. Resist. Infect. Control. 2018, 7, 127. [Google Scholar] [CrossRef] [Green Version]
- Breazeale, S.D.; Ribeiro, A.A.; Raetz, C.R. Oxidative decarboxylation of UDP-glucuronic acid in extracts of polymyxin-resistant Escherichia coli. origin of lipid a species modified with 4-amino-4-deoxy-L-arabinose. J. Biol. Chem. 2002, 277, 2886–2896. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.S.; Lin, T.Y.; Wang, W.B.; Liu, M.C.; Hsueh, P.R.; Liaw, S.J. Characterization of UDP-glucose dehydrogenase and UDP-glucose pyrophosphorylase mutants of Proteus mirabilis: Defectiveness in polymyxin B resistance, swarming, and virulence. Antimicrob. Agents Chemother. 2010, 54, 2000–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwivedi, G.R.; Maurya, A.; Yadav, D.K.; Khan, F.; Gupta, M.K.; Gupta, P.; Darokar, M.P.; Srivastava, S.K. Comparative drug resistance reversal potential of natural glycosides: Potential of synergy niaziridin & niazirin. Curr. Top. Med. Chem. 2019, 19, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Fàbrega, A.; Martin, R.G.; Rosner, J.L.; Tavio, M.M.; Vila, J. Constitutive soxS expression in a fluoroquinolone-resistant strain with a truncated soxR protein and identification of a new member of the marA-soxS-rob regulon, mdtG. Antimicrob. Agents Chemother. 2010, 54, 1218–1225. [Google Scholar] [CrossRef] [Green Version]
- Maunders, E.A.; Ganio, K.; Hayes, A.J.; Neville, S.L.; Davies, M.R.; Strugnell, R.A. The role of zntA in Klebsiella pneumoniae zinc homeostasis. Microbiol. Spectr. 2022, 10, e0177321. [Google Scholar] [CrossRef]
- Chen, J.; Huang, L.; Wang, Q.; Zeng, H.; Xu, J.; Chen, Z. Antibiotics in aquaculture ponds from Guilin, south of China: Occurrence, distribution, and health risk assessment. Environ. Res. 2022, 204, 112084. [Google Scholar] [CrossRef] [PubMed]
- Fatima, S.; Liaqat, F.; Akbar, A.; Sahfee, M.; Samad, A.; Anwar, M. Virulent and multidrug-resistant Klebsiella pneumoniae from clinical samples in Balochistan. Int. Wound J. 2021, 18, 510–518. [Google Scholar] [CrossRef]
- Marques, C.; Menezes, J.; Belas, A.; Aboim, C.; Cavaco-Silva, P.; Trigueiro, G. Klebsiella pneumoniae causing urinary tract infections in companion animals and humans: Population structure, antimicrobial resistance and virulence genes. J. Antimicrob. Chemother. 2019, 74, 594–602. [Google Scholar] [CrossRef]
- Varol, M.; Sunbul, M.R. Organochlorine pesticide, antibiotic and heavy metal residues in mussel, crayfish and fish species from a reservoir on the Euphrates River, Turkey. Environ. Pollut. 2017, 230, 311–319. [Google Scholar] [CrossRef]
- Ni, L.; Chen, D.; Fu, H.; Xie, Q.; Lu, Y.; Wang, X.; Zhao, Y.; Chen, L. Residual levels of antimicrobial agents and heavy metals in 41 species of commonly consumed aquatic products in Shanghai, China, and cumulative exposure risk to children and teenagers. Food Control. 2021, 129, 108225. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, W.; Zhao, Z.; Ye, C.; Zhou, S.; Wu, S. Molecular characterization of carbapenem-resistant Klebsiella pneumoniae isolates with focus on antimicrobial resistance. BMC Genom. 2019, 20, 822. [Google Scholar] [CrossRef] [Green Version]
- Fortier, L.C.; Sekulovic, O. Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 2013, 4, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Bleriot, I.; Trastoy, R.; Blasco, L.; Fernandez-Cuenca, F.; Ambroa, A.; Fernandez-Garcia, L. Genomic analysis of 40 prophages located in the genomes of 16 carbapenemase-producing clinical strains of Klebsiella pneumoniae. Microb. Genom. 2020, 6, e000369. [Google Scholar] [CrossRef] [PubMed]
- Gillings, M.; Boucher, Y.; Labbate, M.; Holmes, A.; Krishnan, S.; Holley, M. The evolution of class 1 integrons and the rise of antibiotic resistance. J. Bacteriol. 2008, 190, 5095–5100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firoozeh, F.; Mahluji, Z.; Khorshidi, A.; Zibaei, M. Molecular characterization of class 1, 2 and 3 integrons in clinical multi-drug resistant Klebsiella pneumoniae isolates. Antimicrob. Resist. Infect. Control. 2019, 8, 59. [Google Scholar] [CrossRef] [Green Version]
- Nzabarushimana, E.; Tang, H. Insertion sequence elements-mediated structural variations in bacterial genomes. Mob. DNA 2018, 9, 29. [Google Scholar] [CrossRef]
- Devi, V.; Harjai, K.; Chhibber, S. CRISPR-Cas systems: Role in cellular processes beyond adaptive immunity. Folia. Microbiol. 2022, 67, 837–850. [Google Scholar] [CrossRef]
- Wu, Y.; Battalapalli, D.; Hakeem, M.J.; Selamneni, V.; Zhang, P.; Draz, M.S. Engineered CRISPR-Cas systems for the detection and control of antibiotic-resistant infections. J. Nanobiotechnol. 2021, 19, 401. [Google Scholar] [CrossRef]
- Remya, P.A.; Shanthi, M.; Sekar, U. Characterisation of virulence genes associated with pathogenicity in Klebsiella pneumoniae. Indian J. Med. Microbiol. 2019, 37, 210–218. [Google Scholar] [CrossRef]
- Kuş, H.; Arslan, U.; Türk Dağı, H.; Fındık, D. Investigation of various virulence factors of Klebsiella pneumoniae strains isolated from nosocomial infections. Mikrobiyol. Bul. 2017, 51, 329–339. [Google Scholar] [CrossRef]
- van Duin, D.; Paterson, D.L. Multidrug-resistant bacteria in the community: An update. Infect. Dis. Clin. N. Am. 2020, 34, 709–722. [Google Scholar] [CrossRef]
- Tan, T.Y.; Ong, M.; Cheng, Y.; Ng, L.S.Y. Hypermucoviscosity, rmpA, and aerobactin are associated with community-acquired Klebsiella pneumoniae bacteremic isolates causing liver abscess in Singapore. J. Microbiol. Immunol. Infect. 2019, 52, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Candan, E.D.; Aksoz, N. Klebsiella pneumoniae: Characteristics of carbapenem resistance and virulence factors. Acta. Biochim. Pol. 2015, 62, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Ssekatawa, K.; Byarugaba, D.K.; Nakavuma, J.L.; Kato, C.D.; Ejobi, F.; Tweyongyere, R.; Eddie, W.M. Prevalence of pathogenic Klebsiella pneumoniae based on PCR capsular typing harbouring carbapenemases encoding genes in Uganda tertiary hospitals. Antimicrob. Resist. Infect. Control. 2021, 10, 57. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Growth of the K. pneumoniae isolates under different pH conditions. The isolates were incubated in the TSB (0.5% NaCl) at 37 °C. (A–G): K. pneumoniae 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, and 8-2-10-5, respectively.
Figure 1.
Growth of the K. pneumoniae isolates under different pH conditions. The isolates were incubated in the TSB (0.5% NaCl) at 37 °C. (A–G): K. pneumoniae 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, and 8-2-10-5, respectively.
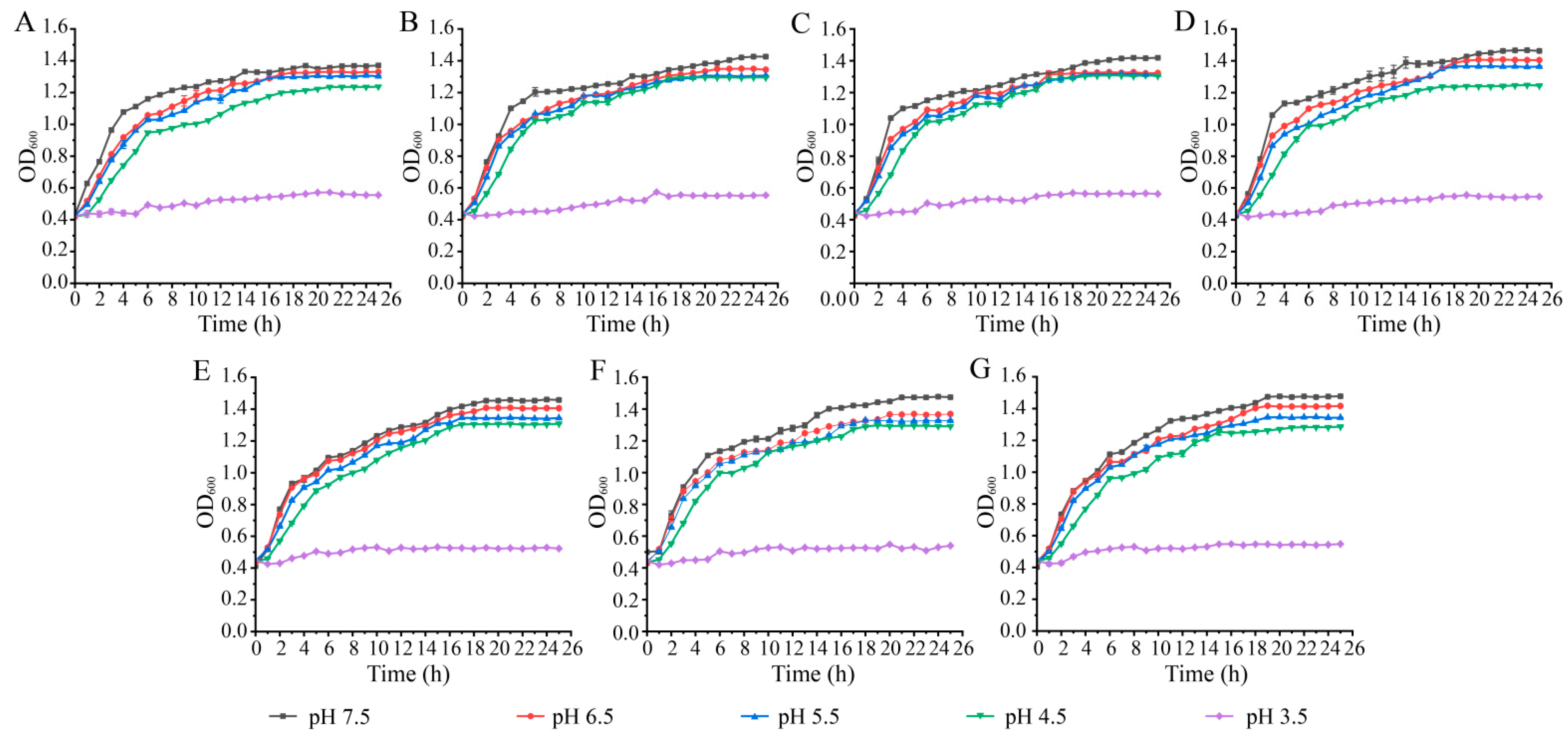
Figure 2.
Growth of the K. pneumoniae isolates under different concentrations of NaCl. The isolates were incubated in the TSB (pH 7.5) at 37 °C. (A–G): K. pneumoniae 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, and 8-2-10-5, respectively.
Figure 2.
Growth of the K. pneumoniae isolates under different concentrations of NaCl. The isolates were incubated in the TSB (pH 7.5) at 37 °C. (A–G): K. pneumoniae 7-5-4, 7-10-14, 7-13-2, 7-17-8, 8-1-12-1, 8-2-5-4, and 8-2-10-5, respectively.

Figure 3.
Gene organizations of the GIs identified in the K. pneumoniae genomes (A,B). Different colors referred to COG classification to mark gene functions, and genes not annotated to COG database were displayed in grey.
Figure 3.
Gene organizations of the GIs identified in the K. pneumoniae genomes (A,B). Different colors referred to COG classification to mark gene functions, and genes not annotated to COG database were displayed in grey.

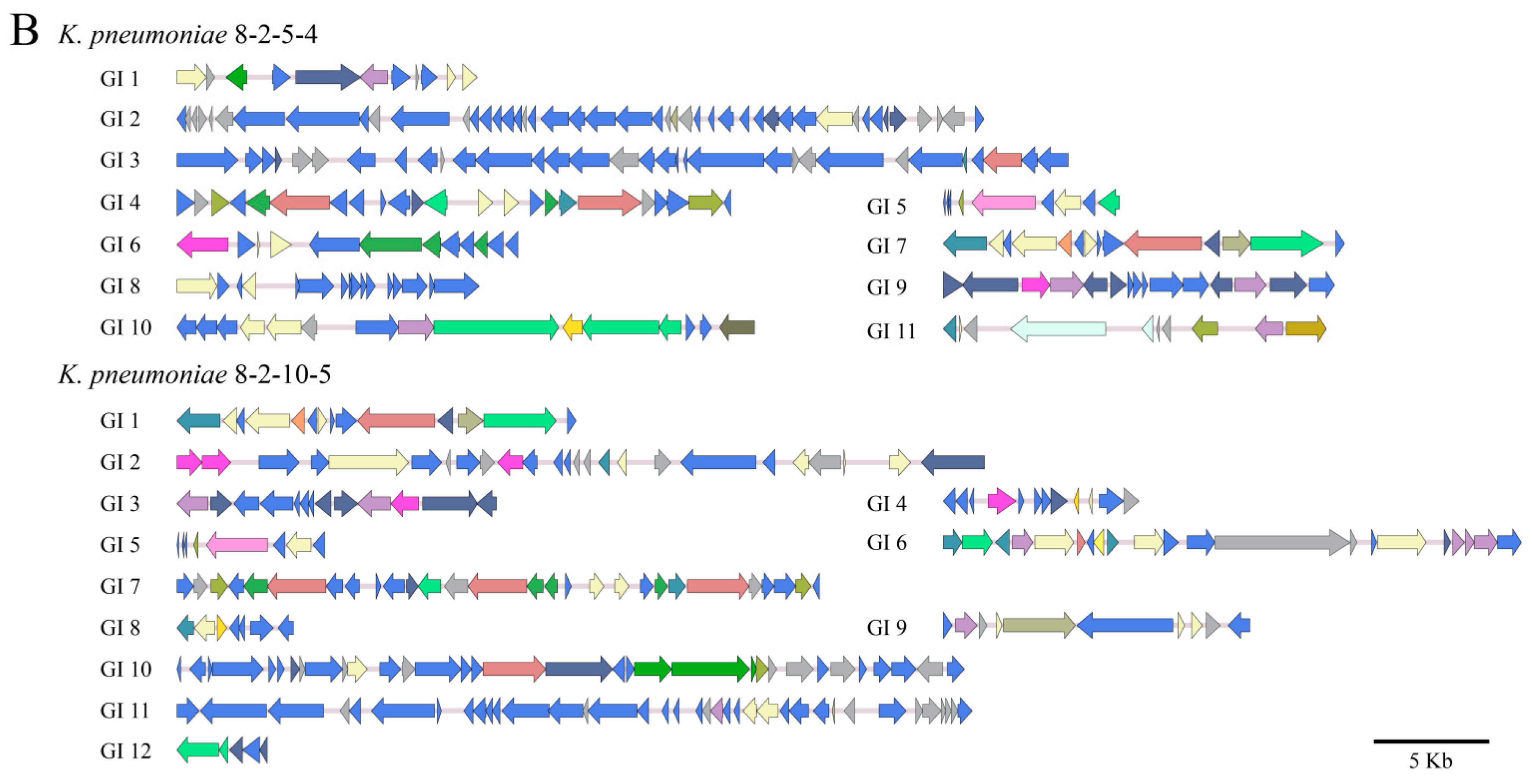
Figure 4.
Gene organizations of the prophages identified in the K. pneumoniae genomes.
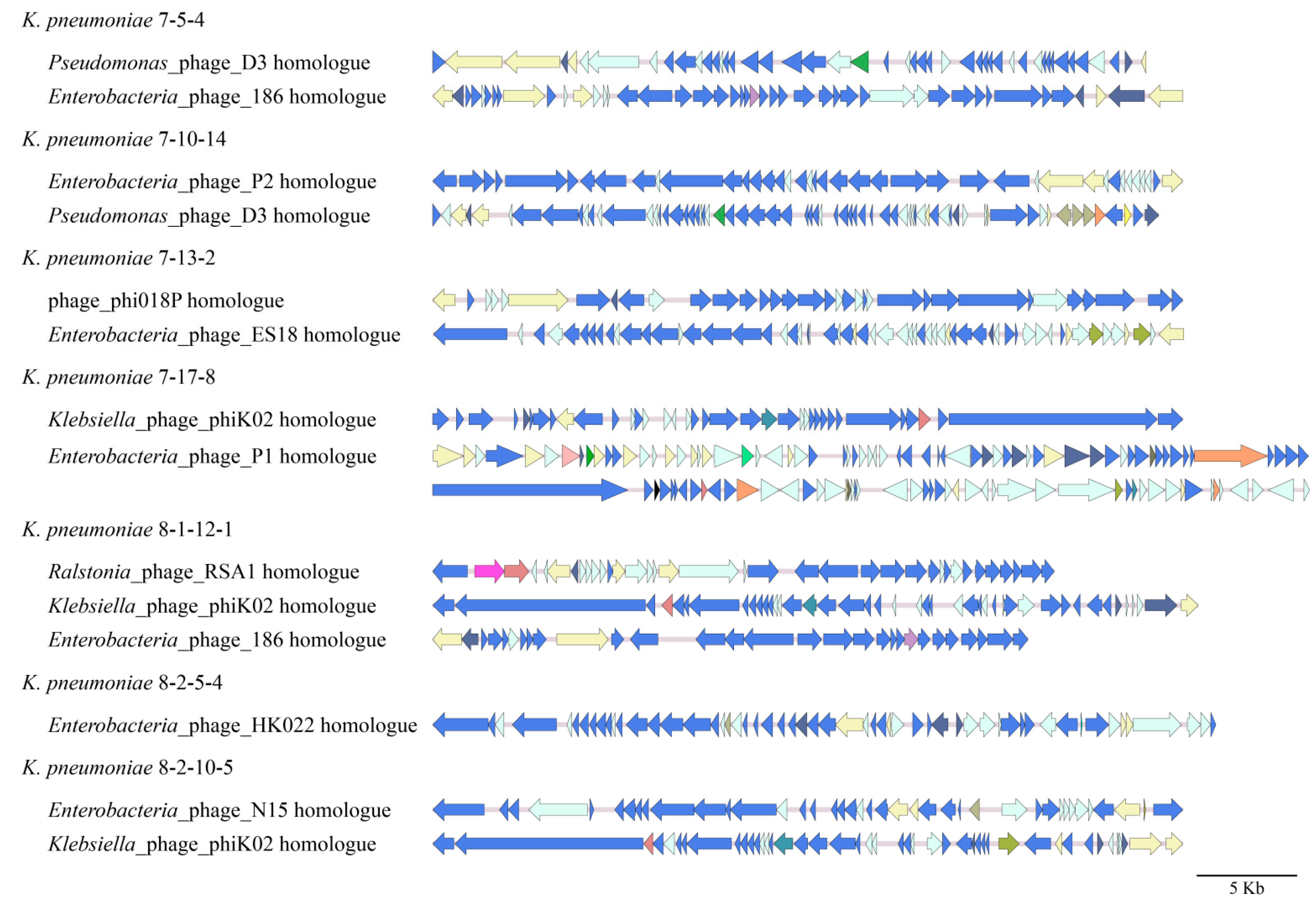
Figure 5.
The structure diagram of the INs identified in the K. pneumoniae genomes.
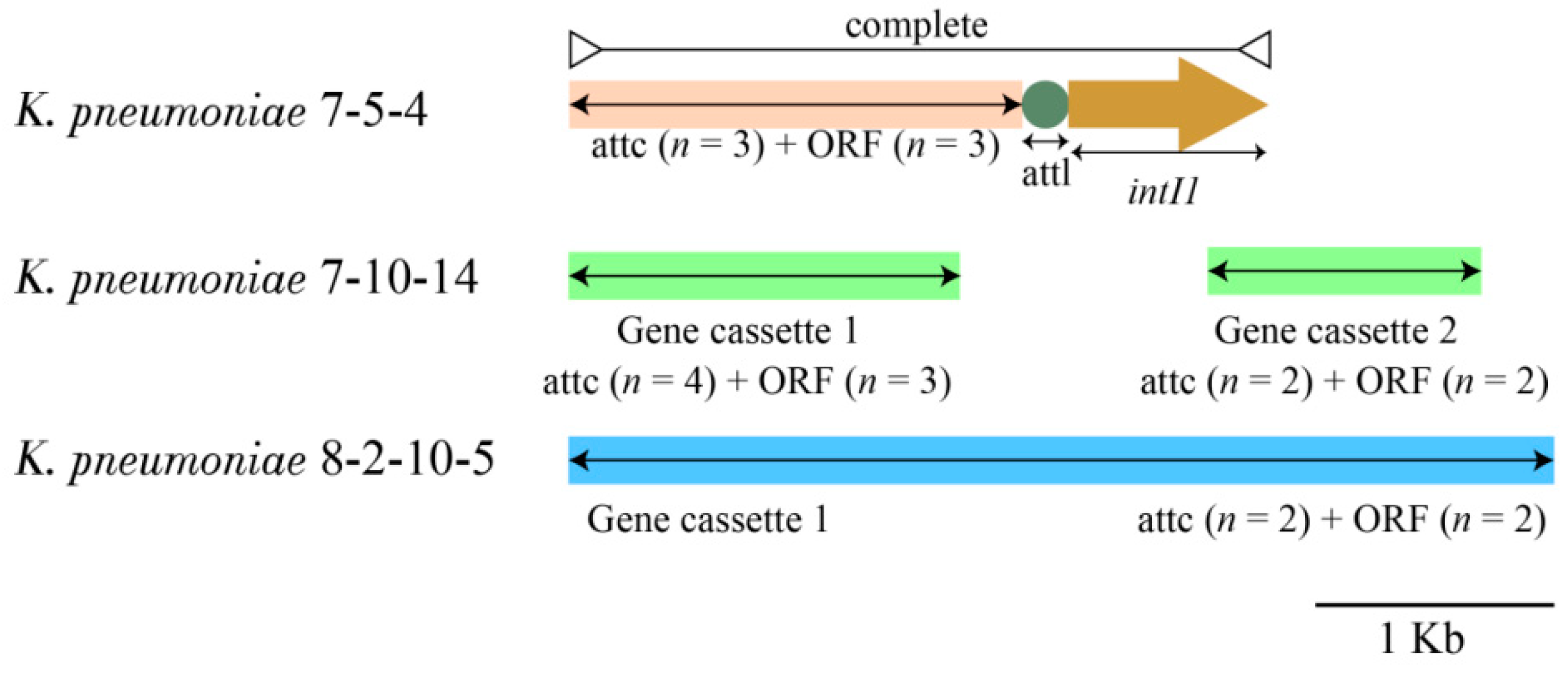
Figure 6.
Structural features of the CRISPR repeats identified in the K. pneumoniae genomes. The repeat sequences were shown by the rectangle in different colors, and the spacer regions were represented by rhombuses in different colors.
Figure 6.
Structural features of the CRISPR repeats identified in the K. pneumoniae genomes. The repeat sequences were shown by the rectangle in different colors, and the spacer regions were represented by rhombuses in different colors.
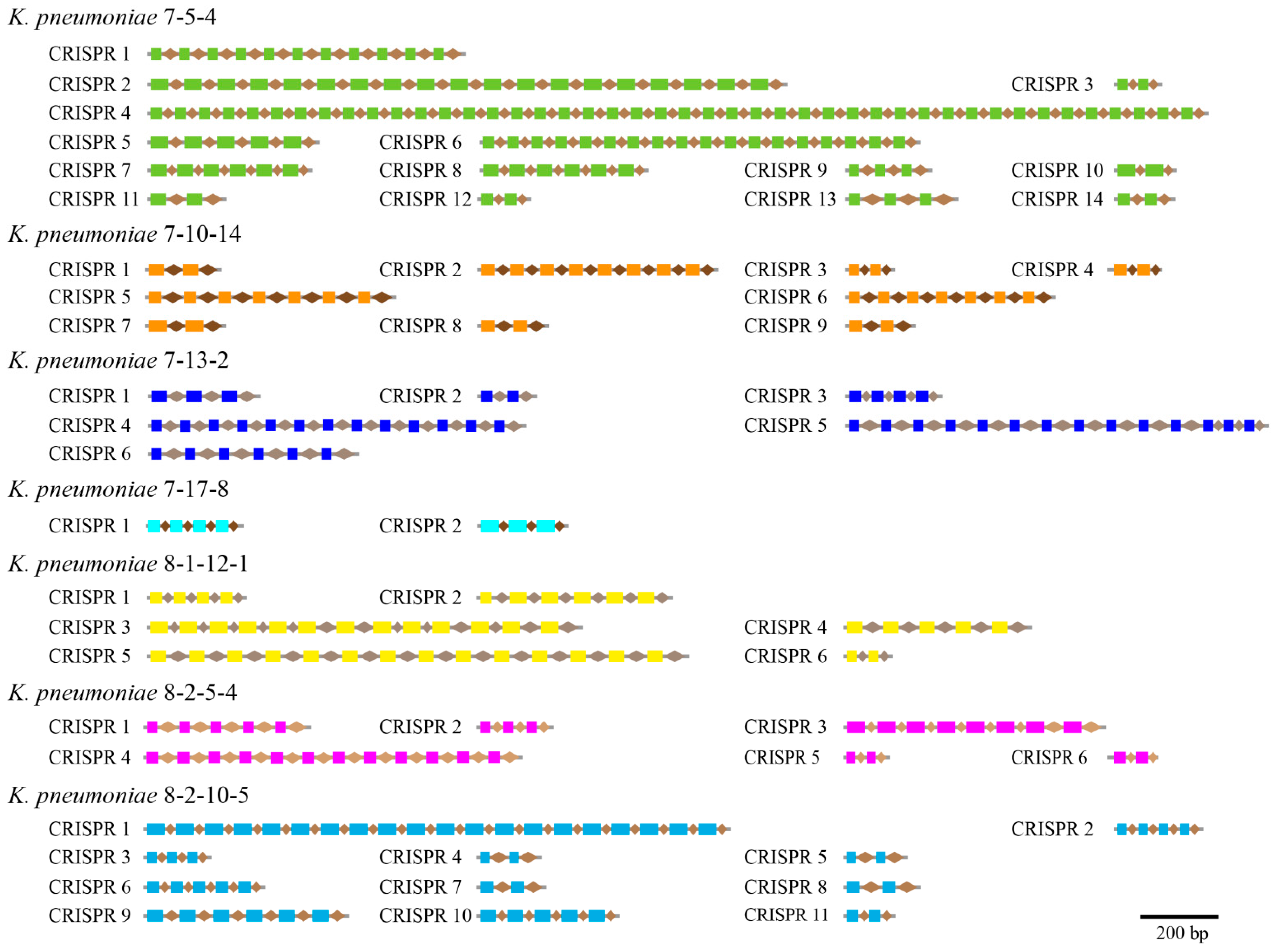
Figure 7.
The Venn diagram showing the number of core genes and strain-specific genes in the seven K. pneumoniae genomes.
Figure 7.
The Venn diagram showing the number of core genes and strain-specific genes in the seven K. pneumoniae genomes.

Figure 8.
The phylogenetic tree showing the relationship of the 72 K. pneumoniae genomes. Complete genome sequences of the 65 K. pneumoniae isolates were retrieved from the GenBank database with accession numbers shown in the tree. The seven K. pneumoniae genomes determined in this study were marked with red dots. The maximum likelihood method was used to build a tree, with 1000 bootstrap replications and a cut-off threshold of ≥50% bootstrap values.
Figure 8.
The phylogenetic tree showing the relationship of the 72 K. pneumoniae genomes. Complete genome sequences of the 65 K. pneumoniae isolates were retrieved from the GenBank database with accession numbers shown in the tree. The seven K. pneumoniae genomes determined in this study were marked with red dots. The maximum likelihood method was used to build a tree, with 1000 bootstrap replications and a cut-off threshold of ≥50% bootstrap values.
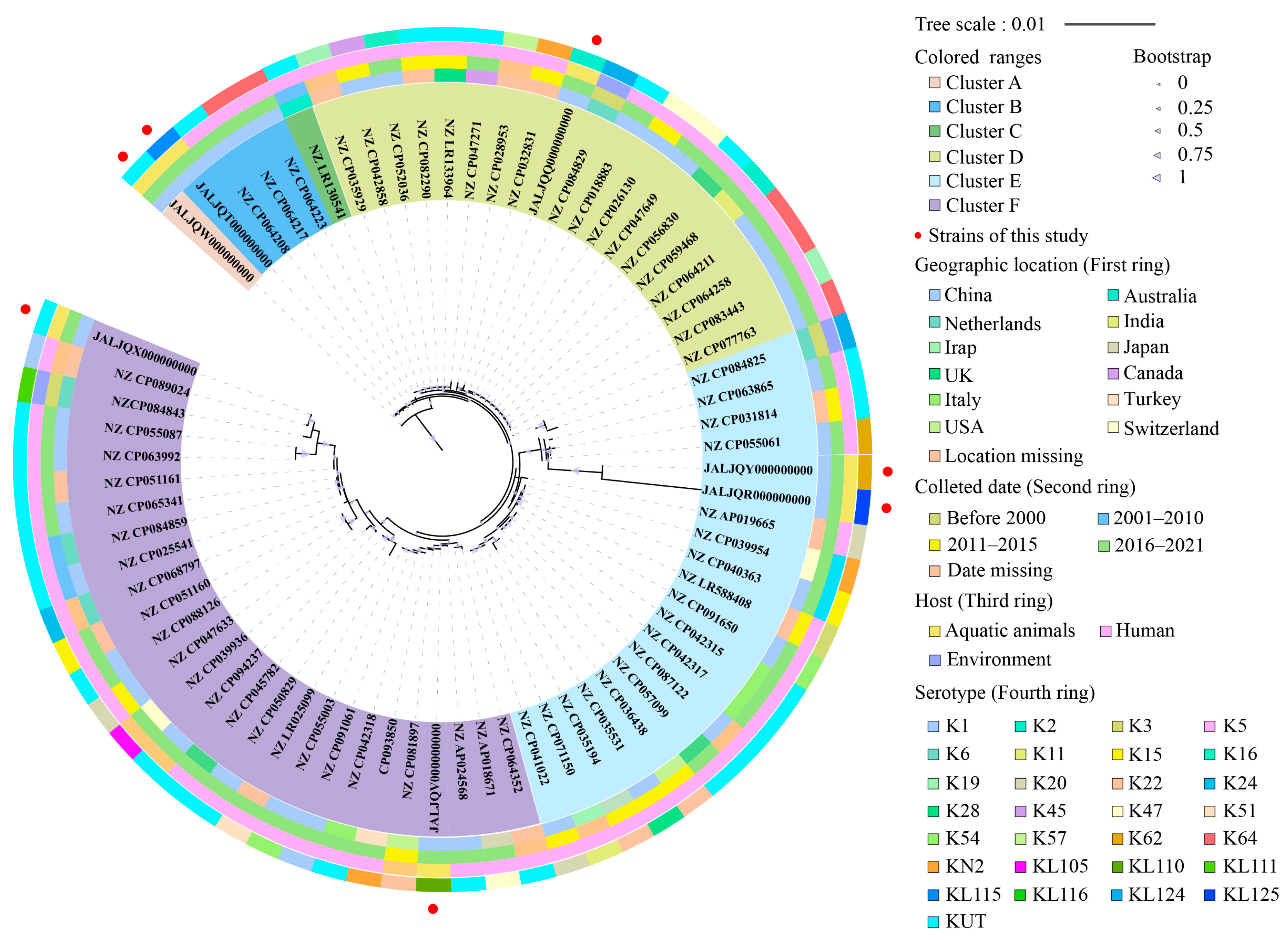
Table 1.
Genome features of the K. pneumoniae isolates of aquatic animal origins.
| Genome Feature | K. pneumoniae Isolate | ||||||
|---|---|---|---|---|---|---|---|
| 7-5-4 | 7-10-14 | 7-13-2 | 7-17-8 | 8-1-12-1 | 8-2-5-4 | 8-2-10-5 | |
| Genome size (bp) | 5,857,823 | 5,376,532 | 5,412,275 | 5,438,640 | 5,593,530 | 5,432,731 | 5,256,522 |
| G + C (%) | 56.35 | 57.28 | 57.29 | 57.12 | 57.21 | 57.32 | 57.81 |
| DNA Scaffold | 81 | 113 | 79 | 50 | 74 | 64 | 45 |
| Total predicted gene | 5662 | 5142 | 5165 | 5189 | 5369 | 5143 | 4986 |
| Protein-coding gene | 5558 | 5044 | 5060 | 5089 | 5260 | 5042 | 4885 |
| RNA gene | 240 | 231 | 238 | 234 | 249 | 224 | 235 |
| Genes assigned to COG | 4986 | 4761 | 4757 | 4770 | 4916 | 4777 | 4639 |
| Genes with unknown function | 1363 | 1230 | 1288 | 1271 | 1357 | 1230 | 1188 |
| GI | 17 | 8 | 8 | 15 | 16 | 11 | 12 |
| Prophage | 2 | 2 | 2 | 2 | 3 | 1 | 2 |
| IN | 1 | 2 | 0 | 0 | 0 | 0 | 1 |
| IS | 2 | 2 | 6 | 2 | 4 | 4 | 2 |
| CRISPR-Cas repeat | 14 | 9 | 6 | 2 | 6 | 6 | 11 |
| Source | This study | This study | This study | This study | This study | [15] | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Guan, H.; Xie, L.; Chen, L. Survival and Genome Evolution Signatures of Klebsiella pneumoniae Isolates Originated in Seven Species of Aquatic Animals. Diversity 2023, 15, 527. https://doi.org/10.3390/d15040527
AMA Style
Guan H, Xie L, Chen L. Survival and Genome Evolution Signatures of Klebsiella pneumoniae Isolates Originated in Seven Species of Aquatic Animals. Diversity. 2023; 15(4):527. https://doi.org/10.3390/d15040527
Chicago/Turabian StyleGuan, Huiqiong, Lu Xie, and Lanming Chen. 2023. "Survival and Genome Evolution Signatures of Klebsiella pneumoniae Isolates Originated in Seven Species of Aquatic Animals" Diversity 15, no. 4: 527. https://doi.org/10.3390/d15040527
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.