Methods to Estimate the Diversity in the Marine Photosynthetic Protist Community with Illustrations from Case Studies: A Review

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Why Use Molecular Techniques?
- Only very small samples (in the range of milliliters up to a liter) can be used for most analyses.
- High sensitivity, enabling the researcher to detect even single specific cells among thousands of others.
- Dead or non-culturable cells can be analyzed.
- Species-specific data (such as sequences) can be obtained without the need to culture or even isolate a species.
3. Which Genes to Select?
- Universally present with the same function in all organisms;
- Variable regions of conservation, which enables design of primers or probes to be designed;
- Many copies, which makes PCR easy or genome application for later PCR also easy;
- No evidence for lateral gene transfer, so vertical descent is analyzed by phylogenetic methods.
4. Methods for Determining Biodiversity in Environmental Samples by Sequence Analysis
4.1. 454 Sequencing and the Rare Biosphere
4.2. Barcoding
4.2.1. Case Studies
4.3. Molecular Probes for Identification and Characterization of Marine Phytoplankton



5. Fingerprinting Methods as Applied to Environmental Samples
- The PCR fragment size is limited to some 500 bp because of the separation capacity of polyacrylamide gels. Therefore, these methods cannot handle full-length SSU genes;
- It is difficult, if not impossible, to compare patterns across gels. Therefore the number of samples that can be reliably compared with one another is limited to the number of slots on the gel;
- The methods are not trivial and have to be optimized for each primer pair and for new sample types. Therefore, the initial help of an experienced person is necessary to establish the methods. However, given the rapidly accumulating dataset on this method in the literature, this problem may become less relevant in the future;
- Sometimes the methods are “too sensitive” because even pure cultures produce more than one band.
5.1. Analysis of Population Structure Using Molecular Markers
5.1.1. Isozymes
5.1.2. PCR-Based Population Markers
6. What Questions Can be Answered with Molecular Techniques?
6.1. Taxonomic Affinities
6.1.1. Case Studies at the Genus and Species Level
Skeletonema costatum
Pseudo-nitzschia delicatissima and Pseudo-nitzschia pseudodelicatissima
Alexandrium tamarense
Sellaphora pupula
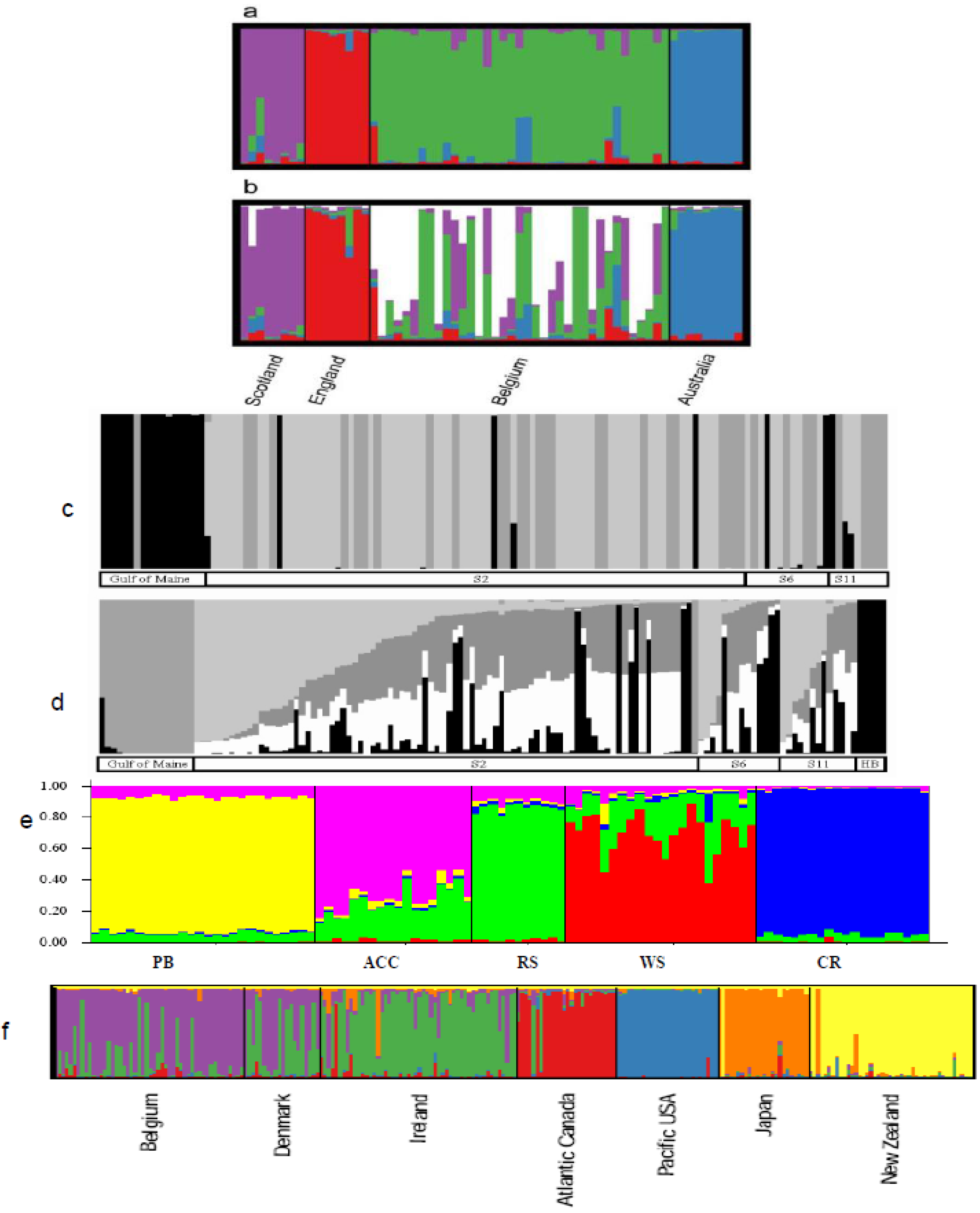
6.2. Population Structure
6.2.1. Case Studies

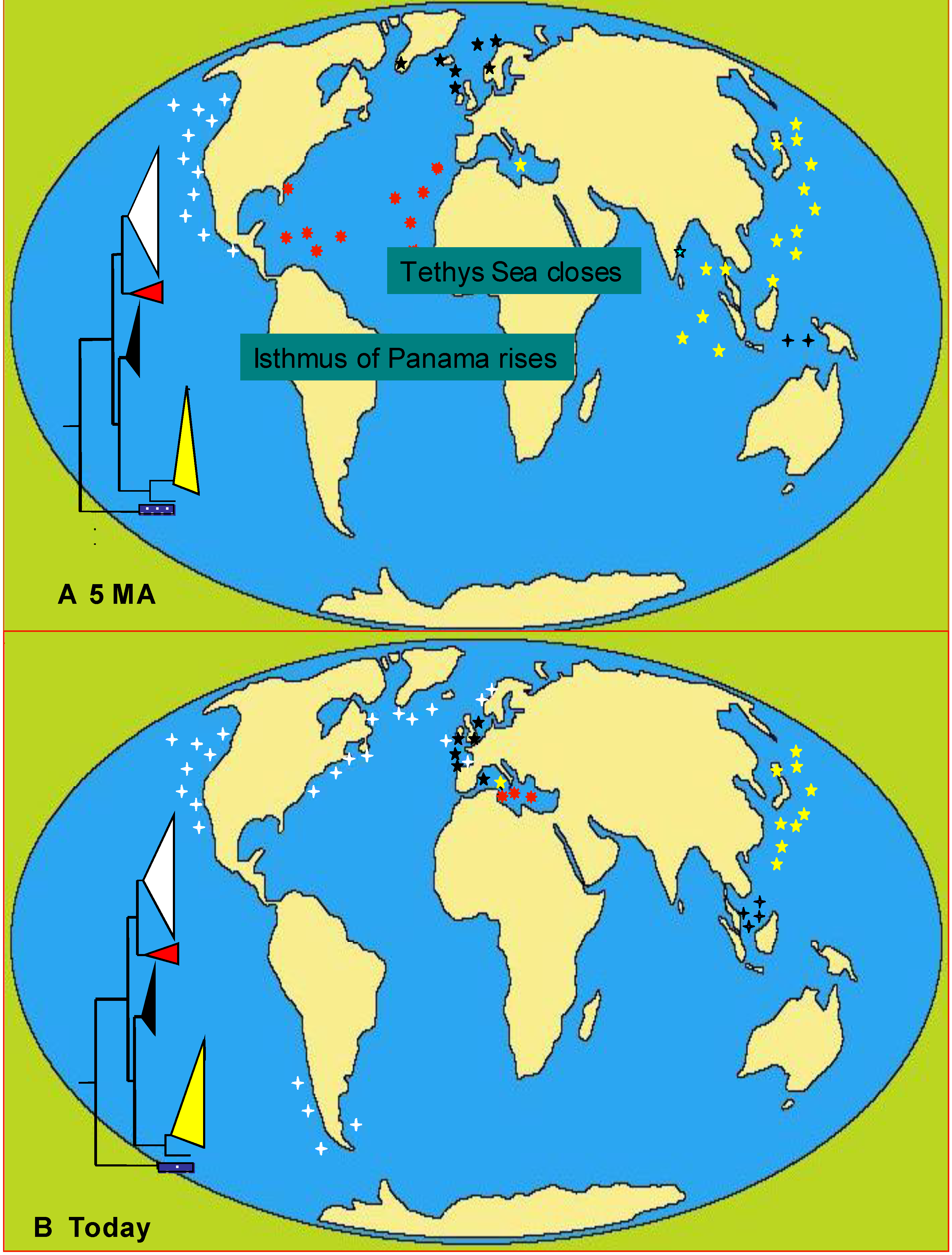
6.3. Phylogeography
6.3.1. Case Studies
Alexandrium tamarense

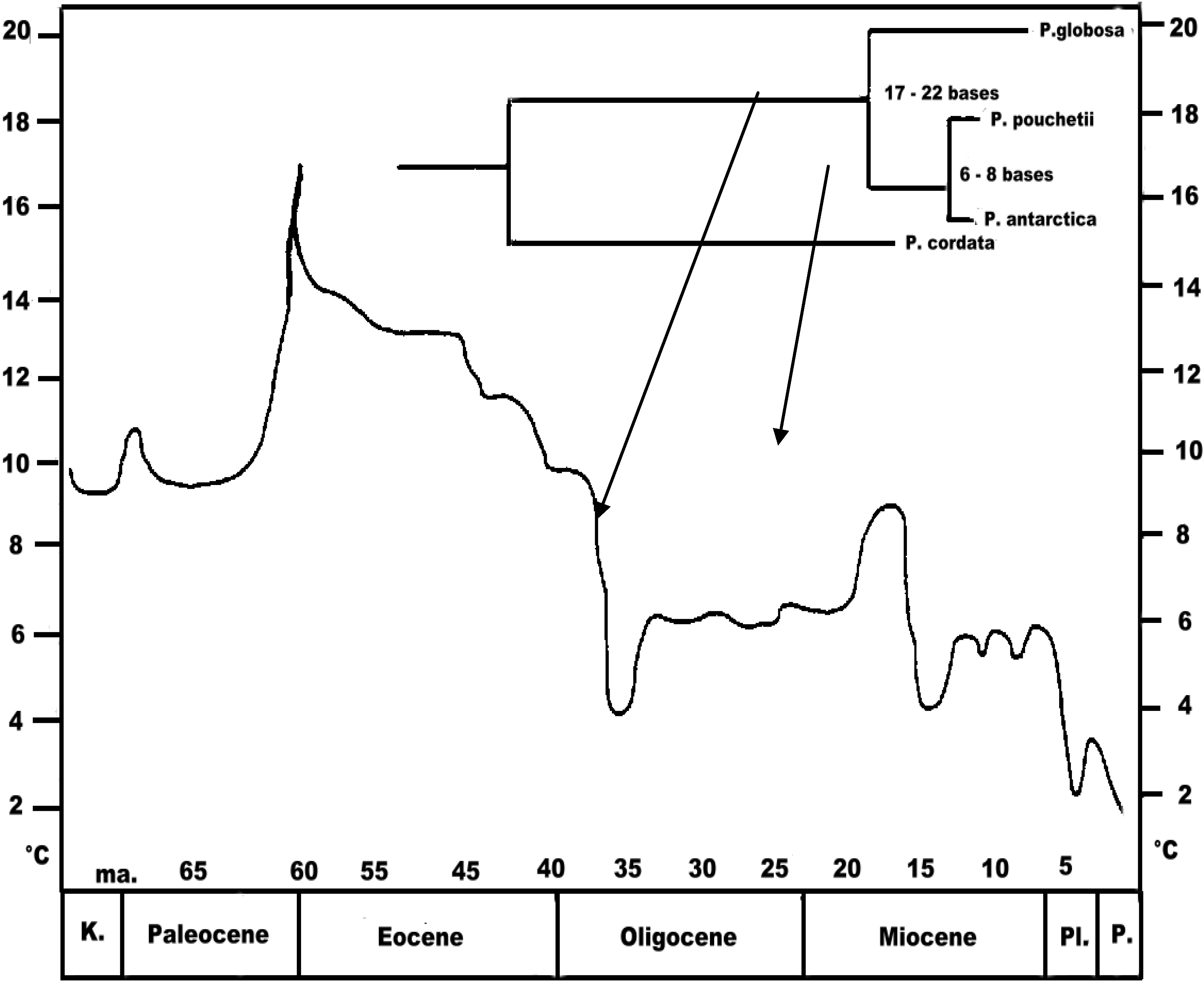
Phaeocystis

6. Conclusions
Acknowledgements
References and Notes
- Ormond, R.F.; Gage, J.D.; Angel, M.V. Marine Biodiversity, Patterns and Processes; Cambridge University Press: Cambridge, UK, 1987; p. 449. [Google Scholar]
- Moestrup, Ø. Further studies of presumedly primitive green algae, including the description of Pedinophyceae class. nov. and Resultor gen. nov. J. Phycol. 1991, 27, 119–133. [Google Scholar]
- Andersen, R.A.; Saunders, G.W.; Paskind, M.P.; Sexton, J.P. Ultrastructure and 18S rRNA gene sequence for Pelagomonas calceolata gen. et sp. nov. and the description of a new algal class, the Pelagophyceae classis nov. J. Phycol. 1993, 29, 701–715. [Google Scholar]
- Guillou, L.; Chretiennot-Dinot, M-J.; Medlin, L.K.; Claustre, H.; Loiseaux-De Goer, S; Vaulot, D. Bolidomonas, a new genus with two species belonging to a new algal class, The Bolidophyceae. J. Phycol. 1999, 35, 368–381. [Google Scholar]
- Kawachi, M.; Inouye, I.; Honda, D.; O’Kelly, C.J.; Bailey, J.C.; Bidigare, R.R.; Andersen, R.A. The Pinguiophyceae classis nova, a new class of chromophyte algae whose members produce large amounts of omega-3 fatty acids. Phycol. Res. 2002, 50, 31–47. [Google Scholar]
- Not, F.; Simon, N.; Biegala, I.C.; Vaulot, D. Application of fluorescent in situ hybridisation coupled with tyramide signal amplification (FISH-TSA) to assess eukaryotic picoplankton composition. Aquat. Microbial. Ecol. 2002, 28, 157–166. [Google Scholar]
- Gosling, E.M. Speciation and species concepts in the marine environment. In Genetics and Evolution of Aquatic Organisms; Beaumont, A.R., Ed.; Chapman & Hall: London, UK, 1994; pp. 1–15. [Google Scholar]
- Waterbury, J.B.; Watson, S.W.; Guillard, R.R.L.; Brand, L.E. Widespread occurrence of a unicellular, marine planktonic cyanobacterium. Nature London 1979, 345, 63–65. [Google Scholar]
- Waterbury, J.B.; Watson, S.W.; Valois, F.W.I.; Franks, D.G. Biological and ecological characterization of the marine unicellular cyanobacterium Synechococcus, in photosynthetic picoplankton. Can. Bull. Fish. Aquat. Sci. 1986, 214, 71–120. [Google Scholar]
- Chisholm, S.W.; Olson, R.J.; Zettler, E.R.; Goericke, R.; Waterbury, J.; Welschmeyer, N. A novel free-living prochlorophyte abundant in the oceanic euphotic zone. Nature 1988, 334, 340–343. [Google Scholar]
- Moon-van der Staay, S.Y.; van der Staay, G.W.M.; Guillou, L.; Claustre, H.; Medlin, L.K.; Vaulot, D. Abundance and diversity of Prymnesiophyceae in the picoplankton community from the equatorial Pacific Ocean inferred from 18S rDNA sequences. Limnol. Oceanogr. 2000, 45, 98–109. [Google Scholar]
- Palumbi, S.R. Marine speciation on a small planet. TREE 1992, 7, 114–118. [Google Scholar]
- Brand, L.E. Genetic variability and spatial patterns of genetic differentiation in the reproductive rates of the marine coccolithophores Emiliania huxleyi and Gephyrocapsa oceanica. Limnol. Oceanogr. 1982, 27, 236–245. [Google Scholar]
- Brand, L.E. Review of genetic variation in marine phytoplankton species and the ecological implications. Biol. Oceanogr. 1989, 6, 397–409. [Google Scholar]
- Gallagher, J.C. Population genetics of Skeletonema costatum (Bacillariophyceae), in Narragansett Bay. J. Phycol. 1980, 16, 464–474. [Google Scholar]
- Hedgecock, D. Population genetics of marine organisms. US GLOBEC News 1994, 6, 1-3, 11. [Google Scholar]
- Carroll, S.P.; Hendry, A.P; Reznick, D.N.; Fox, C.W. Evolution on ecological timescales. Funct. Ecol. 2007, 21, 387–393. [Google Scholar]
- Muyzer, G.; De Wall, E.C.; Uitterlinden, A.G. Profiling on complex microbial populations by denaturing gradient gel electrophoresis analysis by polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Env. Microbiol. 1993, 59, 695–700. [Google Scholar]
- Schwieger, F.; Tebbe, C.C. A new approach to utilize PCR-single-strand-conformation polymorphism for 16S rRNA gene-based microbial community analysis. Appl. Environ. Microbiol. 1998, 64, 4870–4876. [Google Scholar]
- Gillet, E.M. Which DNA Marker for Which Purpose? Final Compendium of the Research Project Development, optimisation and validation of molecular tools for assessment of biodiversity in forest trees in the European Union DGXII Biotechnology FW IV Research Programme Molecular Tools for Biodiversity; University of Groningen: Gronningen, NL, 1999; Available online: http://webdoc.gwdg.de/ebook/y/1999/whichmarker/index.htm (accessed on 15 July 2010).
- Karp, A.; Isaac, P.G.; Ingram, D.S. Molecular Tools for Studying Biodiversity; Chapman & Hall: London, UK, 1998; p. 498. [Google Scholar]
- Medlin, L.K.; Barker G.L.A., G.L.A.; Baumann, M.; Hayes, P.K.; Lange, M. Molecular biology and systematics. In The Haptophyte Algae; Green, J.C., Leadbeater, B.S.C., Eds.; Clarendon Press: Oxford, UK, 1994; pp. 393–412. [Google Scholar]
- Medlin, L.K.; Sáez, A.G.; Young, J.R. Did mixotrophy prevent phytoplankton extinctions across the K/T boundary? Mar. Micropaleon 2007, 67, 69–86. [Google Scholar]
- Woese, C.R. Bacterial evolution. Microbiol. Rev. 1987, 51, 221–271. [Google Scholar]
- Medlin, L.K.; Sato, S.; Mann, D.G.; Kooistra, W.C.H.F. Molecular evidence confirms sister relationship of Ardissonea, Climacosphenia, and Toxarium within the bipolar centric diatoms Bacillariophyta, Mediophyceae, and cladistic analyses confirm that extremely elongated shape has arisen twice in the diatoms. J. Phycol. 2008, 44, 1340–1348. [Google Scholar]
- Watanabe, K.; Kodama, Y.; Harayama, S. Design and evaluation of PCR primers to amplify bacterial 16S ribosomal DNA fragments used for community fingerprinting. J. Microbiol. Methods 2001, 44, 253–262. [Google Scholar]
- Lami, R.; Ghiglione, J-F.; Desdevieses, Y.; West, N.; Lebaron, P. Annual patterns of presence and activity of marine bacteria monitored by 16S rDNA-16SrRNA fingerprints in the coastal NW Mediterranean Sea. Aq. Micro. Ecol. 2009, 54, 199–210. [Google Scholar]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Welch, D.M.; Huse, S.M.; Neal, P.R.; Arieta, J.M.; Herndl, G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. USA 2006, 103, 12115–12120. [Google Scholar]
- Patrick, R. Use of Algae, Especially Diatoms, in the Assessment of Water Quality; American Society for Testing Materials, Special Technical Publication 528; ASTM International: West Conshohocken, PA, USA, 1973; pp. 76–95. [Google Scholar]
- Patrick, R.; Hohn, M.H.; Wallace, J.H. A new method for determine the pattern of the diatom flora. Not. Nat. 1954, 259, 1–12. [Google Scholar]
- McIntyre, C.D.; Overton, W.S. Distributional pattern in assemblages of attached diatoms from Yaquina Estuary, Oregon. Ecology 1987, 52, 758–777. [Google Scholar]
- Evans, K.M.; Wortley, A.H.; Mann, D.G. An assessment of potential Diatom “Barcode” Genes: coxI, rbcL, 18S and ITS rDNA, and their effectiveness in determining relationships in Sellaphora (Bacillariophyta). Protist 2007, 158, 349–364. [Google Scholar]
- Moniz, M.B.J.; Kaczmarska, I. Barcoding diatoms, is there a good marker? Mol. Ecol. Res. 2009, 1, 65–74. [Google Scholar]
- Moniz, M.B.J.; Kaczmarska, I. Barcoding of diatoms, nuclear encoded ITS revisited. Protist 2010, 161, 7–34. [Google Scholar]
- Robba, L.; Russell, S.J.; Barker, G.; Broadie, J. Assessing the use of the mitochondrial Cox1 marker for use in DNA barcoding of Red Algae (Rhodophyta). Am J. Bot. 2006, 93, 1101–1108. [Google Scholar]
- McDevit, D.C.; Saunders, G.W. A DNA barcode examination of the Laminariaceae (Phaeophyceae) in Canada reveals novel biogeographical and evolutionary insights. Phycologia 2010, 49, 235–248. [Google Scholar]
- Amann, R.I. In situ identification of micro-organisms by whole cell hybridisation with rRNA-targeted nucleic acid probes. In Molecular Microbiology and Ecology Manual 3.3.6; Akkermans, A.D.L., van Elsas, J.D., de Brujin, F.J., Eds.; Kluwer Academic Publishers: Dordrecht, the Netherlands, 1995; pp. 1–15. [Google Scholar]
- Amann, R.I.; Ludwig, W.; Schleifer, K.-H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 1995, 95, 143–169. [Google Scholar]
- Stahl, D.A.; Amann, R.I. Development and application of nucleic acid probes. In Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E., Goodfellow, M., Eds.; John Wiley & Sons Ltd.: Chichester, UK, 1991; pp. 205–248. [Google Scholar]
- Knauber, D.C.; Berry, E.S.; Fawley, M.W. Ribosomal RNA-based oligonucleotide probes to identify marine green ultraplankton. J. Euk. Microbiol. 1996, 43, 89–94. [Google Scholar]
- Simon, N.; Lebot, N.; Marie, D.; Partensky, F.; Vaulot, D. Fluorescent in–situ hybridisation with rRNA-targeted oligonucleotide probes to identify small phytoplankton by flow cytometry. Appl. Environ. Microbiol. 1995, 61, 2506–2513. [Google Scholar]
- Simon, N.; Campbell, L.; Ornolfsdottir, E.; Groben, E.; Guilou, L.; Lange, M.; Medlin, L.K. Oligonucleotide probes for the identification of three algal groups by dot blot and fluorescent whole cell hybridisation. J. Euk. Micobiol. 2000, 47, 76–84. [Google Scholar]
- Scholin, C.A.; Anderson, D.M. Identification of group- and strain-specific genetic markers for globally distributed Alexandrium (Dinophyceae). I. RFLP analysis of SSU rRNA genes. J. Phycol. 1994, 29, 209–216. [Google Scholar]
- Scholin, C.A.; Hallegraeff, G.M.; Anderson, D.M. Identification of group- and strain-specific genetic markers for globally distributed Alexandrium (Dinophyceae). II. Sequence analysis of a fragment of the LSU rRNA gene. J. Phycol. 1994, 30, 999–1011. [Google Scholar]
- Scholin, C.A.; Hallegraeff, G.M.; Anderson, D.M. Molecular evolution of the Alexandrium tamarense species complex (Dinophyceae), dispersal in the North American and West Pacific regions. Phycologia 1995, 34, 472–485. [Google Scholar]
- Miller, P.E.; Scholin, C.A. Identification of cultured Pseudo-nitzschia Bacillariophyceae, using species-specific LSU targeted fluorescent probes. J. Phycol. 1996, 32, 646–655. [Google Scholar]
- Medlin, L.K.; Lange, M.; Edvardsen, B.; Larsen, A. Cosmopolitan flagellates and their genetic links. In The Flagellate Algae; Green, J.C., Leadbeater, B.S.C., Eds.; Francis and Taylor: London, UK, 2000; pp. 288–308. [Google Scholar]
- Simon, N.; Brenner, J.; Edvardsen, B.; Medlin, L.K. The identification of Chrysochromulina and Prymnesium species Haptophyta, Prymnesiophyceae, using fluorescent or chemiluminescent oligonucleotide probes, a means for improving studies on toxic algae. Eur. J. Phycol. 1997, 32, 393–401. [Google Scholar]
- Lange, M.; Simon, N.; Guillou, L.; Vaulot, D.; Amann, R.; Ludwig, W.; Medlin, L.K. Identification of the Class Prymnesiophyceae and the genus Phaeocystis with rRNA-targeted nucleic acid probes detected by flow cytometry. J. Phycol. 1996, 32, 858–868. [Google Scholar]
- John, U.; Cembella, A.; Hummert, C.; Elbrächter, M.; Groben, R.; Medlin, L.K. Discrimination of the toxigenic dinoflagellate species Alexandrium tamarense and Alexandrium ostenfeldii in co-occurring natural populations from Scottish Coastal waters. Eur. J. Phycol. 2003, 38, 25–40. [Google Scholar]
- Kemp, P.F.; Lee, S.; LaRoche, J. Estimating the growth rate of slowly growing marine bacteria from RNA content. Appl. Environ. Microbiol. 1993, 59, 2594–2601. [Google Scholar]
- Schönhuber, W.; Fuchs, B.; Juretschko, S.; Amann, R. Improved sensitivity of whole-cell hybridisation by the combination of horseradish perxidase-labeled oligonucleotides and Tyramide Signal Amplification. Appl. Environ. Microbiol. 1997, 63, 3268–3273. [Google Scholar]
- Schönhuber, W.; Zarda, B.; Eix, S.; Rippka, R.; Herdmann, M.; Ludwig, W.; Amann, R. In situ identification of cyanobacteria with horseradish peroxidise-labeled, rRNA-targeted oligonucleotide probes. Appl. Environ. Microbiol. 1999, 65, 1259–1267. [Google Scholar]
- West, N.J.; Schönhuber, W.A.; Fuller, N.; Amann, R.I.; Rippka, R.; Post, A.; Scanlan, D. Closely related Plochlorococcus genotypes show remarkably different depth distributions in two oceanic regions as revealed by in situ hybridisation using 16SrRNA-targeted oligonucleotides. Microbiology 2001, 147, 1731–1744. [Google Scholar]
- Biegala, I.C.; Not, F.; Vaulot, D.; Simon, N. Quantitative assessment of picoeukaryotes in the natural environment by using taxon-specific oligonucleotide probes in association with tyramide signal amplification-fluorescence in situ hybridisation and flow cytometry. Appl. Environ. Microbiol. 2003, 69, 5519–5529. [Google Scholar]
- Not, F.; Latasa, M.; Marie, D.; Cariou, T.; Vaulot, D.; Simon, N. A single species, Micromonas pusilla (Prasinophyceae), dominates the eukaryotic picoplankton in the western English channel. Appl. Environ. Microbiol. 2004, 70, 4063–4072. [Google Scholar]
- Not, F.; Valentin, K.; Romari, K.; Massana, R.; Vaulot, D.; Medlin, L.K. Picobiliphytes: A marine picoplanktonic algal group with unknown affinities to other eukaryotes. Science 2007, 315, 253–255. [Google Scholar]
- Biegala, I.C.; Kennaway, G.; Alverca, E.; Lennon, J.F.; Vaulot, D.; Simon, N. Identification of bacteria associated with dinoflagellates (Dinophyceae), Alexandrium spp. using tyramide signal amplification-fluorescent in situ hybridization and confocal microscopy. J. Phycol. 2002, 38, 404–411. [Google Scholar]
- Alverca, E.; Biegala, I.C.; Kennaway, G.M.; Lewis, J.; Franca, S. In situ identification and localization of bacteria associated with Gyrodinium instriatum (Gymnodiniales, Dinophyceae) by electron and confocal microscopy. Eur. J. Phycol. 2002, 37, 523–530. [Google Scholar]
- Galluzzi, L.; Penna, A.; Bertozzini, E.; Vila, M.; Garces, E.; Magnani, M. Development of a real-time PCR assay for rapid detection and quantification of Alexandrium minutum (a Dinoflagellate). Appl. Environ. Microbiol. 2004, 70, 1199–1206. [Google Scholar]
- Wallner, G.; Amann, R.; Beisker, W. Optimizing fluorescent in situ hybridisation with rRNA targeted probes for flow cytometric identification of microorganisms. Cytometry 1993, 14, 136–143. [Google Scholar]
- Manz, W.; Amann, R.; Ludwig, W.; Wagner, M.; Schliefer, K-H. Phylogenetic oligodeoxynucleotide probes for the major subclasses of proteobacteria, problems and solutions. Syst. Appl. Microbiol 1992, 15, 593–600. [Google Scholar]
- Giovanonni, S.J.; Britschgi, T.; Moyer, C.L.; Field, K.G. Genetic diversity in Sargasso Sea bacterioplankton. Nature 1990, 345, 60–63. [Google Scholar]
- Pace, N.R.; Stahl, D.A.; Lane, D.J; Olsen, G.J. The analysis of microbial populations with ribosomal RNA sequences. Adv. Microb. Ecol. 1986, 9, 1–55. [Google Scholar]
- Rehnstam, A-S.; Bäckman, S.; Smith, D.C.; Azam, F.; Hagström, Å. Blooms of sequence-specific culturable bacteria in the sea. FEMS Microbiol. Ecol. 1993, 102, 161–166. [Google Scholar]
- Metfies, K.; Hujic, S.; Lange, M.; Medlin, L.K. Electrochemical detection of the toxic dinoflagellate A. ostenfeldii with a DNA Biosensor. Biosen. Bioelec 2005, 20, 1349–1357. [Google Scholar]
- Murphy, L.S.; Guillard, R.R.L. Biochemical taxonomy of marine phytoplankton by enzyme electrophoresis. I. The centric diatoms Thalassiosira pseudonana Hasle Heimdal and Thalassiosira fluviatilis Hustedt. J. Phycol. 1976, 12, 9–13. [Google Scholar]
- Brand, L.E.; Murphy, L.S.; Guillard, R.R.L.; Lee, H.-T. Genetic variability and differentiation in the temperature niche component of the diatom Thalassiosira pseudonana. Mar. Biol. 1981, 62, 103–110. [Google Scholar]
- Hasle, G.R. Some fresh water and brackish water species of the diatom genus Thalassiosira Cleve. Phycologia 1978, 17, 263–292. [Google Scholar]
- Hasle, G.R. The marine planktonic diatom Thalassiosira oceanica sp. nov. and T. parthenia. J. Phycol. 1983, 19, 220–229. [Google Scholar]
- Cembella, A.D.; Taylor, F.J.R. Electrophoretic variability within the Protogonyaulax tamarensis/catenella species complex, Pyridine linked dehydrogenases. Biochem. Sys. Ecol. 1986, 143, 311–323. [Google Scholar]
- Cembella, A.D.; Taylor, F.J.R.; Therriault, J.C. Cladistic analysis of electrophoretic variants within the toxic dinoflagellate genus Protogonyaulax. Bot. Mar. 1988, 31, 39–51. [Google Scholar]
- Hayhome, B.A.; Anderson, D.M.; Kulis, D.M.; Whitten, D.J. Variation among congeneric dinoflagellates from the northeastern United States and Canada. I. Enzyme electrophoresis. Mar. Bio. 1989, 101, 427–435. [Google Scholar]
- Hayhome, B.A.; Whitten, D.J.; Harkins, K.R.; Pfiester, L.A. Intraspecific variation in the dinoflagellate Peridinium volzii. J. Phycol. 1987, 23, 573–580. [Google Scholar]
- Chinain, M.; Germain, M.; Sako, Y.; Pauillac, S.; Legrand, A.-M. Intraspecific variation in the dinoflagellate Gambierdiscus toxicus Dinophyceae, I. Isozyme analysis. J. Phycol. 1997, 33, 36–43. [Google Scholar]
- Sako, Y.; Kim, C.H.; Ninomiya, H.; Adachi, M.; Ishida, Y. Isozyme and cross analysis of mating populations in the Alexandrium catenella/tamarense species complex. In Toxic Marine Phytoplankton; Graneli, E., Sundstrom, B., Edler, L., Anderson, D.M., Eds.; Elsevier: New York, NY, USA, 1990; pp. 320–329. [Google Scholar]
- Gallagher, J.C. Physiological variation and electrophoretic banding patterns of genetically different seasonal populations of Skeletonema costatum (Bacillariophyceae). J. Phycol. 1982, 18, 148–162. [Google Scholar]
- Sarno, D.; Kooistra, W.H.C.F.; Medlin, L.K.; Percopo, I.; Zingone, A. Diversity in the genus Skeletonema (Bacillariophyceae), II. An assessment of the taxonomy of S. costatum-like species with the description of four new species. J. Phycol. 2005, 41, 151–176. [Google Scholar]
- Sarno, D.; Kooistra, W.H.C.F.; Hargraves, P.E.; Zingone, A. Diversity in the genus Skeletonema Bacillariophyceae, III. Phylogenetic position and morphology of Skeletonema costatum and Skeletonema grevillei, with the description of Skeletonema ardens sp. nov. J. Phycol. 2007, 43, 156–170. [Google Scholar]
- Kooistra, W.H.C.F.; Sarno, D.; Balzano, S.; Gu, H.; Andersen, R.A.; Zingone, A. Global diversity and biogeography of Skeletonema species (Bacillariophyta). Protist 2008, 59, 177–193. [Google Scholar]
- Hadrys, H.; Balick, M.; Schierwater, B. Application of random amplified polymorphic DNA RAPD. Mol. Ecol. 1992, 1, 55–63. [Google Scholar]
- Lynch, M.; Milligan, G. Analysis of population genetic structure with RAPD markers. Mol. Ecol. 1994, 3, 91–99. [Google Scholar]
- Boalch, C.J.; Haskard, K; Hallegraeff, G.M. Genetic variation among strains of the toxic dinoflagellate, Gymnodinium catenatum (Dinophyceae). J. Phycol. 1999, 35, 356–367. [Google Scholar]
- Barker, G.L.A.; Green, J.C.; Hayes, P.K.; Medlin, L.K. Preliminary results using the RAPD analysis to screen bloom populations of Emiliania huxleyi (Haptophyta). Sarsia 1995, 79, 301–306. [Google Scholar]
- Vos, P.; Hogers, R.; Bleeker, M.; Reijans, M.; Van De Lee, T.; Hornes, M.; Frijters, A.; Pot, J.; Peleman, J.; Kuiper, M.; Zabeau, M. AFLP: a new technique for DNA fingerprinting. Nucl. Acid. Res. 1995, 23, 4407–4414. [Google Scholar]
- John, U.; Groben, R.; Beszteri, B.; Medlin, L.K. Utility of Amplified Fragment Length Polymorphisms (AFLP) to analyse genetic structures within the Alexandrium tamarense species complex. Protist 2004, 155, 169–179. [Google Scholar]
- Alpermann, T. Evolutionary Ecology of Alexandrium (Dinophyceae) with Special Emphasis on Genotypic and Phenotypic Variation in the Toxigenic Species A. tamarense. PhD Thesis, University of Bremen, Bremen, Germany, 2009. [Google Scholar]
- Gaebler, S.; Hayes, P.K.; Medlin, L.K. Methods used to reveal genetic diversity in the colony forming prymnesiophyte Phaeocystis antarctica–preliminary results. Biogeochemistry 2007, 83, 19–27. [Google Scholar]
- Burke, T.; Hanotte, O.; Bruford, M.W. Multilocus and single locus minisatellite analysis in population biological studies. In DNA Fingerprinting, Approaches and Applications; Burke, T., Dolf, G., Jeffreys, A.J., Wolff, R., Eds.; Birkhaeuser: Basel, Switzerland, 1991; pp. 154–168. [Google Scholar]
- Wright, J.M.; Bentzen, P. Microsatellites, genetic markers for the future. Rev. Fish. Biol. Fish. 1994, 4, 384–388. [Google Scholar]
- Doyle, R.W. Upwelling, clone selection and the characteristic shape of nutrient uptake curves. Limnol. Oceanogr. 1975, 20, 487–489. [Google Scholar]
- Partensky, F.; Hoepffner, N.; Li, W.K.W.; Ulloa, O.; Vaulot, D. Photoacclimation of Prochlorococcus sp. Prochlorophyta, strains isolated from the North Atlantic and the Mediterranean Sea. Plant Physiol. 1993, 101, 285–296. [Google Scholar]
- Medlin, L.K. If everything is everywhere, do they share a common gene pool? Gene 2007, 405, 180–183. [Google Scholar]
- Bhattacharya, D.; Medlin, L.; Wainwright, P.O.; Ariztia, E.V.; Bibeau, C.; Stickel, S.K.; Sogin, M.L. Algae containing chlorophylls a + c are paraphyletic, molecular evolutionary analysis of the Chromophyta. Evolution 1992, 46, 1801–1817. [Google Scholar]
- Medlin, L.K.; Kooistra, W.H.C.F.; Potter, D.; Saunders, G.W.; Andersen, R.A. Phylogenetic relationships of the 'golden algae' and their plastids. Pl. Syst. Evol. 1997, 11, 187–219. [Google Scholar]
- Cavalier-Smith, T.; Allsopp, M.T.E.; Chai, E.E. Chimeric conundra, are nucleomorphs and chromists monophyletic or polyphyletic? Proc. Natl. Acad. Sci. USA 1994, 91, 11368–11372. [Google Scholar]
- Bhattacharya, D.; Medlin, L. The phylogeny of plastids, a review based on comparison of small–subunit ribosomal RNA coding regions. J. Phycol. 1995, 31, 489–498. [Google Scholar]
- McFadden, G.I.; Gilson, P.R.; Waller, R.F. Molecular phylogeny of Chlorarachniophytes based on plastid rRNA and rbcL sequences. Arch. für Prot. 1995, 145, 231–239. [Google Scholar]
- Rensing, S.A.; Rober, N.; Obrdlik, D.; Müller, S.B.; Hofman, C.J.B.; Maier, U-G. Molecular phylogeny of the stress—70 protein family with certain emphasis on algal relationships. Eur. J. Phycol. 1997, 3, 279–285. [Google Scholar]
- Bhattacharya, D.; Helmchen, T.; Melkonian, M. Molecular evolutionary analyses of nuclear-encoded small subunit ribosomal RNA identify an independent rhizopod lineage containing the Euglyphina and the Chlorarachniophyta. J. Eukaryot. Microbiol. 1995, 42, 65–69. [Google Scholar]
- Hackett, J.D.; Yoon, H.D.; Li, S.; Reyes-Prieto, A.; Rümmele, S.E.; Bhattacharya, D. Phylogenomic analysis supports the monophyly of cryptophytes and haptophytes and the association of Rhizaria with Chromalveolates. Mol. Biol. Evol. 2007, 24, 1702–1713. [Google Scholar]
- Edvardsen, B.; Eikrem, W.; Green, J.C.; Andersen, R.A.; Moon-Van Der Staay, S.Y.; Medlin, L.K. Phylogenetic reconstructions of the Haptophyta inferred from rRNA sequences and available morphological data. Phycologia 2000, 39, 19–35. [Google Scholar]
- Medlin, L.K.; Kooistra, W.C.H.F.; Schmid, A-M. A review of the evolution of the diatoms—a total approach using molecules, morphology and geology. In The Origin and Early Evolution of the Diatoms; Wittkowski, A., Sieminska, J., Eds.; Polish Academy of Science: Cracow, Poland, 2000; pp. 13–35. [Google Scholar]
- McNally, K.L.; Govind, N.S.; Thomé, P.E.; Trench, R.K. Small subunit ribosomal DNA sequence analyses and a reconstruction of the inferred phylogeny among symbiotic dinoflagellates. J. Phycol. 1994, 30, 316–329. [Google Scholar]
- Kaczmarska, I.; Beaton, M.; Benoit, A.C.; Medlin, L.K. Molecular phylogeny of selected members of the Order Thalassiosirales (Bacillariophyta) and evolution of the fultoportula. J. Phycol. 2006, 42, 121–138. [Google Scholar]
- Nanney, D.L.; Meyer, E.B.; Simon, E.M.; Preparata, R.-M. Comparison of ribosomal and isozymic phylogenies of tetrahymenine ciliates. J. Protist. 1989, 36, 1–8. [Google Scholar]
- Medlin, L.K.; Elwood, H.J.; Stickel, S.; Sogin, M.L. Morphological and genetic variation within the diatom Skeletonema costatum (Bacillariophyta): evidence for a new species Skeletonema pseudocostatum. J. Phycol. 1991, 27, 514–524. [Google Scholar]
- Beam, C.A.; Preparata, R.-M.; Himes, M.; Nanney, D.L. Ribosomal RNA sequencing of members of the Crypthecodinium cohnii (Dinophyceae), species complex, comparison with soluble enzyme studies. J. Euk. Microbiol. 1993, 40, 660–667. [Google Scholar]
- Knowlton, N. Sibling species in the sea. Ann. Rev. Ecol. Syst. 1993, 24, 189–216. [Google Scholar]
- DeYoe, H.R.; Chan, A.M.; Suttle, C.A. Phylogeny of Aureococcus anophagefferens and a morphologically similar bloom–forming alga from Texas as determined by 18S ribosomal RNA sequence analysis. J. Phycol. 1995, 31, 413–418. [Google Scholar]
- Potter, D.; Lajeunesse, T.C.; Saunders, G.W.; Andersen, R.A. Convergent evolution masks extensive biodiversity among marine coccoid picoplankton. Biodiv. Cons. 1996, 5, 1–9. [Google Scholar]
- Zingone, A.; Percopo, I.; Sims, P.A.; Sarno, D. Diversity in the genus Skeletonema (Bacillariophyceae). I. A reexamination of the type material of S. costatum with the description of S. grevillei sp. nov. J. Phycol. 2005, 41, 140–150. [Google Scholar]
- Godhe, A.; Mcquoid, M.R.; Karunasagarb, I.; Karunasagarb, I.; Rehnstam-Holm, N.-S. Comparison of three common molecular tools for distinguishing among geographically separated clones of the diatom Skeletonema marinoi Sarno et Zingone Bacillariophyceae. J. Phycol. 2006, 42, 280–291. [Google Scholar]
- Chen, G.-F.; Wang, G.-C.; Zhang, B.-Y.; Fan, X.-L. Morphological and phylogenetic analysis of Skeletonema costatum—like diatoms (Bacillariophyta) from the China Sea. Eur. J. Phycol. 2007, 42, 163–175. [Google Scholar]
- Skov, J.; Lundholm, N.; Pockington, R.; Rosendahl, S.; Moestrup, O. Studies on the marine planktonic diatom Pseudo-nitzschia. 1. Isozyme variation among isolates of P. pseudodelicatissima during bloom in Danish coastal waters. Phycologia 1997, 36, 374–380. [Google Scholar]
- Lundholm, N.; Moestrup, Ø.; Hasle, G.R.; Hoef-Emden, K. A study of the Pseudo-nitzschia pseudodelicatissima/cuspidata complex Bacillariophyceae,, what is P. pseudodelicatissima? J. Phycol. 2003, 39, 797–813. [Google Scholar]
- Lundholm, N.; Moestrup, Ø.; Kotaki, Y.; Hoef-Emden, K.; Scholin, C.; Miller, P. Inter- and intraspecific variation of the Pseudo-nitzschia delicatissima complex Bacillariophyceae, illustrated by rRNA probes, morphological data and phylogenetic analyses. J. Phycol. 2006, 42, 464–481. [Google Scholar]
- Orsini, L.; Procaccini, G.; Sarno, D.; Montresor, M. Multiple rDNA ITS-types within the diatom Pseudo-nitzschia delicatissima Bacillariophyceae, and their relative abundances across a spring bloom in the Gulf of Naples. Mar. Ecol. Prog. Series 2004, 271, 87–98. [Google Scholar]
- Amato, A.; Kooistra, W.H.C.F.; Levialdi Ghiron, J.H.; Mann, D.G.; Pröschold, T.; Montresor, M. Reproductive isolation among sympatric cryptic species in marine diatoms. Protist 2007, 158, 193–207. [Google Scholar]
- Amato, A.; Montresor, M. Morphology, phylogeny, and sexual cycle of Pseudo-nitzschia mannii sp. nov. Bacillariophyceae,, a pseudo–cryptic species within the P. pseudodelicatissima complex. Phycologia 2008, 47, 487–497. [Google Scholar]
- Orive, E.; Laza-Martinez, A.; Seoane, S.; Alonso, A.; Andrade, R.; Miguel, I. Diversity of Pseudo-nitzschia in the Southeastern Bay of Biscay. Diatom Res. 2010, 25, 125–145. [Google Scholar]
- John, U.; Fensome, R.A.; Medlin, L.K. The application of a molecular clock based on molecular sequences and the fossil record to explain the biogeographic distribution within the Alexandrium tamarense “species complex”. Mol. Biol. Evol. 2003, 20, 1015–1027. [Google Scholar]
- Mann, D.G. The species concept in diatoms. Phycologia 1999, 38, 437–495. [Google Scholar]
- Mann, D.G. The species concept in diatoms, evidence for morphologically distinct, sympatric gamodemes in four epipelic species. Plant Syst. Evol. 1989, 164, 215–237. [Google Scholar]
- Wood, A.M.; Leatham, T. The species concept in phytoplankton ecology. J. Phycol. 1992, 28, 723–729. [Google Scholar]
- Amato, A.; Lüdeking, A.; Kooistra, W.H.C.F. Intracellular domoic acid production in Pseudo-nitzschia multistriata isolated from the Gulf of Naples (Tyrrhenian Sea, Italy). Toxicon 2010, 55, 157–161. [Google Scholar]
- Bakker, F.T.; Olsen, J.L.; Stam, W.T. Global phylogeography in the cosmopolitan species Cladophora vagabunda (Chlorophyta), based on nuclear DNA ITS sequences. Eur.J. Phycol. 1995, 30, 187–210. [Google Scholar]
- Bakker, F.T.; Olsen, J.L.; Stam, W.T.; Van Den Hooke, C. Nuclear ribosomal DNA internal transcribed spacer regions ITS1 and ITS2, define discrete biogeographic groups in Cladophora albida (Chlorophyta). J. Phycol. 1992, 28, 839–845. [Google Scholar]
- Medlin, L.K.; Lange, M.; Barker, G.L.A.; Hayes, P.K. Can molecular techniques change our ideas about the species concept? In Molecular Ecology of Aquatic Microbes; Joint, I., Falkowski, P., Carr, N., Eds.; Springer Verlag: Berlin, Germany, 1995; Volume 38, pp. 133–152. [Google Scholar]
- Cracraft, J. Speciation and its ontology, the empirical consequences of alternative species concepts for understanding patterns and process of differentiation. In Speciation and Its Consequences; Otte, D., Endler, J.A., Eds.; Sinauer Associates: Sunderland, MD, USA, 1989; pp. 28–59. [Google Scholar]
- Costas, E.; Zardoya, R.; Bautista, J.; Garrido, A.; Rojo, C.; Lopez-Rodas, V. Morphospecies vs. genospecies in toxic marine dinoflagellates, an analysis of Gymnodinium catenatum/Gyrodiniumimpudicum and Alexandrium minutum/A. lusitanicum using antibodies, lectins, and gene sequences. J. Phycol. 1995, 31, 801–807. [Google Scholar]
- Parker, P.G.; Snow, A.A.; Schug, M.D.; Booton, G.C.; Fuerst, P.A. What molecules can tell us about populations: choosing and using a molecular marker. Ecology 1998, 79, 361–382. [Google Scholar]
- Wright, S. Variability Within and Among Natural Populations, a Treatise in Four Volumes; University of Chicago Press: Chicago, IL, USA, 1978; Volume 4. [Google Scholar]
- Nei, M. Genetic distance between populations. Amer. Nat. 1972, 106, 283–292. [Google Scholar]
- Takezaki, N.; Nei, M. Genetic distances and reconstruction of phylogenetic trees from microsatellite DNA. Genetics 1996, 144, 389–399. [Google Scholar]
- Medlin, L.K.; Lange, M.; Noethig, E.V. Genetic Diversity of Marine Phytoplankton, A review and a look to Antarctic Phytoplankton. Ant. Sci. 2000, 12, 325–331. [Google Scholar]
- Evans, K.M.; Bates, S.S.; Medlin, L.K.; Hayes, P.K. Microsatellite marker development and genetic variation in the toxic marine diatom Pseudo-nitzschia multiseries (Bacillariophyceae). J. Phycol. 2004, 40, 911–920. [Google Scholar]
- Evans, K.M.; Kühn, S.F.; Hayes, P.K. High levels of genetic diversity and low levels of genetic differentiation in North Sea Pseudo-nitzschia pungens (Bacillariophyceae) populations. J. Phycol. 2005, 41, 506–514. [Google Scholar]
- Evans, K.M.; Chepurnov, V.A.; Sluiman, H.J.; Thomas, .S.J.; Spears, B.M.; Mann, D.G. Highly differentiated populations of the freshwater diatom Sellaphora capitata suggest limited dispersal and opportunities for allopatric speciation. Protist 2009, 160, 386–396. [Google Scholar]
- Iglesias-Rodriguez, M.D.; Schofield, O.M.; Batley, P.J.; Medlin, L.K.; Hayes, P.K. Extensive intraspecific genetic diversity in the marine coccolithophore Emiliania huxleyi: the use of microsatellite analysis in marine phytoplankton population studies. J. Phycol. 2006, 42, 526–536. [Google Scholar]
- Nagai, S.; Lian, C.; Yamaguchi, S.; Hamaguchi, M.; Matsuyama, Y.; Itakura, S.; Shimada, H.; Kaga, S.; Yamauchi, H.; Sonda, Y.; Nishikawa, T.; Kim, C.H.; Hogetsu, T. Microsatellite markers reveal population genetic structure of the toxic dinoflagellates (Dinophyceae) in Japanese coastal waters. J. Phycol. 2007, 43, 43–54. [Google Scholar] [CrossRef]
- Rynearson, T.A.; Armbrust, E.V. DNA fingerprinting reveals extensive genetic diversity in a field population of the centric diatom Ditylum brightwellii. Limnol. Oceanogr. 2000, 45, 1329–1340. [Google Scholar]
- Rynearson, T.A.; Armbrust, E.V. Genetic differentiation among populations of the planktonic marine diatom Ditylum brightwellii (Bacillariophyceae). J. Phycol. 2004, 40, 34–43. [Google Scholar]
- Rynearson, T.A.; Armbrust, E.V. Maintenance of clonal diversity during a spring bloom of the centric diatom Ditylum brightwellii. Mol. Ecol. 2005, 14, 1631–1640. [Google Scholar]
- Rynearson, T.A.; Newton, J.A.; Armbrust, E.V. Spring bloom development, genetic variation, and population succession in the planktonic diatom Ditylum brightwellii. Limnol. Oceanogr. 2006, 51, 1249–1261. [Google Scholar]
- Gaebler-Schwarz, S. Etimation of genetic diversity in the colony forming polar pymnesiophyte species Phaeocystis Antarctica. PhD Thesis, University of Bremen, Bremen, Germany, 2009; p. 2217. [Google Scholar]
- Casteleyn, G.; Leliaert, F.; Backeljau, T.; Debeer, A.E.; Kotaki, Y.; Rhodes, L.; Lundholm, N.; Sabbe, K.; Vyverman, W. Limits to gene flow in a cosmopolitan marine planktonic diatom. In Proc. Natl. Acad. Sci. USA; 2010; in press. [Google Scholar]
- Lange, M.; Chen, Y.-Q.; Medlin, L.K. Molecular genetic delineation of Phaeocystis species (Prymnesiophyceae), using coding and non-coding regions of nuclear and plastid genomes. Eur. J. Phycol. 2002, 37, 77–92. [Google Scholar]
- Medlin, L.K. Molecular clocks and inferring evolutionary milestones and biogeography in the microalgae. In Origin and Evolution of Natural Diversity; H. Okada, S.F., Mawatari, N., Suzuki, P., Gautam, P., Eds.; 21st Century COE: Sapporo, Japan, 2008; pp. 31–42. [Google Scholar]
- Medlin, L.K.; Zingone, A. A taxonomic review of the genus Phaeocystis. Biogeochem. 2007, 83, 3–18. [Google Scholar]
- Kooistra, W.H.C.F.; Medlin, L.K. The evolution of the diatoms. IV. The age of the diatoms (Bacillariophyta), based on 18S rDNA sequences. Mol. Phylogen. Evol. 1996, 6, 391–407. [Google Scholar]
- Avise, J.C.; Arnold, J.; Ball, R.M.; Bermingham, E.; Lamb, T.; Neigel, J.E.; Reeb, C.A.; Saunders, N.C. Intraspecific phylogeography, The mitochondrial DNA bridge between population genetics and systematics. Ann. Rev. Syst. Ecol. 1987, 18, 489–522. [Google Scholar]
- Costas, E.; Løpez-Rodas, V. Did the majority of the dinoflagellates perish in the great Permian extinction? Harmful Algal News 1995, 10/11, 8–9. [Google Scholar]
- Sáez, A.G.; Probert, I.; Geisen, M.; Quinn, P.; Young, J.R.; Medlin, L.K. Pseudo-cryptic speciation in Coccolithophores. Proc. Natl. Acad. Sci. USA 2003, 100, 6893–7418. [Google Scholar]
- Wu, C.-I.; Li, W.-H. Evidence for higher rates of nucleotide substitution in rodents than in man. Proc. Natl. Acad. Sci. USA 1985, 82, 1741–1745. [Google Scholar]
- Ochman, H.; Wilson, A.C. Evolution in bacteria, evidence for a universal substitution rate in cellular genomes. J. Mol. Evol. 1987, 26, 74–86. [Google Scholar]
- Olsen, J.L.; Stam, W.T.; Berger, S.; Menzel, D. 18S rDNA and evolution in the Dasycladales Chlorophyta, modern living fossils. J. Phycol. 1994, 30, 729–744. [Google Scholar]
- Vaulot, D.; Birrien, J-L.; Marie, D.; Casotti, R.; Velhuis, M.; Kraay, G.; Chretiennet-Dinet, M-J. Morphology, ploidy, pigment composition, and genome size of cultured strains of Phaeocystis Prymnesiophyceae. J. Phycol. 1994, 30, 1022–1035. [Google Scholar]
- Crame, J.A. Latitudinal range fluctuations in the marine realm through geological times. Trends Ecol. Evol. 1993, 8, 162–166. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Medlin, L.K.; Kooistra, W.H.C.F. Methods to Estimate the Diversity in the Marine Photosynthetic Protist Community with Illustrations from Case Studies: A Review. Diversity 2010, 2, 973-1014. https://doi.org/10.3390/d2070973
Medlin LK, Kooistra WHCF. Methods to Estimate the Diversity in the Marine Photosynthetic Protist Community with Illustrations from Case Studies: A Review. Diversity. 2010; 2(7):973-1014. https://doi.org/10.3390/d2070973
Chicago/Turabian StyleMedlin, Linda K., and Wiebe H.C. F. Kooistra. 2010. "Methods to Estimate the Diversity in the Marine Photosynthetic Protist Community with Illustrations from Case Studies: A Review" Diversity 2, no. 7: 973-1014. https://doi.org/10.3390/d2070973
APA StyleMedlin, L. K., & Kooistra, W. H. C. F. (2010). Methods to Estimate the Diversity in the Marine Photosynthetic Protist Community with Illustrations from Case Studies: A Review. Diversity, 2(7), 973-1014. https://doi.org/10.3390/d2070973