
Nanopore Sequencing Technology as an Emerging Tool for Diversity Studies of Plant Organellar Genomes

 , , , and
, , , and 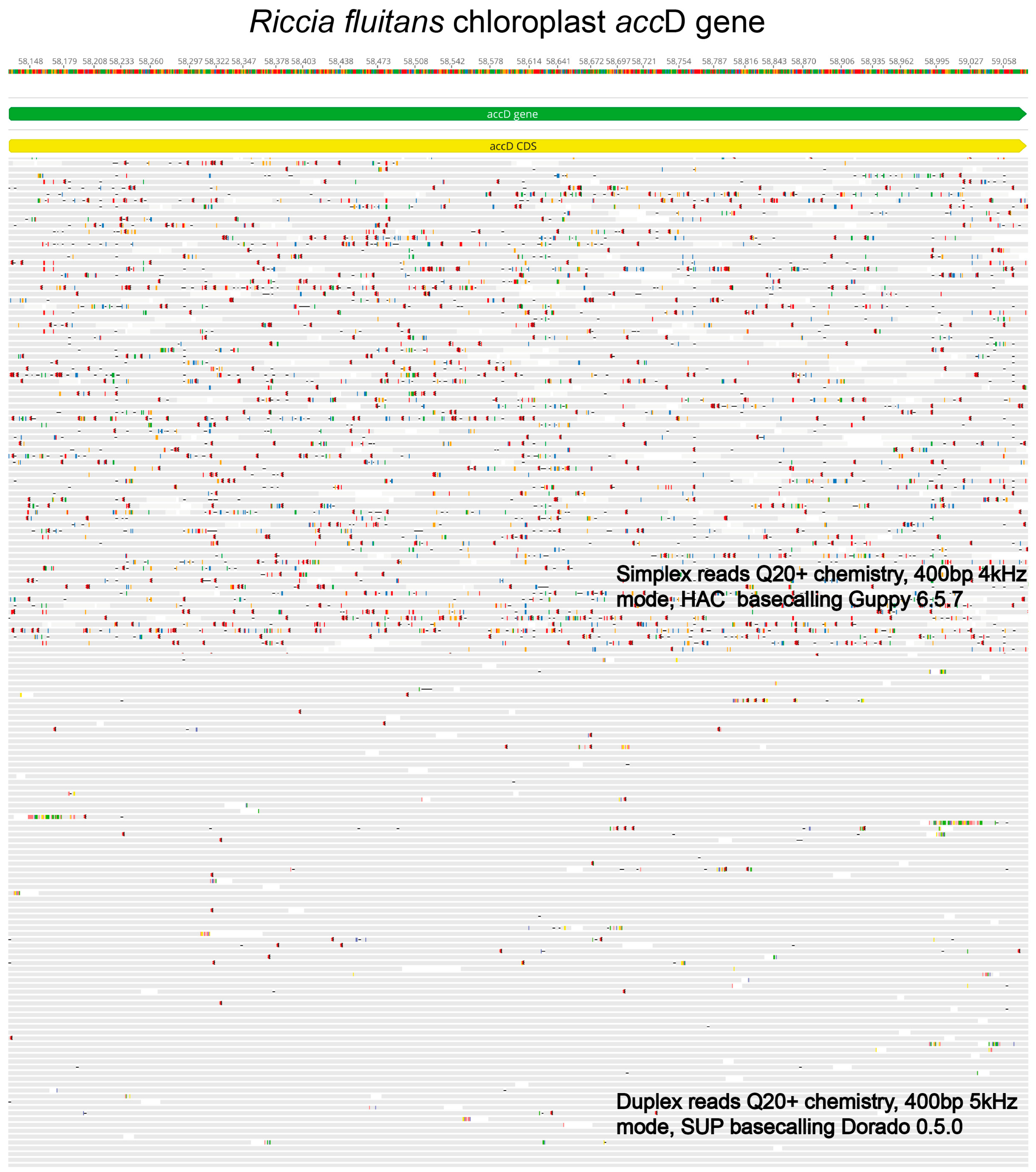{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Challenges, Limitations, and Recent Improvements in Nanopore Sequencing Technology
3. Organellar Genomes Sequenced Using Nanopore Technology
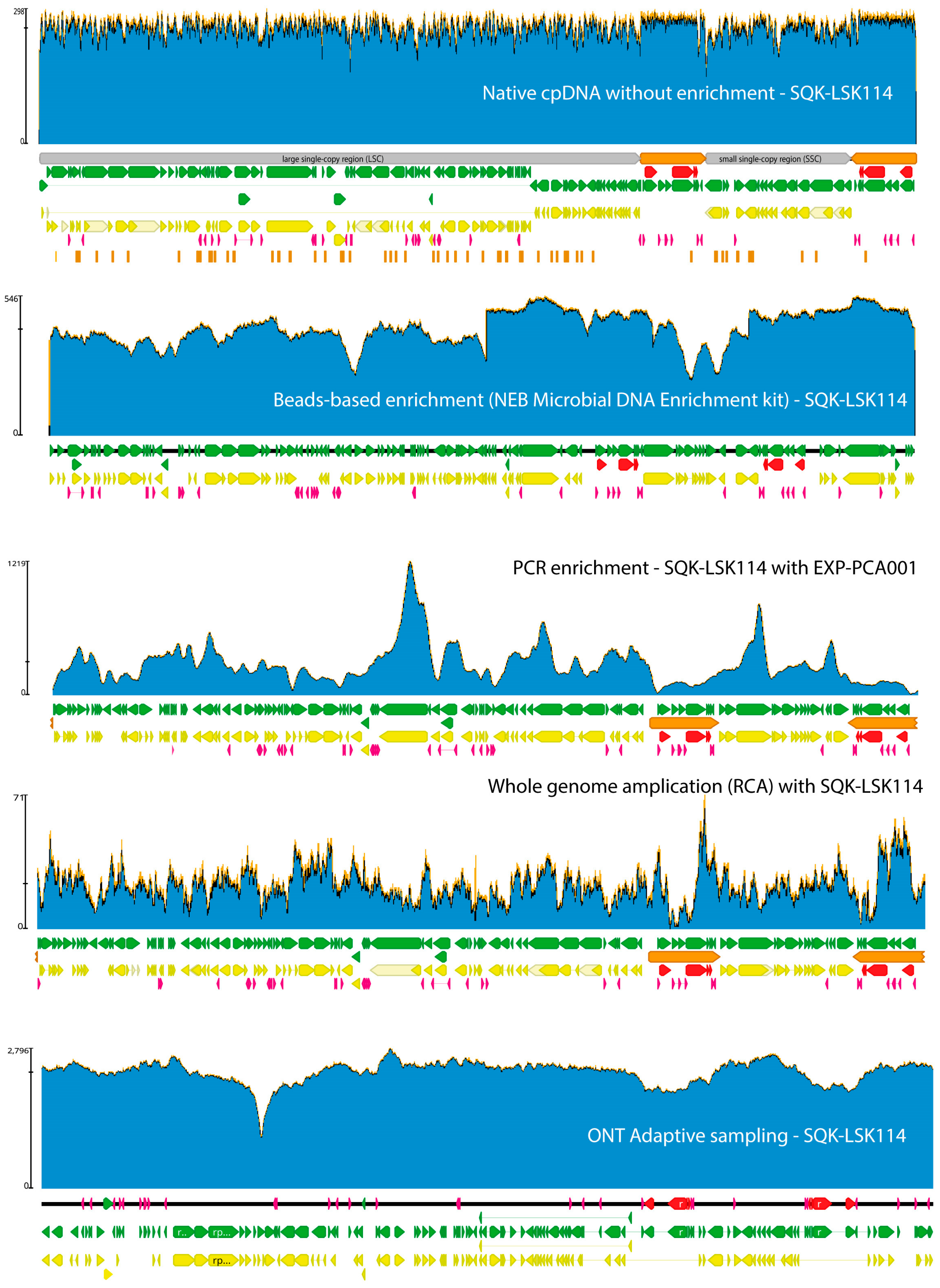
4. Plastid and Mitochondrial DNA Extraction and Enrichment
5. Dedicated Long-Reads Assemblers for Organellar Genomes
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Gray, M.W. The Endosymbiont Hypothesis Revisited. Int. Rev. Cytol. 1992, 141, 233–357. [Google Scholar] [CrossRef] [PubMed]
- Reconstructing Evolution: Gene Transfer from Plastids to the Nucleus—Bock—2008—BioEssays—Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/10.1002/bies.20761 (accessed on 1 January 2024).
- Palmer, J.D.; Herbon, L.A. Plant Mitochondrial DNA Evolves Rapidly in Structure, but Slowly in Sequence. J. Mol. Evol. 1988, 28, 87–97. [Google Scholar] [CrossRef]
- Mower, J.P.; Stefanović, S.; Hao, W.; Gummow, J.S.; Jain, K.; Ahmed, D.; Palmer, J.D. Horizontal Acquisition of Multiple Mitochondrial Genes from a Parasitic Plant Followed by Gene Conversion with Host Mitochondrial Genes. BMC Biol. 2010, 8, 150. [Google Scholar] [CrossRef] [PubMed]
- Knoop, V. When You Can’t Trust the DNA: RNA Editing Changes Transcript Sequences. Cell. Mol. Life Sci. 2011, 68, 567–586. [Google Scholar] [CrossRef] [PubMed]
- Green, B.R. Chloroplast Genomes of Photosynthetic Eukaryotes. Plant J. 2011, 66, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; dePamphilis, C.W.; Leebens-Mack, J.; Müller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 Genes from 64 Plastid Genomes Resolves Relationships in Angiosperms and Identifies Genome-Scale Evolutionary Patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed]
- The First 50 Plant Genomes—Michael—2013—The Plant Genome—Wiley Online Library. Available online: https://acsess.onlinelibrary.wiley.com/doi/10.3835/plantgenome2013.03.0001in (accessed on 14 January 2024).
- Sugiura, M. The Chloroplast Genome. Plant Mol. Biol. 1992, 19, 149–168. [Google Scholar] [CrossRef]
- Smith, D.R. Plastid Genomes Hit the Big Time. New Phytol. 2018, 219, 491–495. [Google Scholar] [CrossRef]
- Wang, W.; Lanfear, R. Long-Reads Reveal That the Chloroplast Genome Exists in Two Distinct Versions in Most Plants. Genome Biol. Evol. 2019, 11, 3372–3381. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore Sequencing Technology, Bioinformatics and Applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- Prudnikow, L.; Pannicke, B.; Wünschiers, R. A Primer on Pollen Assignment by Nanopore-Based DNA Sequencing. Front. Ecol. Evol. 2023, 11, 1112929. [Google Scholar] [CrossRef]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore Sequencing and Assembly of a Human Genome with Ultra-Long Reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of Neural Network Basecalling Tools for Oxford Nanopore Sequencing. Genome Biol. 2019, 20, 129. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and Challenges in Long-Read Sequencing Data Analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef]
- Samarakoon, H.; Ferguson, J.M.; Gamaarachchi, H.; Deveson, I.W. Accelerated Nanopore Basecalling with SLOW5 Data Format. Bioinformatics 2023, 39, btad352. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.J. Progress in Plant Genome Sequencing. Appl. Biosci. 2022, 1, 113–128. [Google Scholar] [CrossRef]
- Pucker, B.; Irisarri, I.; de Vries, J.; Xu, B. Plant Genome Sequence Assembly in the Era of Long Reads: Progress, Challenges and Future Directions. Quant. Plant Biol. 2022, 3, e5. [Google Scholar] [CrossRef]
- Sanderson, N.D.; Kapel, N.; Rodger, G.; Webster, H.; Lipworth, S.; Street, T.L.; Peto, T.; Crook, D.; Stoesser, N. Comparison of R9.4.1/Kit10 and R10/Kit12 Oxford Nanopore Flowcells and Chemistries in Bacterial Genome Reconstruction. Microb. Genom. 2023, 9, 000910. [Google Scholar] [CrossRef]
- Ni, Y.; Liu, X.; Simeneh, Z.M.; Yang, M.; Li, R. Benchmarking of Nanopore R10.4 and R9.4.1 Flow Cells in Single-Cell Whole-Genome Amplification and Whole-Genome Shotgun Sequencing. Comput. Struct. Biotechnol. J. 2023, 21, 2352–2364. [Google Scholar] [CrossRef]
- Sawicki, J.; Krawczyk, K.; Kurzyński, M.; Maździarz, M.; Paukszto, Ł.; Sulima, P.; Szczecińska, M. Nanopore Sequencing of Organellar Genomes Revealed Heteroplasmy in Simple Thalloid and Leafy Liverworts. Acta Soc. Bot. Pol. 2023, 92, 172516. [Google Scholar] [CrossRef]
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Schranz, M.E.; Michelmore, R.W.; Christensen, A.C. The Alternative Reality of Plant Mitochondrial DNA: One Ring Does Not Rule Them All. PLoS Genet. 2019, 15, e1008373. [Google Scholar] [CrossRef] [PubMed]
- Wynn, E.L.; Christensen, A.C. Repeats of Unusual Size in Plant Mitochondrial Genomes: Identification, Incidence and Evolution. G3 GenesGenomesGenetics 2019, 9, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Szandar, K.; Krawczyk, K.; Myszczyński, K.; Ślipiko, M.; Sawicki, J.; Szczecińska, M. Breaking the Limits—Multichromosomal Structure of an Early Eudicot Pulsatilla Patens Mitogenome Reveals Extensive RNA-Editing, Longest Repeats and Chloroplast Derived Regions among Sequenced Land Plant Mitogenomes. BMC Plant Biol. 2022, 22, 109. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Zhu, W.; Sloan, D.B.; Wu, Z. Long-Read Sequencing Characterizes Mitochondrial and Plastid Genome Variants in Arabidopsis Msh1 Mutants. Plant J. Cell Mol. Biol. 2022, 112, 738–755. [Google Scholar] [CrossRef]
- Masutani, B.; Arimura, S.; Morishita, S. Investigating the Mitochondrial Genomic Landscape of Arabidopsis Thaliana by Long-Read Sequencing. PLoS Comput. Biol. 2021, 17, e1008597. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, J.; Bączkiewicz, A.; Buczkowska, K.; Górski, P.; Krawczyk, K.; Mizia, P.; Myszczyński, K.; Ślipiko, M.; Szczecińska, M. The Increase of Simple Sequence Repeats during Diversification of Marchantiidae, An Early Land Plant Lineage, Leads to the First Known Expansion of Inverted Repeats in the Evolutionarily-Stable Structure of Liverwort Plastomes. Genes 2020, 11, 299. [Google Scholar] [CrossRef]
- Liu, J.; Ni, Y.; Liu, C. Polymeric Structure of the Cannabis Sativa L. Mitochondrial Genome Identified with an Assembly Graph Model. Gene 2023, 853, 147081. [Google Scholar] [CrossRef]
- Wang, W.; Schalamun, M.; Morales-Suarez, A.; Kainer, D.; Schwessinger, B.; Lanfear, R. Assembly of Chloroplast Genomes with Long- and Short-Read Data: A Comparison of Approaches Using Eucalyptus Pauciflora as a Test Case. BMC Genom. 2018, 19, 977. [Google Scholar] [CrossRef]
- Scheunert, A.; Dorfner, M.; Lingl, T.; Oberprieler, C. Can We Use It? On the Utility of de Novo and Reference-Based Assembly of Nanopore Data for Plant Plastome Sequencing. PLoS ONE 2020, 15, e0226234. [Google Scholar] [CrossRef]
- The First Genome for the Cape Primrose Streptocarpus Rexii (Gesneriaceae), a Model Plant for Studying Meristem-Driven Shoot Diversity—Nishii—2022—Plant Direct—Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/10.1002/pld3.388 (accessed on 1 January 2024).
- Plášek, V.; Sawicki, J.; Seppelt, R.D.; Cave, L.H. Orthotrichum Cupulatum Hoffm. Ex Brid. Var. Lithophilum, a New Variety of Epilithic Bristle Moss from Tasmania. Acta Soc. Bot. Pol. 2023, 92, 1–8. [Google Scholar] [CrossRef]
- Ciborowski, K.; Skierkowski, B.; Żukowska, K.; Krawczyk, K.; Sawicki, J. Nanopore Sequencing of Chloroplast Genome of Scapania Undulata (L.) Dumort., 1835 (Scapaniaceae, Jungermanniales). Mitochondrial DNA Part B Resour. 2022, 7, 1424–1426. [Google Scholar] [CrossRef]
- López, K.E.R.; Armijos, C.E.; Parra, M.; Torres, M.d.L. The First Complete Chloroplast Genome Sequence of Mortiño (Vaccinium floribundum) and Comparative Analyses with Other Vaccinium Species. Horticulturae 2023, 9, 302. Available online: https://www.mdpi.com/2311-7524/9/3/302 (accessed on 15 January 2024). [CrossRef]
- Fulton, T.M.; Chunwongse, J.; Tanksley, S.D. Microprep Protocol for Extraction of DNA from Tomato and Other Herbaceous Plants. Plant Mol. Biol. Rep. 1995, 13, 207–209. [Google Scholar] [CrossRef]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA Extraction Protocol for Plants Containing High Polysaccharide and Polyphenol Components. Plant Mol. Biol. Rep. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Russo, A.; Mayjonade, B.; Frei, D.; Potente, G.; Kellenberger, R.T.; Frachon, L.; Copetti, D.; Studer, B.; Frey, J.E.; Grossniklaus, U.; et al. Low-Input High-Molecular-Weight DNA Extraction for Long-Read Sequencing From Plants of Diverse Families. Front. Plant Sci. 2022, 13, 1494. [Google Scholar] [CrossRef]
- Kang, M.; Chanderbali, A.; Lee, S.; Soltis, D.E.; Soltis, P.S.; Kim, S. High-Molecular-Weight DNA Extraction for Long-Read Sequencing of Plant Genomes: An Optimization of Standard Methods. Appl. Plant Sci. 2023, 11, e11528. [Google Scholar] [CrossRef]
- Bock, R. The Give-and-Take of DNA: Horizontal Gene Transfer in Plants. Trends Plant Sci. 2010, 15, 11–22. [Google Scholar] [CrossRef]
- Smith, D.R.; Keeling, P.J. Mitochondrial and Plastid Genome Architecture: Reoccurring Themes, but Significant Differences at the Extremes. Proc. Natl. Acad. Sci. USA 2015, 112, 10177–10184. [Google Scholar] [CrossRef] [PubMed]
- Bensasson, D.; Zhang, D.-X.; Hartl, D.L.; Hewitt, G.M. Mitochondrial Pseudogenes: Evolution’s Misplaced Witnesses. Trends Ecol. Evol. 2001, 16, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Alverson, A.J.; Chuckalovcak, J.P.; Wu, M.; McCauley, D.E.; Palmer, J.D.; Taylor, D.R. Rapid Evolution of Enormous, Multichromosomal Genomes in Flowering Plant Mitochondria with Exceptionally High Mutation Rates. PLoS Biol. 2012, 10, e1001241. [Google Scholar] [CrossRef]
- Mariac, C.; Scarcelli, N.; Pouzadou, J.; Barnaud, A.; Billot, C.; Faye, A.; Kougbeadjo, A.; Maillol, V.; Martin, G.; Sabot, F.; et al. Cost-effective Enrichment Hybridization Capture of Chloroplast Genomes at Deep Multiplexing Levels for Population Genetics and Phylogeography Studies. Mol. Ecol. Resour. 2014, 14, 1103–1113. [Google Scholar] [CrossRef]
- Wang, J.; Mu, W.; Yang, T.; Song, Y.; Hou, Y.G.; Wang, Y.; Gao, Z.; Liu, X.; Liu, H.; Zhao, H. Targeted Enrichment of Novel Chloroplast-Based Probes Reveals a Large-Scale Phylogeny of 412 Bamboos. BMC Plant Biol. 2021, 21, 76. [Google Scholar] [CrossRef]
- Uribe-Convers, S.; Duke, J.R.; Moore, M.J.; Tank, D.C. A Long PCR–Based Approach for DNA Enrichment Prior to next-Generation Sequencing for Systematic Studies. Appl. Plant Sci. 2014, 2, 1300063. [Google Scholar] [CrossRef]
- Goremykin, V.V.; Viola, R.; Hellwig, F.H. Removal of Noisy Characters from Chloroplast Genome-Scale Data Suggests Revision of Phylogenetic Placements of Amborella and Ceratophyllum. J. Mol. Evol. 2009, 68, 197–204. [Google Scholar] [CrossRef]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative Chloroplast Genomics: Analyses Including New Sequences from the Angiosperms Nuphar Advena and Ranunculus Macranthus. BMC Genomics 2007, 8, 174. [Google Scholar] [CrossRef]
- Potapov, V.; Ong, L.J. Examining Sources of Error in PCR by Single-Molecule Sequencing. PLoS ONE 2017, 12, e0169774. [Google Scholar] [CrossRef] [PubMed]
- Acinas, G.A.; Sarma-Rupavtarm, R.; Klepac-Ceraj, V.; Polz, M.F. PCR-Induced Sequence Artifacts and Bias: Insights from Comparison of Two 16S rRNA Clone Libraries Constructed from the Same Sample. App. Environ. Microb. 2005, 71, 8966–8969. [Google Scholar] [CrossRef]
- Yigit, E.; Hernandez, D.I.; Trujillo, J.T.; Dimalanta, E.; Bailey, C.D. Genome and Metagenome Sequencing: Using the Human Methyl-Binding Domain to Partition Genomic DNA Derived from Plant Tissues. Appl. Plant Sci. 2014, 2, 1400064. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.; Holmes, N.; Clarke, T.; Munro, R.; Debebe, B.J.; Loose, M. Readfish Enables Targeted Nanopore Sequencing of Gigabase-Sized Genomes. Nat. Biotechnol. 2021, 39, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Loose, M.; Malla, S.; Stout, M. Real-Time Selective Sequencing Using Nanopore Technology. Nat. Methods 2016, 13, 751–754. [Google Scholar] [CrossRef]
- Wanner, N.; Larsen, P.A.; McLain, A.; Faulk, C. The Mitochondrial Genome and Epigenome of the Golden Lion Tamarin from Fecal DNA Using Nanopore Adaptive Sequencing. BMC Genom. 2021, 22, 726. [Google Scholar] [CrossRef] [PubMed]
- Gan, M.; Wu, B.; Yan, G.; Li, G.; Sun, L.; Lu, G.; Zhou, W. Combined Nanopore Adaptive Sequencing and Enzyme-Based Host Depletion Efficiently Enriched Microbial Sequences and Identified Missing Respiratory Pathogens. BMC Genom. 2021, 22, 732. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Heavens, D.; Lan, Y.; Horsfield, S.; Clark, M.D.; Leggett, R.M. Nanopore Adaptive Sampling: A Tool for Enrichment of Low Abundance Species in Metagenomic Samples. Genome Biol. 2022, 23, 11. [Google Scholar] [CrossRef] [PubMed]
- Kipp, E.J.; Lindsey, L.L.; Milstein, M.S.; Blanco, C.M.; Baker, J.P.; Faulk, C.; Oliver, J.D.; Larsen, P.A. Nanopore Adaptive Sampling for Targeted Mitochondrial Genome Sequencing and Bloodmeal Identification in Hematophagous Insects. Parasit. Vectors 2023, 16, 68. [Google Scholar] [CrossRef] [PubMed]
- Weilguny, L.; De Maio, N.; Munro, R.; Manser, C.; Birney, E.; Loose, M.; Goldman, N. Dynamic, Adaptive Sampling during Nanopore Sequencing Using Bayesian Experimental Design. Nat. Biotechnol. 2023, 41, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Soorni, A.; Haak, D.; Zaitlin, D.; Bombarely, A. Organelle_PBA, a Pipeline for Assembling Chloroplast and Mitochondrial Genomes from PacBio DNA Sequencing Data. BMC Genom. 2017, 18, 49. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and Accurate Long-Read Assembly via Adaptive k-Mer Weighting and Repeat Separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving Bacterial Genome Assemblies from Short and Long Sequencing Reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of Long, Error-Prone Reads Using Repeat Graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Miyamoto, M.; Motooka, D.; Gotoh, K.; Imai, T.; Yoshitake, K.; Goto, N.; Iida, T.; Yasunaga, T.; Horii, T.; Arakawa, K.; et al. Performance Comparison of Second- and Third-Generation Sequencers Using a Bacterial Genome with Two Chromosomes. BMC Genom. 2014, 15, 699. [Google Scholar] [CrossRef]
- Denisov, G.; Walenz, B.; Halpern, A.L.; Miller, J.; Axelrod, N.; Levy, S.; Sutton, G. Consensus Generation and Variant Detection by Celera Assembler. Bioinformatics 2008, 24, 1035–1040. [Google Scholar] [CrossRef]
- Syme, A.E.; McLay, T.G.B.; Udovicic, F.; Cantrill, D.J.; Murphy, D.J.; McLay, T.G.B.; Udovicic, F.; Cantrill, D.J.; Murphy, D.J. Long-Read Assemblies Reveal Structural Diversity in Genomes of Organelles—An Example with Acacia Pycnantha. Gigabyte 2021, 2021, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Shafin, K.; Pesout, T.; Lorig-Roach, R.; Haukness, M.; Olsen, H.E.; Bosworth, C.; Armstrong, J.; Tigyi, K.; Maurer, N.; Koren, S.; et al. Nanopore Sequencing and the Shasta Toolkit Enable Efficient de Novo Assembly of Eleven Human Genomes. Nat. Biotechnol. 2020, 38, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-Resolved de Novo Assembly Using Phased Assembly Graphs with Hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xie, P.; Guo, Y.; Zhou, W.; Liu, E.; Yu, Y. Easy353: A Tool to Get Angiosperms353 Genes for Phylogenomic Research. Mol. Biol. Evol. 2022, 39, msac261. [Google Scholar] [CrossRef]
- Zhou, W.; Armijos, C.E.; Lee, C.; Lu, R.; Wang, J.; Ruhlman, T.A.; Jansen, R.K.; Jones, A.M.; Jones, C.D. Plastid Genome Assembly Using Long-Read Data. Mol. Ecol. Resour. 2023, 23, 1442–1457. [Google Scholar] [CrossRef]
- Lee, C.; Ruhlman, T.A.; Jansen, R.K. Unprecedented Intraindividual Structural Heteroplasmy in Eleocharis (Cyperaceae, Poales) Plastomes. Genome Biol. Evol. 2020, 12, 641–655. [Google Scholar] [CrossRef]
- Ruhlman, T.A.; Zhang, J.; Blazier, J.C.; Sabir, J.S.M.; Jansen, R.K. Recombination-Dependent Replication and Gene Conversion Homogenize Repeat Sequences and Diversify Plastid Genome Structure. Am. J. Bot. 2017, 104, 559–572. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sawicki, J.; Krawczyk, K.; Paukszto, Ł.; Maździarz, M.; Kurzyński, M.; Szablińska-Piernik, J.; Szczecińska, M. Nanopore Sequencing Technology as an Emerging Tool for Diversity Studies of Plant Organellar Genomes. Diversity 2024, 16, 173. https://doi.org/10.3390/d16030173
Sawicki J, Krawczyk K, Paukszto Ł, Maździarz M, Kurzyński M, Szablińska-Piernik J, Szczecińska M. Nanopore Sequencing Technology as an Emerging Tool for Diversity Studies of Plant Organellar Genomes. Diversity. 2024; 16(3):173. https://doi.org/10.3390/d16030173
Chicago/Turabian StyleSawicki, Jakub, Katarzyna Krawczyk, Łukasz Paukszto, Mateusz Maździarz, Mateusz Kurzyński, Joanna Szablińska-Piernik, and Monika Szczecińska. 2024. "Nanopore Sequencing Technology as an Emerging Tool for Diversity Studies of Plant Organellar Genomes" Diversity 16, no. 3: 173. https://doi.org/10.3390/d16030173