Culture-Independent Molecular Tools for Soil and Rhizosphere Microbiology

Abstract
:1. Introduction
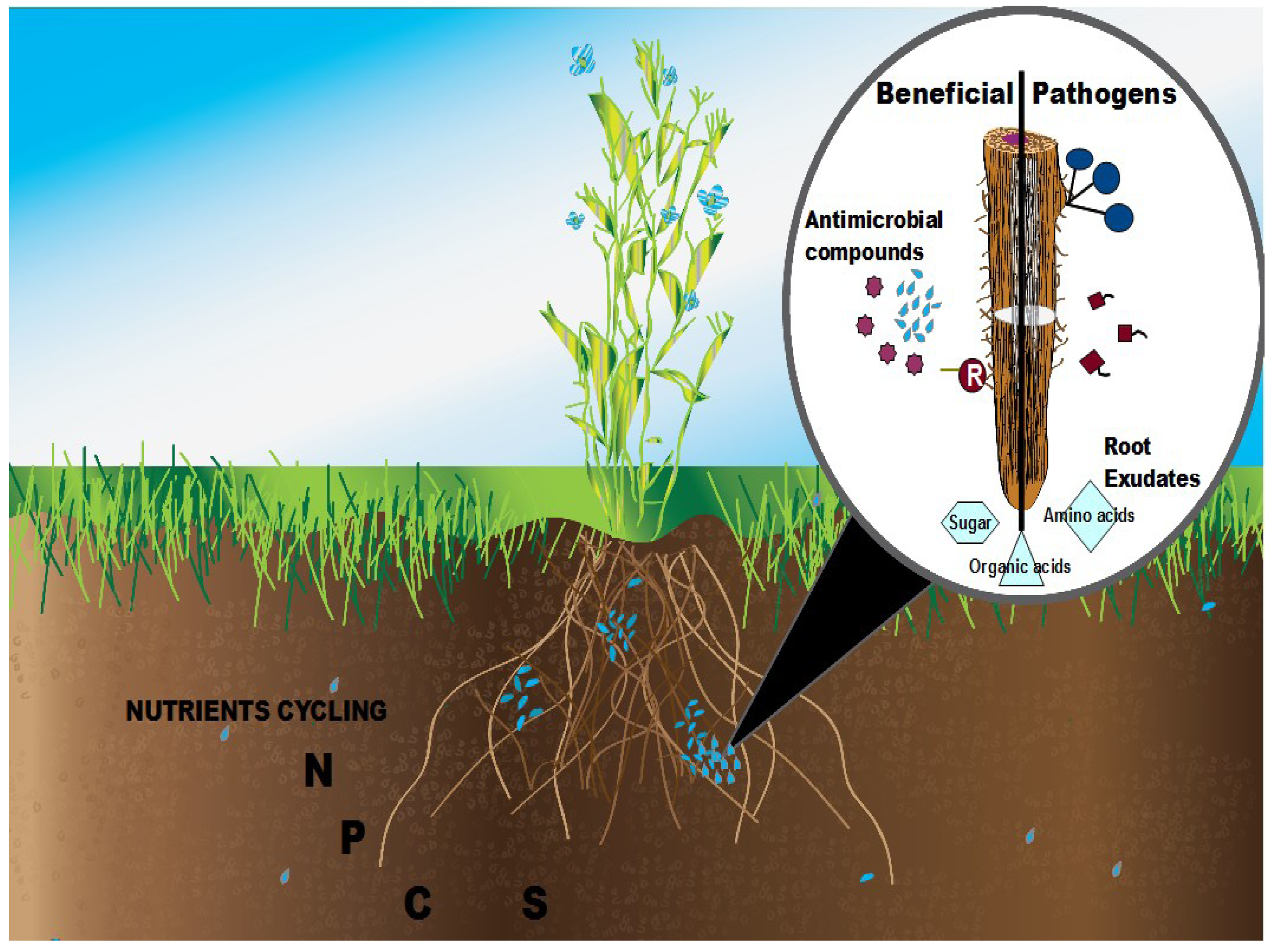
2. Rhizosphere Plant-Microbe Interactions
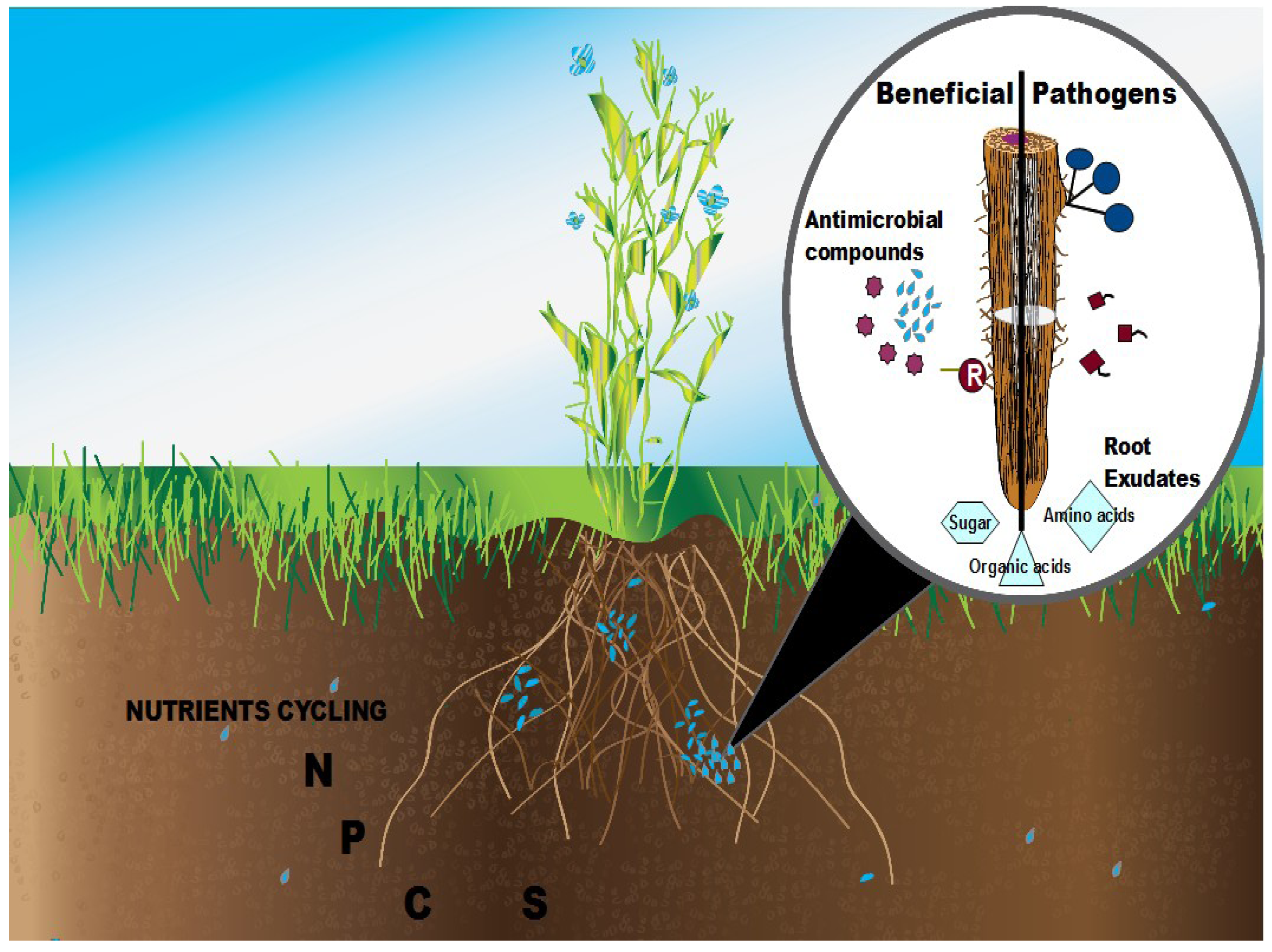
3. Methods for Studying Microbial Communities

{kind=link}
| Methods | Advantages | Disadvantages | Ref. | |
|---|---|---|---|---|
| BIOMASS | CFE |
|
| [45,46] |
| PLFA |
|
| [47] | |
| Q-PCR |
|
| [48] | |
| DIVERSITY | DGGE/TGGE |
|
| [49] |
| DIVERSITY | SSCP |
|
| [50] |
| T-RFLP |
|
| [49,51,52] | |
| RISA/ARISA |
|
| [49] | |
| LH-PCR |
|
| [52,53,54] | |
| RAPD |
|
| [55,56] | |
| ARDRA |
|
| [51] | |
| FISH |
|
| [57] | |
| DIVERSITY | DNA ARRAY |
|
| [58,59] |
| DIVERSITY | Next Generation Sequencing (16S rRNA amplicon sequencing) |
|
| |
| Next Generation Sequencing (metagenomics) |
|
| [60,61] | |
| ACTIVITY | FDA |
|
| [62,63] |
| SIP |
|
| [64] | |
| Functional Gene Arrays (RNA-based) |
|
| [58,59] | |
| Next Generation Sequencing (Metatranscriptomics) |
|
| [43,65] |
3.1. Low to Medium Resolution Fingerprinting Methods Based on PCR Analysis
3.1.1. DGGE/TGGE
3.1.2. T-RFLP
3.1.3. SSCP
3.1.4. ARISA/RISA
3.1.5. LH-PCR
3.1.6. RAPD
3.1.7. ARDRA
3.1.8. Q-PCR
3.2. Non-PCR Based Methods
3.2.1. CFE
3.2.2. PLFA
3.2.3. FDA
3.2.4. SIP
3.2.5. DNA Arrays
3.2.6. FISH
3.3. High-Throughput Sequencing Technologies
| Technology | Cost | Read length | Run time | Error rate | Output per run | Notes | Ref. |
|---|---|---|---|---|---|---|---|
| 454 | Low | Up to 1000 bp (GS FLX+). | 23 h | Low | 0.7 Gb |
| [152,153] |
| Illumina | Low | 2×100 bp | 3 to 11 days | Low | 120 Gb to 600 Gb |
| [152] |
| SOLiD | High | 50 bp | Up to 8 days | Moderate | 150 Gb |
| [152,154,155] |
| PGM | Moderate | 400 bp | 3 h | High | From 20 Mb to 400 Mb |
| [156,157,158] |
| HeliScope | High | 35 bp | 30 days | High | Over 1 Gb per day |
| [154,155] |
| SMRT | Low | 1100 bp | 2 h | High | 230 Gb |
| [159] |
3.3.1. Roche 454 FLX Pyrosequencer
3.3.2. Illumina Genome Analyzer
3.3.3. Applied Biosystems Sequencing by Oligonucleotide Ligation and Detection (SOLiD) Sequencer
3.3.4. Ion Personal Genome Machine (PGM)
3.3.5. Heliscope Single Molecule Sequencer
3.3.6. Pacific Biosciences SMRT DNA Sequencer
4. Choice of Methods and Complimentary Approaches
5. Conclusions
Acknowledgements
Conflict of Interest
References and Notes
- Evans, A. The feeding of the nine billion. Available online: http://www.chathamhouse.org/sites/default/files/public/Research/Energy,%20Environment%20and%20Development/r0109food.pdf (accessed on 6 May 2013).
- FAO. World agriculture: Towards 2030/2050. 2050. Available online: http://www.fao.org/fileadmin/user_upload/esag/docs/Interim_report_AT2050web.pdf (accessed on 6 May 2013).
- Herrick, J.E. Soil quality: An indicator of sustainable land management? Appl. Soil Ecol. 2000, 15, 75–83. [Google Scholar] [CrossRef]
- Karlen, D.L.; Mausbach, M.J.; Doran, J.W.; Cline, R.G.; Harris, R.F.; Schuman, G.E. Soil quality: A concept, definition, and framework for evaluation (a guest editorial). Soil Sci. Soc. Am. J. 1997, 61, 4–10. [Google Scholar] [CrossRef]
- Lal, R. Soil degradation by erosion. Land Degrad. Dev. 2001, 12, 519–539. [Google Scholar] [CrossRef]
- White, P.J.; Brown, P.H. Plant nutrition for sustainable development and global health. Ann. Bot. 2010, 105, 1073–1080. [Google Scholar] [CrossRef]
- Fageria, N.K.; Baligar, V.C.; Jones, C.A. Growth and Mineral Nutrition of Field Crops, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Compant, S.; Clément, C.; Sessitsch, A. Plant growth-promoting bacteria in the rhizo- and endosphere of plants: Their role, colonization, mechanisms involved and prospects for utilization. Soil Biol. Biochem. 2010, 42, 669–678. [Google Scholar] [CrossRef] [Green Version]
- Lugtenberg, B.; Kamilova, F. Plant-growth-promoting rhizobacteria. Ann. Rev. Microbiol. 2009, 63, 541–556. [Google Scholar] [CrossRef]
- Adesemoye, A.O.; Kloepper, J.W. Plant–microbes interactions in enhanced fertilizer-use efficiency. Appl. Microbiol. Biotechnol. 2009, 85, 1–12. [Google Scholar] [CrossRef]
- Yang, J.; Kloepper, J.W.; Ryu, C.M. Rhizosphere bacteria help plants tolerate abiotic stress. Trends Plant Sci. 2009, 14, 1–4. [Google Scholar] [CrossRef]
- Berendsen, R.L.; Pieterse, C.M.J.; Bakker, P.A.H.M. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef]
- Smalla, K.; Sessitsch, A.; Hartmann, A. The rhizosphere: “Soil compartment influenced by the root”. FEMS Microbiol. Ecol. 2006, 56, 165. [Google Scholar] [CrossRef]
- Dennis, P.G.; Miller, A.J.; Hirsch, P.R. Are root exudates more important than other sources of rhizodeposits in structuring rhizosphere bacterial communities? FEMS Microbiol. Ecol. 2010, 72, 313–327. [Google Scholar] [CrossRef]
- Berg, G.; Smalla, K. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol. Ecol. 2009, 68, 1–13. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K.; van Themaat, E.V.L.; Ahmadinejad, N.; Assenza, F.; Rauf, P.; Huettel, B.; Reinhardt, R.; Schmelzer, E.; et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef]
- Houlden, A.; Timms-Wilson, T.M.; Day, M.J.; Bailey, M.J. Influence of plant developmental stage on microbial community structure and activity in the rhizosphere of three field crops. FEMS Microbiol. Ecol. 2008, 65, 193–201. [Google Scholar] [CrossRef]
- Carvalhais, L.C.; Dennis, P.G.; Fedoseyenko, D.; Hajirezaei, M.R.; Borriss, R.; von Wirén, N. Root exudation of sugars, amino acids, and organic acids by maize as affected by nitrogen, phosphorus, potassium, and iron deficiency. J. Plant Nutr. Soil Sci. 2011, 174, 3–11. [Google Scholar] [CrossRef]
- Badri, D.V.; Vivanco, J.M. Regulation and function of root exudates. Plant Cell Environ. 2009, 32, 666–681. [Google Scholar] [CrossRef]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; del Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef]
- Mendes, R.; Kruijt, M.; de Bruijn, I.; Dekkers, E.; van der Voort, M.; Schneider, J.H.; Piceno, Y.M.; DeSantis, T.Z.; Andersen, G.L.; Bakker, P.A.; et al. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 2011, 332, 1097–1100. [Google Scholar] [CrossRef]
- Nihorimbere, V.; Ongena, M.; Smargiassi, M.; Thonart, P. Beneficial effect of the rhizosphere microbial community for plant growth and health. Biotechnol. Agron. Soc. Environ. 2011, 15, 327–337. [Google Scholar]
- Raaijmakers, J.M.; Paulitz, T.C.; Steinberg, C.; Alabouvette, C.; Moënne-Loccoz, Y. The rhizosphere: A playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant Soil 2009, 321, 341–361. [Google Scholar] [CrossRef]
- Vessey, J.K. Plant growth promoting rhizobacteria as biofertilizers. Plant Soil 2003, 255, 571–586. [Google Scholar] [CrossRef]
- Berg, G. Plant–microbe interactions promoting plant growth and health: Perspectives for controlled use of microorganisms in agriculture. Appl. Microbiol. Biotechnol. 2009, 84, 11–18. [Google Scholar] [CrossRef]
- Davies, P.J. The plant hormones: Their nature, occurrence, and functions. In Plant Hormones—Biosynthesis, Signal Transduction, Action, 3rd ed.; Davies, P.J., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2010; pp. 1–15. [Google Scholar]
- Lemanceau, P.; Mazurier, S.; Avoscan, L.; Robin, A.; Briat, J.f. Reciprocal interactions between plants and fluorescent Pseudomonas in relation to iron in the rhizosphere. In Molecular Microbial Ecology of the Rhizosphere; de Bruijn, F.J., Ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2013; pp. 1181–1189. [Google Scholar]
- Hadar, Y.; Papadopoulou, K.K. Suppressive composts: Microbial ecology links between abiotic environments and healthy plants. Ann. Rev. Phytopathol. 2012, 50, 133–153. [Google Scholar] [CrossRef]
- Damiani, I.; Baldacci-Cresp, F.; Hopkins, J.; Andrio, E.; Balzergue, S.; Lecomte, P.; Puppo, A.; Abad, P.; Favery, B.; Hérouart, D. Plant genes involved in harbouring symbiotic rhizobia or pathogenic nematodes. New Phytol. 2012, 194, 511–522. [Google Scholar] [CrossRef]
- Dechorgnat, J.; Patrit, O.; Krapp, A.; Fagard, M.; Daniel-Vedele, F. Characterization of the NRT2.6 gene in Arabidopsis thaliana: A link with plant response to biotic and abiotic stress. PLoS One 2012, 7, e42491. [Google Scholar]
- Hann, D.; Boller, T. Microbial effectors and their role in plant defense suppression. In Effectors in Plant-Microbe Interactions; Martin, F., Kamoun, S., Eds.; Wiley-Blackwell: Chichester, UK, 2012; pp. 33–52. [Google Scholar]
- López-Fuentes, E.; Ruíz-Valdiviezo, V.M.; Martínez-Romero, E.; Gutiérrez-Miceli, F.A.; Dendooven, L.; Rincón-Rosales, R. Bacterial community in the roots and rhizosphere of hypericum silenoides juss. 1804. Afr. J. Microbiol. Res. 2012, 6, 2704–2711. [Google Scholar]
- Doornbos, R.F.; Geraats, B.P.J.; Kuramae, E.E.; van Loon, L.; Bakker, P.A.H.M. Effects of jasmonic acid, ethylene, and salicylic acid signaling on the rhizosphere bacterial community of Arabidopsis thaliana. Mol. Plant-Microbe Interact. 2011, 24, 395–407. [Google Scholar] [CrossRef]
- Compant, S.; Mitter, B.; Colli-Mull, J.G.; Gangl, H.; Sessitsch, A. Endophytes of grapevine flowers, berries, and seeds: Identification of cultivable bacteria, comparison with other plant parts, and visualization of niches of colonization. Microb. Ecol. 2011, 62, 188–197. [Google Scholar] [CrossRef]
- Lecomte, J.; St-Arnaud, M.; Hijri, M. Isolation and identification of soil bacteria growing at the expense of arbuscular mycorrhizal fungi. FEMS Microbiol. Lett. 2011, 317, 43–51. [Google Scholar] [CrossRef]
- Jones, D.L.; Nguyen, C.; Finlay, R.D. Carbon flow in the rhizosphere: Carbon trading at the soil–root interface. Plant Soil 2009, 321, 5–33. [Google Scholar] [CrossRef]
- Marschner, P.; Crowley, D.; Rengel, Z. Rhizosphere interactions between microorganisms and plants govern iron and phosphorus acquisition along the root axis–model and research methods. Soil Biol. Biochem. 2011, 43, 883–894. [Google Scholar] [CrossRef]
- Pellegrini, A.; Corneo, P.E.; Camin, F.; Ziller, L.; Tosi, S.; Pertot, I. Studying trophic interactions between a plant pathogen and two different antagonistic microorganisms using a 13C-labeled compound and isotope ratio mass spectrometry. Rapid Commun. Mass Spectrom. 2012, 26, 510–516. [Google Scholar] [CrossRef]
- Opel, K.L.; Chung, D.; McCord, B.R. A study of PCR inhibition mechanisms using real time PCR. J. Forensic Sci. 2010, 55, 25–33. [Google Scholar] [CrossRef]
- Arbeli, Z.; Fuentes, C.L. Improved purification and PCR amplification of DNA from environmental samples. FEMS Microbiol. Lett. 2007, 272, 269–275. [Google Scholar] [CrossRef]
- Lakay, F.M.; Botha, A.; Prior, B.A. Comparative analysis of environmental DNA extraction and purification methods from different humic acid-rich soils. J. Appl. Microbiol. 2007, 102, 265–273. [Google Scholar] [CrossRef]
- Proshkin, S.; Rahmouni, A.R.; Mironov, A.; Nudler, E. Cooperation between translating ribosomes and RNA polymerase in transcription elongation. Science 2010, 328, 504–508. [Google Scholar] [CrossRef]
- Carvalhais, L.C.; Dennis, P.G.; Tyson, G.W.; Schenk, P.M. Application of metatranscriptomics to soil environments. J. Microbiol. Methods 2012, 91, 246–251. [Google Scholar] [CrossRef]
- Killham, K. Integrated soil management–moving towards globally sustainable agriculture. J. Agric. Sci.-Lond. 2011, 149, 29–36. [Google Scholar] [CrossRef]
- Alessi, D.S.; Walsh, D.M.; Fein, J.B. Uncertainties in determining microbial biomass C using the chloroform fumigation–extraction method. Chem. Geol. 2011, 280, 58–64. [Google Scholar] [CrossRef]
- Ocio, J.; Brookes, P. An evaluation of methods for measuring the microbial biomass in soils following recent additions of wheat straw and the characterization of the biomass that develops. Soil Biol. Biochem. 1990, 22, 685–694. [Google Scholar] [CrossRef]
- Kaur, A.; Chaudhary, A.; Kaur, A.; Choudhary, R.; Kaushik, R. Phospholipid fatty acid-a bioindicator of environment monitoring and assessment in soil ecosystem. Curr. Sci. Bangalore 2005, 89, 1103. [Google Scholar]
- Smith, C.J.; Osborn, A.M. Advantages and limitations of quantitative PCR (qPCR)-based approaches in microbial ecology. FEMS Microbiol. Ecol. 2009, 67, 6–20. [Google Scholar] [CrossRef]
- Kirk, J.L.; Beaudette, L.A.; Hart, M.; Moutoglis, P.; Klironomos, J.N.; Lee, H.; Trevors, J.T. Methods of studying soil microbial diversity. J. Microbiol. Methods 2004, 58, 169–188. [Google Scholar] [CrossRef]
- Konstantinos, K.V.; Panagiotis, P.; Antonios, V.T.; Agelos, P.; Argiris, N.V. PCR–SSCP: A method for the molecular analysis of genetic diseases. Mol. Biotechnol. 2008, 38, 155–163. [Google Scholar] [CrossRef]
- Nocker, A.; Burr, M.; Camper, A.K. Genotypic microbial community profiling: A critical technical review. Microb. Ecol. 2007, 54, 276–289. [Google Scholar] [CrossRef]
- Okubo, A.; Sugiyama, S. Comparison of molecular fingerprinting methods for analysis of soil microbial community structure. Ecol. Res. 2009, 24, 1399–1405. [Google Scholar] [CrossRef]
- Mills, D.E.K.; Entry, J.A.; Gillevet, P.M.; Mathee, K. Assessing microbial community diversity using amplicon length heterogeneity polymerase chain reaction. Soil Sci. Soc. Am. J. 2007, 71, 572–578. [Google Scholar] [CrossRef]
- Ritchie, N.J.; Schutter, M.E.; Dick, R.P.; Myrold, D.D. Use of length heterogeneity PCR and fatty acid methyl ester profiles to characterize microbial communities in soil. Appl. Environ. Microbiol. 2000, 66, 1668–1675. [Google Scholar] [CrossRef]
- Fritsch, P.; Rieseberg, L.H. The use of random amplified polymorphic DNA (RAPD) in conservation genetics. Mol. Genet. Approaches Conserv. 1996, 1996, 54–73. [Google Scholar]
- Newbury, J.; Ford-Lloyd, B. The use of RAPD for assessing variation in plants. Plant Growth Regul. 1993, 12, 43–51. [Google Scholar] [CrossRef]
- Moter, A.; Göbel, U.B. Fluorescence in situ hybridization (FISH) for direct visualization of microorganisms. J. Microbiol. Methods 2000, 41, 85–112. [Google Scholar] [CrossRef]
- Li, E.S.Y.; Liu, W.T. DNA microarray technology in microbial ecology studies-principle, applications and current limitations. Microbes Environ. 2003, 18, 175–187. [Google Scholar] [CrossRef]
- Everett, K.; Rees-George, J.; Pushparajah, I.; Janssen, B.; Luo, Z. Advantages and disadvantages of microarrays to study microbial population dynamics - a minireview. N. Z. Plant Prot. 2010, 63, 1–6. [Google Scholar]
- Manichanh, C.; Chapple, C.E.; Frangeul, L.; Gloux, K.; Guigo, R.; Dore, J. A comparison of random sequence reads versus 16S rDNA sequences for estimating the biodiversity of a metagenomic library. Nucleic Acids Res. 2008, 36, 5180–5188. [Google Scholar]
- Podar, M.; Abulencia, C.B.; Walcher, M.; Hutchison, D.; Zengler, K.; Garcia, J.A.; Holland, T.; Cotton, D.; Hauser, L.; Keller, M. Targeted access to the genomes of low-abundance organisms in complex microbial communities. Appl. Environ. Microbiol. 2007, 73, 3205–3214. [Google Scholar] [CrossRef]
- Adam, G.; Duncan, H. Development of a sensitive and rapid method for the measurement of total microbial activity using fluorescein diacetate (FDA) in a range of soils. Soil Biol. Biochem. 2001, 33, 943–951. [Google Scholar] [CrossRef]
- Green, V.; Stott, D.; Diack, M. Assay for fluorescein diacetate hydrolytic activity: Optimization for soil samples. Soil Biol. Biochem. 2006, 38, 693–701. [Google Scholar] [CrossRef]
- Dumont, M.G.; Murrell, J.C. Stable isotope probing—linking microbial identity to function. Nat. Rev. Microbiol. 2005, 3, 499–504. [Google Scholar] [CrossRef]
- Carvalhais, L.C.; Dennis, P.G.; Tyson, G.W.; Schenk, P.M. Rhizosphere metatranscriptomics: Challenges and opportunities. In Molecular Microbial Ecology of the Rhizosphere; de Bruijn, F.J., Ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2013; pp. 1137–1144. [Google Scholar]
- Ranjard, L.; Poly, F.; Nazaret, S. Monitoring complex bacterial communities using culture-independent molecular techniques: Application to soil environment. Res. Microbiol. 2000, 151, 167–177. [Google Scholar] [CrossRef]
- McGrath, K.C.; Thomas-Hall, S.R.; Cheng, C.T.; Leo, L.; Alexa, A.; Schmidt, S.; Schenk, P.M. Isolation and analysis of mRNA from environmental microbial communities. J. Microbiol. Methods 2008, 75, 172–176. [Google Scholar] [CrossRef]
- Dowd, S.E.; Sun, Y.; Secor, P.R.; Rhoads, D.D.; Wolcott, B.M.; James, G.A.; Wolcott, R.D. Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol. 2008, 8, 43. [Google Scholar] [CrossRef]
- Nübel, U.; Garcia-Pichel, F.; Muyzer, G. PCR primers to amplify 16S rRNA genes from cyanobacteria. Appl. Environ. Microbiol. 1997, 63, 3327–3332. [Google Scholar]
- Cleary, D.F.R.; Smalla, K.; Mendonca-Hagler, L.C.S.; Gomes, N.C.M. Assessment of variation in bacterial composition among microhabitats in a mangrove environment using DGGE fingerprints and barcoded pyrosequencing. PLoS One 2012, 7, e29380. [Google Scholar]
- Wakelin, S.; Mander, C.; Gerard, E.; Jansa, J.; Erb, A.; Young, S.; Condron, L.; O’Callaghan, M. Response of soil microbial communities to contrasted histories of phosphorus fertilisation in pastures. Appl. Soil Ecol. 2012, 61, 40–48. [Google Scholar] [CrossRef]
- Caliz, J.; Montserrat, G.; Martí, E.; Sierra, J.; Cruañas, R.; Garau, M.A.; Triadó-Margarit, X.; Vila, X. The exposition of a calcareous mediterranean soil to toxic concentrations of Cr, Cd and Pb produces changes in the microbiota mainly related to differential metal bioavailability. Chemosphere 2012, 89, 494–504. [Google Scholar] [CrossRef]
- Babin, D.; Ding, G.C.; Pronk, G.J.; Heister, K.; Kögel-Knabner, I.; Smalla, K. Metal oxides, clay minerals and charcoal determine the composition of microbial communities in matured artificial soils and their response to phenanthrene. FEMS Microbiol. Ecol. 2013. [Google Scholar] [CrossRef]
- Zhou, X.; Wu, F. P-coumaric acid influenced cucumber rhizosphere soil microbial communities and the growth of Fusarium oxysporum f. Sp. Cucumerinum owen. PLoS One 2012, 7, e48288. [Google Scholar] [CrossRef]
- Frerichs, J.; Oppermann, B.I.; Gwosdz, S.; Möller, I.; Herrmann, M.; Krüger, M. Microbial community changes at a terrestrial volcanic CO2 vent induced by soil acidification and anaerobic microhabitats within the soil column. FEMS Microbiol. Ecol. 2012, 84, 60–74. [Google Scholar]
- Carcer, D.A.; Martin, M.; Mackova, M.; Macek, T.; Karlson, U.; Rivilla, R. The introduction of genetically modified microorganisms designed for rhizoremediation induces changes on native bacteria in the rhizosphere but not in the surrounding soil. ISME J. 2007, 1, 215–223. [Google Scholar] [CrossRef]
- Osborn, A.M.; Moore, E.R.; Timmis, K.N. An evaluation of terminal-restriction fragment length polymorphism (T-RFLP) analysis for the study of microbial community structure and dynamics. Environ. Microbiol. 2000, 2, 39–50. [Google Scholar] [CrossRef]
- Dunbar, J.; Ticknor, L.O.; Kuske, C.R. Phylogenetic specificity and reproducibility and new method for analysis of terminal restriction fragment profiles of 16s rRNA genes from bacterial communities. Appl. Environ. Microbiol. 2001, 67, 190–197. [Google Scholar] [CrossRef]
- Aiken, J.T. Terminal restriction fragment length polymorphism for soil microbial community fingerprinting. Soil Sci. Soc. Am. J. 2011, 75, 102–111. [Google Scholar] [CrossRef]
- Tipayno, S.; Kim, C.-G.; Sa, T. T-RFLP analysis of structural changes in soil bacterial communities in response to metal and metalloid contamination and initial phytoremediation. Appl. Soil Ecol. 2012, 61, 137–146. [Google Scholar] [CrossRef]
- Gough, H.L.; Stahl, D.A. Microbial community structures in anoxic freshwater lake sediment along a metal contamination gradient. ISME J. 2011, 5, 543–558. [Google Scholar] [CrossRef]
- Hilton, S.; Bennett, A.J.; Keane, G.; Bending, G.D.; Chandler, D.; Stobart, R.; Mills, P. Impact of shortened crop rotation of oilseed rape on soil and rhizosphere microbial diversity in relation to yield decline. PLoS One 2013, 8, e59859. [Google Scholar]
- Toljander, J.F.; Lindahl, B.D.; Paul, L.R.; Elfstrand, M.; Finlay, R.D. Influence of arbuscular mycorrhizal mycelial exudates on soil bacterial growth and community structure. FEMS Microbiol. Ecol. 2007, 61, 295–304. [Google Scholar] [CrossRef]
- Stefanis, C.; Alexopoulos, A.; Voidarou, C.; Vavias, S.; Bezirtzoglou, E. Principal methods for isolation and identification of soil microbial communities. Folia Microbiol. 2013, 58, 61–68. [Google Scholar] [CrossRef]
- Rossmann, B.; Müller, H.; Smalla, K.; Mpiira, S.; Tumuhairwe, J.B.; Staver, C.; Berg, G. Banana-associated microbial communities in Uganda are highly diverse but dominated by Enterobacteriaceae. Appl. Environ. Microbiol. 2012, 78, 4933–4941. [Google Scholar] [CrossRef]
- Badin, A.L.; Mustafa, T.; Bertrand, C.; Monier, A.; Delolme, C.; Geremia, R.A.; Bedell, J.P. Microbial communities of urban stormwater sediments: The phylogenetic structure of bacterial communities varies with porosity. FEMS Microbiol. Ecol. 2012, 81, 324–338. [Google Scholar] [CrossRef]
- Gasser, I.; Müller, H.; Berg, G. Ecology and characterization of polyhydroxyalkanoate-producing microorganisms on and in plants. FEMS Microbiol. Ecol. 2009, 70, 142–150. [Google Scholar] [CrossRef]
- Zachow, C.; Berg, C.; Müller, H.; Meincke, R.; Komon-Zelazowska, M.; Druzhinina, I.S.; Kubicek, C.P.; Berg, G. Fungal diversity in the rhizosphere of endemic plant species of Tenerife (Canary Islands): Relationship to vegetation zones and environmental factors. ISME J. 2009, 3, 79–92. [Google Scholar] [CrossRef]
- Nai, Y.H.; Zemb, O.; Gutierrez-Zamora, M.L.; Manefield, M.; Powell, S.M.; Breadmore, M.C. Capillary electrophoresis ribosomal RNA single-stranded conformation polymorphism: A new approach for characterization of low-diversity microbial communities. Anal. Bioanal. Chem. 2012, 404, 1897–1906. [Google Scholar] [CrossRef]
- Fuhrman, J.A.; Steele, J.A.; Hewson, I.; Schwalbach, M.S.; Brown, M.V.; Green, J.L.; Brown, J.H. A latitudinal diversity gradient in planktonic marine bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 7774–7778. [Google Scholar]
- Danovaro, R.; Luna, G.M.; Dell’Anno, A.; Pietrangeli, B. Comparison of two fingerprinting techniques, terminal restriction fragment length polymorphism and automated ribosomal intergenic spacer analysis, for determination of bacterial diversity in aquatic environments. Appl. Environ.l Microbiol. 2006, 72, 5982–5989. [Google Scholar] [CrossRef]
- Sepehri, S.; Kotlowski, R.; Bernstein, C.N.; Krouse, D.O. Microbial diversity of inflamed and noninflamed gut biopsy tissues in inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 675–683. [Google Scholar] [CrossRef]
- Mathew, R.P.; Feng, Y.; Githinji, L.; Ankumah, R.; Balkcom, K.S. Impact of no-tillage and conventional tillage systems on soil microbial communities. Appl. Environ. Soil Sci. 2012. [Google Scholar] [CrossRef]
- Quilliam, R.S.; Marsden, K.A.; Gertler, C.; Rousk, J.; DeLuca, T.H.; Jones, D.L. Nutrient dynamics, microbial growth and weed emergence in biochar amended soil are influenced by time since application and reapplication rate. Agric. Ecosyst. Environ. 2012, 158, 192–199. [Google Scholar] [CrossRef]
- Pascal, J.; Pierre-Alain, M.; Virginie, N.; Toan, T.D. Utilization of microbial abundance and diversity as indicators of the origin of soil aggregates produced by earthworms. Soil Biol. Biochem. 2013, 57, 950–952. [Google Scholar] [CrossRef]
- Zancarini, A.; Mougel, C.; Voisin, A.-S.; Prudent, M.; Salon, C.; Munier-Jolain, N. Soil nitrogen availability and plant genotype modify the nutrition strategies of Medicago truncatula and the associated rhizosphere microbial communities. PLoS One 2012, 7, e47096. [Google Scholar]
- Baudoin, E.; Nazaret, S.; Mougel, C.; Ranjard, L.; Moënne-Loccoz, Y. Impact of inoculation with the phytostimulatory PGPR Azospirillum lipoferum crt1 on the genetic structure of the rhizobacterial community of field-grown maize. Soil Biol. Biochem. 2009, 41, 409–413. [Google Scholar] [CrossRef]
- Kovacs, A.; Yacoby, K.; Gophna, U. A systematic assessment of automated ribosomal intergenic spacer analysis (ARISA) as a tool for estimating bacterial richness. Res. Microbiol. 2010, 161, 192–197. [Google Scholar] [CrossRef]
- Chaudhary, D.R.; Saxena, J.; Lorenz, N.; Dick, L.K.; Dick, R.P. Microbial profiles of rhizosphere and bulk soil microbial communities of biofuel crops switchgrass (Panicum virgatum L.) and jatropha (Jatropha curcas L.). Appl. Environ. Soil Sci. 2012, 906864. [Google Scholar]
- Wallenius, K.; Rita, H.; Mikkonen, A.; Lappi, K.; Lindstrom, K.; Hartikainen, H.; Raateland, A.; Niemi, R.M. Effects of land use on the level, variation and spatial structure of soil enzyme activities and bacterial communities. Soil Biol. Biochem. 2011, 43, 1464–1473. [Google Scholar] [CrossRef]
- Wu, T.; Chellemi, D.O.; Graham, J.H.; Rosskopf, E.N. Assessment of fungal communities in soil and tomato roots subjected to diverse land and crop management systems. Soil Biol. Biochem. 2008, 40, 1967–1970. [Google Scholar] [CrossRef]
- Hadrys, H.; Balick, M.; Schierwater, B. Applications of random amplified polymorphic DNA (RAPD) in molecular ecology. Mol. Ecol. 1992, 1, 55–63. [Google Scholar] [CrossRef]
- Amorim, J.; Vidal, R.; Lacerda-Junior, G.; Dias, J.; Brendel, M.; Rezende, R.; Cascardo, J. A simple boiling-based DNA extraction for RAPD profiling of landfarm soil to provide representative metagenomic content. Genet. Mol. Res. 2012, 11, 182–189. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ying, Y.X.; Zhao, D.Y.; Jin, S.; Ding, W.L. Genetic diversity analysis on rhizosphere soil microbial population of Panax ginseng and Panax quinquefolium by RAPD. Chin. Tradit. Herb. Drugs 2010, 41, 1871–1875. [Google Scholar]
- Gao, Y.; Miao, C.Y.; Xia, J.; Mao, L.; Wang, Y.F.; Zhou, P. Plant diversity reduces the effect of multiple heavy metal pollution on soil enzyme activities and microbial community structure. Front. Environ. Sci. Eng. 2012, 6, 213–223. [Google Scholar] [CrossRef]
- Singh, S.K.; Rai, M.K.; Sahoo, L. An improved and efficient micropropagation of Eclipta alba through transverse thin cell layer culture and assessment of clonal fidelity using RAPD analysis. Ind. Crop. Prod. 2012, 37, 328–333. [Google Scholar] [CrossRef]
- Yang, M.-M.; Xu, L.-P.; Xue, Q.-Y.; Yang, J.-H.; Xu, Q.; Liu, H.-X.; Guo, J.-H. Screening potential bacterial biocontrol agents towards Phytophthora capsici in pepper. Eur. J. Plant Pathol. 2012, 134, 811–820. [Google Scholar] [CrossRef]
- Liu, G.-X.; Hu, P.; Zhang, W.; Wu, X.; Yang, X.; Chen, T.; Zhang, M.; Li, S.-W. Variations in soil culturable bacteria communities and biochemical characteristics in the Dongkemadi glacier forefield along a chronosequence. Folia Microbiol. 2012, 57, 485–494. [Google Scholar] [CrossRef]
- Guerrero-Molina, M.F.; Winik, B.C.; Pedraza, R.O. More than rhizosphere colonization of strawberry plants by Azospirillum brasilense. Appl. Soil Ecol. 2012, 61, 205–212. [Google Scholar] [CrossRef]
- Lee, M.S.; Do, J.O.; Park, M.S.; Jung, S.; Lee, K.H.; Bae, K.S.; Park, S.J.; Kim, S.B. Dominance of Lysobacter sp in the rhizosphere of two coastal sand dune plant species, Calystegia soldanella and Elymus mollis. Antonie van Leeuwenhoek 2006, 90, 19–27. [Google Scholar] [CrossRef]
- Hagerberg, D.; Manique, N.; Brandt, K.K.; Larsen, J.; Nybroe, O.; Olsson, S. Low concentration of copper inhibits colonization of soil by the arbuscular mycorrhizal fungus Glomus intraradices and changes the microbial community structure. Microb.Ecol. 2011, 61, 844–852. [Google Scholar] [CrossRef]
- Higuchi, R.; Fockler, C.; Dollinger, G.; Watson, R. Kinetic PCR analysis: Real-time monitoring of DNA amplification reactions. Biotechnology 1993, 11, 1026–1030. [Google Scholar] [CrossRef]
- Navarrete, A.A.; Kuramae, E.E.; Hollander, M.; Pijl, A.S.; van Veen, J.A.; Tsai, S.M. Acidobacterial community responses to agricultural management of soybean in Amazon forest soils. FEMS Microbiol. Ecol. 2013, 83, 607–621. [Google Scholar] [CrossRef]
- Ai, C.; Liang, G.; Sun, J.; Wang, X.; He, P.; Zhou, W. Different roles of rhizosphere effect and long-term fertilization in the activity and community structure of ammonia oxidizers in a calcareous fluvo-aquic soil. Soil Biol. Biochem. 2013, 57, 30–42. [Google Scholar] [CrossRef]
- Fierer, N.; Jackson, J.A.; Vilgalys, R.; Jackson, R.B. Assessment of soil microbial community structure by use of taxon-specific quantitative PCR assays. Appl. Environ. Microbiol. 2005, 71, 4117–4120. [Google Scholar] [CrossRef]
- Bacchetti De Gregoris, T.; Aldred, N.; Clare, A.S.; Burgess, J.G. Improvement of phylum- and class-specific primers for real-time PCR quantification of bacterial taxa. J. Microbiol. Methods 2011, 86, 351–356. [Google Scholar] [CrossRef]
- Heid, C.A.; Stevens, J.; Livak, K.J.; Williams, P.M. Real-time quantitative PCR. Genome Res. 1996, 6, 986–994. [Google Scholar] [CrossRef]
- McGrath, K.C.; Dombrecht, B.; Manners, J.M.; Schenk, P.M.; Edgar, C.I.; Maclean, D.J.; Scheible, W.R.; Udvardi, M.K.; Kazan, K. Repressor- and activator-type ethylene response factors functioning in jasmonate signaling and disease resistance identified via a genome-wide screen of Arabidopsis transcription factor gene expression. Plant Physiol. 2005, 139, 949–959. [Google Scholar] [CrossRef]
- Beck, T.; Joergensen, R.; Kandeler, E.; Makeschin, F.; Nuss, E.; Oberholzer, H.; Scheu, S. An inter-laboratory comparison of ten different ways of measuring soil microbial biomass C. Soil Biol. Biochem. 1997, 29, 1023–1032. [Google Scholar] [CrossRef]
- Brookes, P.; Landman, A.; Pruden, G.; Jenkinson, D. Chloroform fumigation and the release of soil nitrogen: A rapid direct extraction method to measure microbial biomass nitrogen in soil. Soil Biol. Biochem. 1985, 17, 837–842. [Google Scholar] [CrossRef]
- Joergensen, R.G. The fumigation-extraction method to estimate soil microbial biomass: Calibration of the kec value. Soil Biol. Biochem. 1996, 28, 25–31. [Google Scholar] [CrossRef]
- Setia, R.; Verma, S.L.; Marschner, P. Measuring microbial biomass carbon by direct extraction–comparison with chloroform fumigation-extraction. Eur. J. Soil Biol. 2012, 53, 103–106. [Google Scholar] [CrossRef]
- Philippot, L.; Ritz, K.; Pandard, P.; Hallin, S.; Martin-Laurent, F. Standardisation of methods in soil microbiology: Progress and challenges. FEMS Microbiol. Ecol. 2012, 82, 1–10. [Google Scholar] [CrossRef]
- Malik, A.; Blagodatskaya, E.; Gleixner, G. Soil microbial carbon turnover decreases with increasing molecular size. Soil Biol. Biochem. 2013, 62, 115–118. [Google Scholar] [CrossRef]
- Liu, Y.; Li, X.; Xing, Z.; Zhao, X.; Pan, Y. Responses of soil microbial biomass and community composition to biological soil crusts in the revegetated areas of the tengger desert. Appl. Soil Ecol. 2013, 65, 52–59. [Google Scholar] [CrossRef]
- Buyer, J.S.; Sasser, M. High throughput phospholipid fatty acid analysis of soils. Appl. Soil Ecol. 2012, 61, 127–130. [Google Scholar] [CrossRef]
- Chodak, M.; Gołębiewski, M.; Morawska-Płoskonka, J.; Kuduk, K.; Niklińska, M. Diversity of microorganisms from forest soils differently polluted with heavy metals. Appl. Soil Ecol. 2013, 64, 7–14. [Google Scholar] [CrossRef]
- Jiang, Y.; Sun, B.; Jin, C.; Wang, F. Soil aggregate stratification of nematodes and microbial communities affects the metabolic quotient in an acid soil. Soil Biol. Biochem. 2013, 60, 1–9. [Google Scholar] [CrossRef]
- Sipila, T.P.; Yrjala, K.; Alakukku, L.; Palojarvi, A. Cross-site soil microbial communities under tillage regimes: Fungistasis and microbial biomarkers. Appl. Environ. Microbiol. 2012, 78, 8191–8201. [Google Scholar] [CrossRef]
- Schurig, C.; Smittenberg, R.H.; Berger, J.; Kraft, F.; Woche, S.K.; Goebel, M.-O.; Heipieper, H.J.; Miltner, A.; Kaestner, M. Microbial cell-envelope fragments and the formation of soil organic matter: A case study from a glacier forefield. Biogeochemistry 2013, 113, 595–612. [Google Scholar]
- Frostegard, A.; Tunlid, A.; Baath, E. Use and misuse of PLFA measurements in soils. Soil Biolol. Biochem. 2011, 43, 1621–1625. [Google Scholar] [CrossRef]
- Swisher, R.; Carroll, G.C. Fluorescein diacetate hydrolysis as an estimator of microbial biomass on coniferous needle surfaces. Microb.Ecol. 1980, 6, 217–226. [Google Scholar] [CrossRef]
- Finkenbein, P.; Kretschmer, K.; Kuka, K.; Klotz, S.; Heilmeier, H. Soil enzyme activities as bioindicators for substrate quality in revegetation of a subtropical coal mining dump. Soil Biol. Biochem. 2013, 56, 87–89. [Google Scholar] [CrossRef]
- Zumsteg, A.; Bååth, E.; Stierli, B.; Zeyer, J.; Frey, B. Bacterial and fungal community responses to reciprocal soil transfer along a temperature and soil moisture gradient in a glacier forefield. Soil Biol. Biochem. 2013, 61, 121–132. [Google Scholar] [CrossRef]
- Bhattacharyya, P.; Roy, K.; Neogi, S.; Manna, M.; Adhya, T.; Rao, K.; Nayak, A. Influence of elevated carbon dioxide and temperature on belowground carbon allocation and enzyme activities in tropical flooded soil planted with rice. Environ. Monit. Assess. 2013, 1–13. [Google Scholar]
- Radajewski, S.; Ineson, P.; Parekh, N.R.; Murrell, J.C. Stable-isotope probing as a tool in microbial ecology. Nature 2000, 403, 646–649. [Google Scholar] [CrossRef]
- Lu, Y.; Abraham, W.R.; Conrad, R. Spatial variation of active microbiota in the rice rhizosphere revealed by in situ stable isotope probing of phospholipid fatty acids. Environ. Microbiol. 2007, 9, 474–481. [Google Scholar] [CrossRef]
- Bodé, S.; Fancy, R.; Boeckx, P. Stable isotope probing of amino sugars–a promising tool to assess microbial interactions in soils. Rapid Commun. Mass Spectrom. 2013, 27, 1367–1379. [Google Scholar] [CrossRef]
- Zhou, J.; Kang, S.; Schadt, C.W.; Garten, C.T., Jr. Spatial scaling of functional gene diversity across various microbial taxa. Proc. Natl. Acad. Sci. USA 2008, 105, 7768–7773. [Google Scholar]
- He, Z.; Gentry, T.J.; Schadt, C.W.; Wu, L.; Liebich, J.; Chong, S.C.; Huang, Z.; Wu, W.; Gu, B.; Jardine, P.; et al. Geochip: A comprehensive microarray for investigating biogeochemical, ecological and environmental processes. ISME J. 2007, 1, 67–77. [Google Scholar] [CrossRef]
- Yergeau, E.; Kang, S.; He, Z.; Zhou, J.; Kowalchuk, G.A. Functional microarray analysis of nitrogen and carbon cycling genes across an antarctic latitudinal transect. ISME J. 2007, 1, 163–179. [Google Scholar] [CrossRef]
- Fan, B.; Carvalhais, L.C.; Becker, A.; Fedoseyenko, D.; von Wiren, N.; Borriss, R. Transcriptomic profiling of Bacillus amyloliquefaciens fzb42 in response to maize root exudates. BMC Microbiol. 2012, 12, 116. [Google Scholar] [CrossRef]
- Hayden, H.L.; Mele, P.M.; Bougoure, D.S.; Allan, C.Y.; Norng, S.; Piceno, Y.M.; Brodie, E.L.; DeSantis, T.Z.; Andersen, G.L.; Williams, A.L.; et al. Changes in the microbial community structure of bacteria, archaea and fungi in response to elevated CO2 and warming in an Australian native grassland soil. Environ. Microbiol. 2012, 14, 3081–3096. [Google Scholar] [CrossRef]
- Russo, S.E.; Legge, R.; Weber, K.A.; Brodie, E.L.; Goldfarb, K.C.; Benson, A.K.; Tan, S. Bacterial community structure of contrasting soils underlying bornean rain forests: Inferences from microarray and next-generation sequencing methods. Soil Biol. Biochem. 2012, 55, 48–59. [Google Scholar] [CrossRef]
- Cruz-Martinez, K.; Rosling, A.; Zhang, Y.; Song, M.Z.; Andersen, G.L.; Banfield, J.F. Effect of rainfall-induced soil geochemistry dynamics on grassland soil microbial communities. Appl. Environ. Microbiol. 2012, 78, 7587–7595. [Google Scholar] [CrossRef]
- Zysko, A.; Sanguin, H.; Hayes, A.; Wardleworth, L.; Zeef, L.A.H.; Sim, A.; Paterson, E.; Singh, B.K.; Kertesz, M.A. Transcriptional response of Pseudomonas aeruginosa to a phosphate-deficient lolium perenne rhizosphere. Plant Soil 2012, 359, 25–44. [Google Scholar] [CrossRef]
- Khudyakov, J.I.; D’haeseleer, P.; Borglin, S.E.; DeAngelis, K.M.; Woo, H.; Lindquist, E.A.; Hazen, T.C.; Simmons, B.A.; Thelen, M.P. Global transcriptome response to ionic liquid by a tropical rain forest soil bacterium, Enterobacter lignolyticus. Proc. Natl. Acad. Sci. USA 2012, 109, E2173–E2182. [Google Scholar] [CrossRef]
- He, Z.; Deng, Y.; van Nostrand, J.D.; Tu, Q.; Xu, M.; Hemme, C.L.; Li, X.; Wu, L.; Gentry, T.J.; Yin, Y.; et al. Geochip 3.0 as a high-throughput tool for analyzing microbial community composition, structure and functional activity. ISME J. 2010, 4, 1167–1179. [Google Scholar] [CrossRef]
- Waldron, P.J.; Wu, L.; van Nostrand, J.O.Y.D.; Schadt, C.W.; He, Z.; Watson, D.B.; Jardine, P.M.; Palumbo, A.V.; Hazen, T.C.; Zhou, J. Functional gene array-based analysis of microbial community structure in groundwaters with a gradient of contaminant levels. Environtal Sci. Technol. 2009, 43, 3529–3534. [Google Scholar] [CrossRef]
- Wu, L.; Kellogg, L.; Devol, A.H.; Tiedje, J.M.; Zhou, J. Microarray-based characterization of microbial community functional structure and heterogeneity in marine sediments from the gulf of Mexico. Appl. Environ. Microbiol. 2008, 74, 4516–4529. [Google Scholar] [CrossRef]
- Simon, C.; Daniel, R. Metagenomic analyses: Past and future trends. Appl. Environ. Microbiol. 2011, 77, 1153–1161. [Google Scholar] [CrossRef]
- Loman, N.J.; Constantinidou, C.; Chan, J.Z.; Halachev, M.; Sergeant, M.; Penn, C.W.; Robinson, E.R.; Pallen, M.J. High-throughput bacterial genome sequencing: An embarrassment of choice, a world of opportunity. Nat. Rev. Microbiol. 2012, 10, 599–206. [Google Scholar] [CrossRef]
- Mardis, E.R. Next-generation DNA sequencing methods. Ann. Rev. Genomics Hum. Genet. 2008, 9, 387–402. [Google Scholar] [CrossRef]
- Shokralla, S.; Spall, J.L.; Gibson, J.F.; Hajibabaei, M. Next-generation sequencing technologies for environmental DNA research. Mol. Ecol. 2012, 21, 1794–1805. [Google Scholar] [CrossRef]
- Magi, A.; Benelli, M.; Gozzini, A.; Girolami, F.; Torricelli, F.; Brandi, M.L. Bioinformatics for next generation sequencing data. Genes 2010, 1, 294–307. [Google Scholar] [CrossRef]
- Loman, N.J.; Misra, R.V.; Dallman, T.J.; Constantinidou, C.; Gharbia, S.E.; Wain, J.; Pallen, M.J. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 2012, 30, 434–439. [Google Scholar] [CrossRef] [Green Version]
- Egan, A.N.; Schlueter, J.; Spooner, D.M. Applications of next-generation sequencing in plant biology. Am. J. Bot. 2012, 99, 175–185. [Google Scholar] [CrossRef]
- Diaz-Sanchez, S.; Hanning, I.; Pendleton, S.; D’Souza, D. Next-generation sequencing: The future of molecular genetics in poultry production and food safety. Poult. Sci. 2013, 92, 562–572. [Google Scholar] [CrossRef]
- Schadt, E.E.; Turner, S.; Kasarskis, A. A window into third-generation sequencing. Hum. Mol. Genet. 2010, 19, R227–R240. [Google Scholar] [CrossRef]
- Carvalhais, L.C.; Dennis, P.G.; Badri, D.V.; Tyson, G.W.; Vivanco, J.M.; Schenk, P.M. Activation of the jasmonic acid plant defence pathway alters the composition of rhizosphere bacterial communities. PLoS One 2013, 8, e56457. [Google Scholar]
- Dohrmann, A.B.; Kuting, M.; Junemann, S.; Jaenicke, S.; Schluter, A.; Tebbe, C.C. Importance of rare taxa for bacterial diversity in the rhizosphere of Bt- and conventional maize varieties. ISME J. 2013, 7, 37–49. [Google Scholar] [CrossRef]
- Lami, R.; Jones, L.C.; Cottrell, M.T.; Lafferty, B.J.; Ginder-Vogel, M.; Sparks, D.L.; Kirchman, D.L. Arsenite modifies structure of soil microbial communities and arsenite oxidization potential. FEMS Microbiol. Ecol. 2013, 84, 270–279. [Google Scholar] [CrossRef]
- Eilers, K.G.; Debenport, S.; Anderson, S.; Fierer, N. Digging deeper to find unique microbial communities: The strong effect of depth on the structure of bacterial and archaeal communities in soil. Soil Biol. Biochem. 2012, 50, 58–65. [Google Scholar] [CrossRef]
- Sutton, N.B.; Maphosa, F.; Morillo, J.A.; Al-Soud, W.A.; Langenhoff, A.A.M.; Grotenhuis, T.; Rijnaarts, H.H.M.; Smidt, H. Impact of long-term diesel contamination on soil microbial community structure. Appl. Environ. Microbiol. 2013, 79, 619–630. [Google Scholar] [CrossRef]
- Suleiman, A.; Manoeli, L.; Boldo, J.; Pereira, M.; Roesch, L. Shifts in soil bacterial community after eight years of land-use change. Syst. Appl. Microbiol. 2013, 36, 137–144. [Google Scholar] [CrossRef]
- Li, R.; Khafipour, E.; Krause, D.O.; Entz, M.H.; de Kievit, T.R.; Fernando, W.D. Pyrosequencing reveals the influence of organic and conventional farming systems on bacterial communities. PLoS One 2012, 7, e51897. [Google Scholar]
- Sequencing systems. Available online: http://www.illumina.com/systems/sequencing.ilmn (accessed on 6 May 2013).
- Fierer, N.; Leff, J.W.; Adams, B.J.; Nielsen, U.N.; Bates, S.T.; Lauber, C.L.; Owens, S.; Gilbert, J.A.; Wall, D.H.; Caporaso, J.G. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl. Acad. Sci. USA 2012, 109, 21390–21395. [Google Scholar] [CrossRef]
- Bartram, A.K.; Lynch, M.D.J.; Stearns, J.C.; Moreno-Hagelsieb, G.; Neufeld, J.D. Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end Illumina reads. Appl. Environ. Microbiol. 2011, 77, 3846–3852. [Google Scholar] [CrossRef]
- Dorr de Quadros, P.; Zhalnina, K.; Davis-Richardson, A.; Fagen, J.R.; Drew, J.; Bayer, C.; Camargo, F.A.; Triplett, E.W. The effect of tillage system and crop rotation on soil microbial diversity and composition in a subtropical Acrisol. Diversity 2012, 4, 375–395. [Google Scholar] [CrossRef]
- McGuire, K.L.; Payne, S.G.; Palmer, M.I.; Gillikin, C.M.; Keefe, D.; Kim, S.J.; Gedallovich, S.M.; Discenza, J.; Rangamannar, R.; Koshner, J.A.; et al. Digging the new york city skyline: Soil fungal communities in green roofs and city parks. PLoS One 2013, 8, e58020. [Google Scholar] [CrossRef]
- Uroz, S.; Ioannidis, P.; Lengelle, J.; Cébron, A.; Morin, E.; Buée, M.; Martin, F. Functional assays and metagenomic analyses reveals differences between the microbial communities inhabiting the soil horizons of a norway spruce plantation. PLoS One 2013, 8, e55929. [Google Scholar]
- Koskinen, J.P.; Laine, P.; Niemi, O.; Nykyri, J.; Harjunpää, H.; Auvinen, P.; Paulin, L.; Pirhonen, M.; Palva, T.; Holm, L. Genome sequence of Pectobacterium sp. Strain SCC3193. J. Bacteriol. 2012, 194, 6004–6004. [Google Scholar] [CrossRef]
- Whiteley, A.S.; Jenkins, S.; Waite, I.; Kresoje, N.; Payne, H.; Mullan, B.; Allcock, R.; O’Donnell, A. Microbial 16S rRNA ion tag and community metagenome sequencing using the Ion Torrent (PGM) platform. J. Microbiol. Methods 2012, 91, 80–88. [Google Scholar] [CrossRef]
- Bell, T.H.; Yergeau, E.; Juck, D.; Whyte, L.; Greer, C. Alteration of microbial community structure affects diesel biodegradation in an arctic soil. FEMS Microbiol. Ecol. 2013, 85, 51–61. [Google Scholar] [CrossRef]
- Bell, T.H.; Yergeau, E.; Maynard, C.; Juck, D.; Whyte, L.G.; Greer, C.W. Predictable bacterial composition and hydrocarbon degradation in arctic soils following diesel and nutrient disturbance. ISME J. 2013, 7, 1200–1210. [Google Scholar] [CrossRef]
- Kapranov, P.; Ozsolak, F.; Milos, P.M. Profiling of short RNAs using helicos single-molecule sequencing. In Next-Generation microRNA Expression Profiling Technology; Springer-Verlag: Berlin, Germany, 2012; pp. 219–232. [Google Scholar]
- Myllykangas, S.; Buenrostro, J.; Ji, H.P. Overview of sequencing technology platforms. In Bioinformatics for High Throughput Sequencing; Springer-Verlag: Berlin, Germany, 2012; pp. 11–25. [Google Scholar]
- English, A.C.; Richards, S.; Han, Y.; Wang, M.; Vee, V.; Qu, J.; Qin, X.; Muzny, D.M.; Reid, J.G.; Worley, K.C.; et al. Mind the gap: Upgrading genomes with pacific biosciences RS long-read sequencing technology. PLoS One 2012, 7, e47768. [Google Scholar] [CrossRef]
- Chin, C.-S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Maron, L.G.; Guimarães, C.T.; Kirst, M.; Albert, P.S.; Birchler, J.A.; Bradbury, P.J.; Buckler, E.S.; Coluccio, A.E.; Danilova, T.V.; Kudrna, D.; et al. Aluminum tolerance in maize is associated with higher MATE1 gene copy number. Proc. Natl. Acad. Sci. USA 2013, 110, 5241–5246. [Google Scholar] [CrossRef]
- Bradford, M.A.; Newington, J.E. With the worms: Soil biodiversity and ecosystem functioning. Biologist 2002, 49, 127–130. [Google Scholar]
- Lavelle, P.; Decaëns, T.; Aubert, M.; Barot, S.; Blouin, M.; Bureau, F.; Margerie, P.; Mora, P.; Rossi, J.-P. Soil invertebrates and ecosystem services. Eur. J. Soil Biol. 2006, 42, S3–S15. [Google Scholar] [CrossRef]
- Barrios, E. Soil biota, ecosystem services and land productivity. Ecol. Econ. 2007, 64, 269–285. [Google Scholar] [CrossRef]
- Dominati, E.; Patterson, M.; Mackay, A. A framework for classifying and quantifying the natural capital and ecosystem services of soils. Ecol. Econ. 2010, 69, 1858–1868. [Google Scholar] [CrossRef]
- Xu, L.; Ravnskov, S.; Larsen, J.; Nilsson, R.H.; Nicolaisen, M. Soil fungal community structure along a soil health gradient in pea fields examined using deep amplicon sequencing. Soil Biol. Biochem. 2012, 46, 26–32. [Google Scholar] [CrossRef]
- Avidano, L.; Gamalero, E.; Cossa, G.P.; Carraro, E. Characterization of soil health in an Italian polluted site by using microorganisms as bioindicators. Appl. Soil Ecol. 2005, 30, 21–33. [Google Scholar] [CrossRef]
- Garside, A.; Bell, M.; Robotham, B.; Magarey, R.; Stirling, G. Managing yield decline in sugarcane cropping systems. Int. Sugar J. 2005, 107, 16–26. [Google Scholar]
- Pietramellara, G.; Ascher, J.; Borgogni, F.; Ceccherini, M.; Guerri, G.; Nannipieri, P. Extracellular DNA in soil and sediment: Fate and ecological relevance. Biol. Fertil. Soils 2009, 45, 219–235. [Google Scholar] [CrossRef]
- Gözdereliler, E.; Boon, N.; Aamand, J.; de Roy, K.; Granitsiotis, M.S.; Albrechtsen, H.-J.; Sørensen, S.R. Comparing metabolic functionalities, community structures, and dynamics of herbicide-degrading communities cultivated with different substrate concentrations. Appl. Environ. Microbiol. 2013, 79, 367–375. [Google Scholar] [CrossRef]
- Drigo, B.; Pijl, A.S.; Duyts, H.; Kielak, A.M.; Gamper, H.A.; Houtekamer, M.J.; Boschker, H.T.; Bodelier, P.L.; Whiteley, A.S.; van Veen, J.A.; et al. Shifting carbon flow from roots into associated microbial communities in response to elevated atmospheric CO2. Proc. Natl. Acad. Sci. USA 2010, 107, 10938–10942. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rincon-Florez, V.A.; Carvalhais, L.C.; Schenk, P.M. Culture-Independent Molecular Tools for Soil and Rhizosphere Microbiology. Diversity 2013, 5, 581-612. https://doi.org/10.3390/d5030581
Rincon-Florez VA, Carvalhais LC, Schenk PM. Culture-Independent Molecular Tools for Soil and Rhizosphere Microbiology. Diversity. 2013; 5(3):581-612. https://doi.org/10.3390/d5030581
Chicago/Turabian StyleRincon-Florez, Vivian A., Lilia C. Carvalhais, and Peer M. Schenk. 2013. "Culture-Independent Molecular Tools for Soil and Rhizosphere Microbiology" Diversity 5, no. 3: 581-612. https://doi.org/10.3390/d5030581