
DNA Barcoding of Pygmy Hoppers—The First Comprehensive Overview of the BOLD Systems’ Data Shows Promise for Species Identification

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
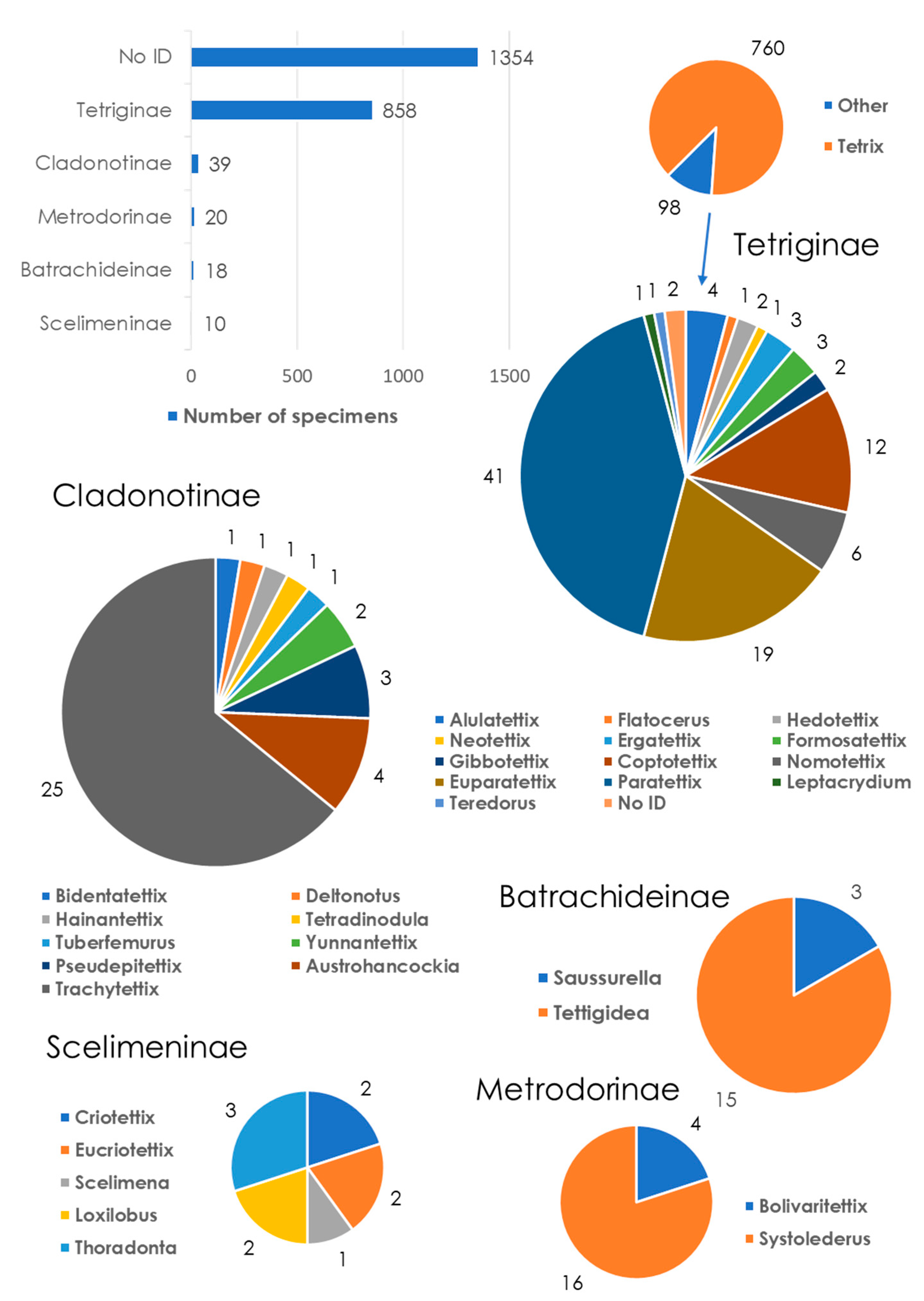
3.1. General Description of the BOLD Dataset
3.2. Review of the BOLD Metadata
3.2.1. Taxonomy
3.2.2. Identification Methods
3.2.3. Geography
3.2.4. Photographic Documentation
3.3. Sequence Analysis
4. Discussion
4.1. Strengths and Shortcomings of the BOLD Database
4.2. DNA Barcoding in Tetrigidae
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


References
- Pentinsaari, M.; Salmela, H.; Mutanen, M.; Roslin, T. Molecular evolution of a widely-adopted taxonomic marker (COI) across the animal tree of life. Sci. Rep. 2016, 6, 35275. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.; Cywinska, A.; Ball, S.L.; DeWaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. B. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed]
- DeSalle, R.; Goldstein, P. Review and interpretation of trends in DNA barcoding. Front. Ecol. Evol. 2019, 7, 302. [Google Scholar] [CrossRef]
- Ratnasingham, S.; Hebert, P.D.N. BOLD: The barcode of life data system (http://www.barcodinglife.org). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef]
- Fernandez-Triana, J.L. Turbo taxonomy approaches: Lessons from the past and recommendations for the future based on the experience with Braconidae (Hymenoptera) parasitoid wasps. ZooKeys 2022, 1087, 199–220. [Google Scholar] [CrossRef]
- Paz, A.; Crawford, A.J. Molecular-based rapid inventories of sympatric diversity: A comparison of DNA barcode clustering methods applied to geography-based vs clade-based sampling of amphibians. J. Biosci. 2012, 37, 887–896. [Google Scholar] [CrossRef]
- Rossini, B.C.; Oliveira, C.A.M.; Melo, F.A.G.D.; Bertaco, V.D.A.; Astarloa, J.M.D.D.; Rosso, J.J.; Foresti, F.; Oliveira, C. Highlighting Astyanax Species Diversity through DNA Barcoding. PLoS ONE 2016, 11, e0167203. [Google Scholar] [CrossRef]
- Sales, N.G.; Mariani, S.; Salvador, G.N.; Pessali, T.C.; Carvalho, D.C. Hidden diversity hampers conservation efforts in a highly impacted Neotropical river system. Front. Genet. 2018, 9, 271. [Google Scholar] [CrossRef]
- Puillandre, N.; Brouillet, S.; Achaz, G. ASAP: Assemble species by automatic partitioning. Mol. Ecol. Resour. 2021, 21, 609–620. [Google Scholar] [CrossRef]
- Song, H.; Moulton, M.J.; Whiting, M.F. Rampant Nuclear Insertion of mtDNA across Diverse Lineages within Orthoptera (Insecta). PLoS ONE 2014, 9, e110508. [Google Scholar] [CrossRef]
- Lehmann, A.W.; Devriese, H.; Tumbrinck, T.; Skejo, J.; Lehmann, G.U.C.; Hochkirch, A. The importance of validated alpha taxonomy for phylogenetic and DNA barcoding studies: A comment on species identification of pygmy grasshoppers (Orthoptera, Tetrigidae). ZooKeys 2017, 679, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Nugent, C.M.; Elliott, T.A.; Ratnasingham, S.; Adamowicz, S.J. coil: An R package for cytochrome c oxidase I (COI) DNA barcode data cleaning, translation, and error evaluation. Genome 2020, 63, 291–305. [Google Scholar] [CrossRef] [PubMed]
- Nugent, C.M. coil: Contextualization and Evaluation of COI-5P Barcode Data. R Package Version 1.2.3. CRAN. Available online: https://CRAN.R-project.org/package=coil (accessed on 10 November 2022).
- Song, H.; Béthoux, O.; Shin, S.; Donath, A.; Letsch, H.; Liu, S.; McKenna, D.D.; Meng, G.; Misof, B.; Podsiadlowski, L.; et al. Phylogenomic analysis sheds light on the evolutionary pathways towards acoustic communication in Orthoptera. Nat. Commun. 2020, 11, 4939. [Google Scholar] [CrossRef]
- Cigliano, M.M.; Braun, H.; Eades, D.C.; Otte, D. Orthoptera Species File. Version 5.0/5.0. Available online: http://Orthoptera.SpeciesFile.org (accessed on 12 March 2023).
- Bolívar, I. Essai sur les Acridiens de la tribu des Tettigidae. Ann. Soc. Ent. Belg. 1887, 31, 175–313. [Google Scholar]
- Tumbrinck, J. Taxonomic revision of the Cladonotinae (Orthoptera: Tetrigidae) from the islands of South-East Asia and from Australia, with general remarks to the classification and morphology of the Tetrigidae and descriptions of new genera and species from New Guinea and New Caledonia. In Biodiversity, Biogeography and Nature Conservation in Wallacea and New Guinea. Vol. II; Telnov, D., Ed.; The Entomological Society of Latvia: Riga, Latvia, 2014; pp. 345–396. [Google Scholar]
- Ahnesjö, J.; Forsman, A. Correlated evolution of colour pattern and body size in polymorphic pygmy grasshoppers, Tetrix undulata. J. Evol. Biol. 2003, 16, 1308–1318. [Google Scholar] [CrossRef]
- Steenman, A.; Lehmann, A.W.; Lehmann, G.U.C. Life-history trade-off between macroptery and reproduction in the wing-dimorphic pygmy grasshopper Tetrix subulata (Orthoptera Tetrigidae). Ethol. Ecol. Evol. 2015, 27, 93–100. [Google Scholar] [CrossRef]
- Zhao, L.; Lin, L.-L.; Zheng, Z.-M. DNA barcoding reveals polymorphism in the pygmy grasshopper Tetrix bolivari (Orthoptera, Tetrigidae). ZooKeys 2016, 582, 111–120. [Google Scholar] [CrossRef]
- Tan, M.K.; Tumbrinck, J.; Baroga-Barbecho, J.B.; Yap, S.A. A new species and morphometric analysis of Cladonotella (Tetrigidae: Cladonotinae). J. Orthoptera Res. 2019, 28, 129–135. [Google Scholar] [CrossRef]
- Itrac-Bruneau, R.; Doucet, G. Contribution à la connaissance des Tetrigidae (Orthoptera) de Guyane. Zoosystema 2022, 44, 405–421. [Google Scholar] [CrossRef]
- Tumbrinck, J. Taxonomic and biogeographic revision of the genus Lamellitettigodes (Orthoptera: Tetrigidae) with description of two new species and additional notes on Lamellitettix, Probolotettix, and Scelimena. J. Orthoptera Res. 2019, 28, 167–180. [Google Scholar] [CrossRef]
- Kasalo, N.; Naskrecki, P.; Rebrina, F.; Skejo, J. Central American Tetrigidae Rambur, 1838 (Orthoptera): A preliminary catalogue. Zoosystema 2023, 45, 177–212. [Google Scholar] [CrossRef]
- Chen, A.-H.; Jiang, G.-F. Phylogenetic relationships among 12 species of Tetrigidae (Orthoptera: Tetrigoidea) based on partial sequences of 12S and 16S ribosomal RNA. Zool. Res. 2004, 25, 510–514. [Google Scholar]
- Chen, Y.-Z.; Deng, W.-A.; Wang, J.-M.; Lin, L.-L.; Zhou, S.-Y. Phylogenetic relationships of Scelimeninae genera (Orthoptera: Tetrigoidea) based on COI, 16S rRNA and 18S rRNA gene sequences. Zootaxa 2018, 4482, 392–400. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022; Available online: https://www.R-project.org/ (accessed on 11 November 2022).
- Firke, S.; Denney, B.; Haid, C.; Knight, R.; Grosser, M.; Zadra, J. janitor: Simple Tools for Examining and Cleaning Dirty Data. R Package Version 2.1.0. Available online: https://CRAN.R-project.org/package=janitor (accessed on 11 November 2022).
- Devriese, H. Bijdrage tot systematiek, morfologie en biologie van de West-Palearktische Tetrigidae. Nieuwsbr. Saltabel. 1996, 15, 1–38. [Google Scholar]
- Silva, D.S.M.; Cadena-Castaneda, O.J.; Pereira, M.R. Batrachideinae (Orthoptera: Caelifera: Tetrigidae): An overview of the most diverse tetrigids of the Neotropical region. Zootaxa 2021, 4946, 1–84. [Google Scholar] [CrossRef]
- Linnaeus, C.N. Systema Naturae per Regna Tria Naturae, 10th ed.; Holmiae, Impensis Direct. Laurentii Salvii: Stockholm, Sweden, 1758; p. 824. [Google Scholar]
- Thunberg, C.P. Hemipterorum maxillosorum genera illustrata plurimisque novis speciebus ditata ac descripta. Mém. Acad. Imp. Sci. St.-Pétersbg. Hist. Acad. 1815, 5, 211–301. [Google Scholar]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2017, 35, 518–522. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree v1.4.4. 2018. Available online: https://github.com/rambaut/figtree/releases (accessed on 12 March 2023).
- Xia, X.; Xie, Z.; Salemi, M.; Chen, L.; Wang, Y. An index of substitution saturation and its application. Mol. Phylogenet. Evol. 2003, 26, 1–7. [Google Scholar] [CrossRef]
- Xia, X.; Lemey, P. Assessing substitution saturation with DAMBE. In The Phylogenetic Handbook: A Practical Approach to DNA and Protein Phylogeny, 2nd ed.; Lemey, P., Salemi, M., Vandamme, A.-M., Eds.; Cambridge University Press: Cambridge, UK, 2009; pp. 615–630. [Google Scholar] [CrossRef]
- Xia, X. DAMBE7: New and improved tools for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef] [PubMed]
- Hancock, J.L. Further studies of the Tetriginae (Orthoptera) in the Oxford University Museum. Trans. Ent. Soc. Lond. 1909, 3, 387–426. [Google Scholar] [CrossRef]
- Latreille, P.A. Histoire Naturelle, Genérale et Particuliere, des Crustacés et des Insectes; Wentworth Press: London, UK, 1802; Volume 3, 467p. [Google Scholar] [CrossRef]
- Janzen, D.H.; Hallwachs, W.; Blandin, P.; Burns, J.M.; Cadiou, J.; Chacón, I.; Dapkey, T.; Deans, A.R.; Epstein, M.E.; Espinoza, B.; et al. Integration of DNA barcoding into an ongoing inventory of complex tropical biodiversity. Mol. Ecol. Resour. 2009, 9, 1–26. [Google Scholar] [CrossRef]
- Say, T. American Entomology or Descriptions of the Insects of North America; Augustus Mitchell, Philadelphia Museum: Philadelphia, PA, USA, 1824; Plate V. [Google Scholar]
- Rehn, J.A.G. The Stanford expedition to Brazil. 1911, Dermaptera and Orthoptera I. Trans. Am. Entomol. 1916, 42, 215–308. [Google Scholar]
- Hawlitschek, O.; Morinière, J.; Lehmann, G.U.C.; Lehmann, A.W.; Kropf, M.; Dunz, A.; Glaw, F.; Detcharoen, M.; Schmidt, S.; Hausmann, A.; et al. DNA barcoding of crickets, katydids and grasshoppers (Orthoptera) from Central Europe with focus on Austria, Germany and Switzerland. Mol. Ecol. Resour. 2017, 17, 1037–1053. [Google Scholar] [CrossRef]
- Devriese, H.; Husemann, M. Afrosystolederus garmsi (Orthoptera, Tetrigidae), a new genus and species from Mount Gibi (Liberia) with remarks on Systolederus, Pseudosystolederus and Teredorus. Zootaxa 2023, 5258, 331–341. [Google Scholar] [CrossRef]
- Günther, K. Revision der Acrydiinae (Orthoptera), III. Sectio Amorphopi (Metrodorae Bol. 1887, aut.). Abh. Ber. Staatl. Mus. Tierkd. Völkerkunde Dresd. (Ser. A Zool.) (N.F.) 1939, 20, 16–335. [Google Scholar]
- Hebard, M. Acrydiinae (Orthoptera, Acrididae) of Southern India. Rev. Suisse Zool. 1930, 36, 565–592. [Google Scholar] [CrossRef]
- Regul, J. Sistematika Skakavaca Veslača (Insecta: Orthoptera: Scelimeninae) Temeljena na Morfološkim Svojstvima [Systematics of Pygmy Rowerhoppers (Insecta: Orthoptera: Scelimeninae) Based on Morphological Characters]. Master’s Thesis, University of Zagreb, Faculty of Science, Department of Biology, Zagreb, Croatia, 2022. [Google Scholar]
- Remigio, E.A.; Hebert, P.D. Testing the utility of partial COI sequences for phylogenetic estimates of gastropod relationships. Mol. Phylogenet. Evol. 2003, 29, 641–647. [Google Scholar] [CrossRef]
- Ros, V.I.; Breeuwer, J.A. Spider mite (Acari: Tetranychidae) mitochondrial COI phylogeny reviewed: Host plant relationships, phylogeography, reproductive parasites and barcoding. Exp. Appl. Acarol. 2007, 42, 239–262. [Google Scholar] [CrossRef] [PubMed]
- Macher, J.N.; Wideman, J.G.; Girard, E.B.; Langerak, A.; Duijm, E.; Jompa, J.; Sadekov, A.; Vos, R.; Wissels, R.; Renema, W. First report of mitochondrial COI in foraminifera and implications for DNA barcoding. Sci. Rep. 2021, 11, 22165. [Google Scholar] [CrossRef]
- Eberle, J.; Ahrens, D.; Mayer, C.; Niehuis, O.; Misof, B. A plea for standardized nuclear markers in metazoan DNA taxonomy. Trends Ecol. Evol. 2020, 35, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Dietz, L.; Eberle, J.; Mayer, C.; Kukowka, S.; Bohacz, C.; Baur, H.; Espeland, M.; Huber, B.A.; Hutter, C.; Mengual, X.; et al. Standardized nuclear markers improve and homogenize species delimitation in Metazoa. Methods Ecol. Evol. 2023, 14, 543–555. [Google Scholar] [CrossRef]
- Strimmer, K.; von Haeseler, A.; Salemi, M. Genetic distances and nucleotide substitution models. In The Phylogenetic Handbook: A Practical Approach to Phylogenetic Analysis and Hypothesis Testing; Lemey, P., Salemi, M., Vandamme, A.-M., Eds.; Cambridge University Press: Cambridge, UK, 2009; pp. 111–141. [Google Scholar] [CrossRef]
- Sherwood, A.R.; Sauvage, T.; Kurihara, A.; Conklin, K.Y.; Presting, G.G. A comparative analysis of COI, LSU and UPA marker data for the Hawaiian florideophyte Rhodophyta: Implications for DNA barcoding of red algae. Cryptogam. Algol. 2010, 31, 451. [Google Scholar]
- Stål, C. Nya Orthoptera. Öfvers. Kongl. Vetensk.-Akad. Förh. 1855, 12, 348–353. [Google Scholar]
- Hancock, J.L. Studies of Tetriginae (Acrydinae) from Sarawak Museum, Borneo. Sarawak Mus. J. 1913, 1, 39–54. [Google Scholar]
- Storozhenko, S.Y. Review of the pygmy grasshoppers of the tribe Cleostratini (Orthoptera: Tetrigidae). Far East. Entomol. 2016, 326, 1–44. [Google Scholar]
- Adžić, K.; Deranja, M.; Franjević, D.; Skejo, J. Are Scelimeninae (Orthoptera: Tetrigidae) monophyletic and why it remains a question? Entomol. News 2020, 129, 128–146. [Google Scholar] [CrossRef]
- Scudder, S.H. Materials for a monograph of the North American Orthoptera including a catalogue of the known New England species. Boston J. Nat. Hist. 1862, 7, 409–480. [Google Scholar] [CrossRef]
- Krauss, H.A. Tettix Türki nov. spec. (Orthopt.). Entomol. Mon. 1876, 1, 103–104. [Google Scholar]
- Bazyluk, W.; Kis, B. Mesotettix (Uvarovitettix) transsylvanicus subgen. n. et sp. n. de Roumanie (Orthoptera, Tetrigidae). Ann. Zool. 1960, 18, 357–363. [Google Scholar]
- Saulcy, F.H.C.D. Notice sur le genre Tetrix, Latr. Bull. Soc. Entomol. Fr. 1888, 6, 135–136. [Google Scholar]
- Barneville, L. Brisout de Catalogue des Acrididés qui se trouvent aux environs de Paris. Ann. Soc. Entomol. Fr. 1848, 6, 411–445. [Google Scholar]
- Moser, V.; Baur, H.; Lehmann, A.W.; Lehmann, G.U.C. Two species?—Limits of the species concepts in the pygmy grasshoppers of the Tetrix bipunctata complex (Orthoptera, Tetrigidae). ZooKeys 2021, 1043, 33–59. [Google Scholar] [CrossRef]
- Morse, A.P. The Acrididae. Subfamily Tettiginae. In Biologia Centrali-Americana. Insecta. Orthoptera; Bruner, L., Morse, A.P., Shelford, R., Eds.; Taylor & Francis: London, UK, 1900; Volume 2, pp. 3–19. [Google Scholar]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasalo, N.; Skejo, J.; Husemann, M. DNA Barcoding of Pygmy Hoppers—The First Comprehensive Overview of the BOLD Systems’ Data Shows Promise for Species Identification. Diversity 2023, 15, 696. https://doi.org/10.3390/d15060696
Kasalo N, Skejo J, Husemann M. DNA Barcoding of Pygmy Hoppers—The First Comprehensive Overview of the BOLD Systems’ Data Shows Promise for Species Identification. Diversity. 2023; 15(6):696. https://doi.org/10.3390/d15060696
Chicago/Turabian StyleKasalo, Niko, Josip Skejo, and Martin Husemann. 2023. "DNA Barcoding of Pygmy Hoppers—The First Comprehensive Overview of the BOLD Systems’ Data Shows Promise for Species Identification" Diversity 15, no. 6: 696. https://doi.org/10.3390/d15060696