Unexpected Binding Mode of a Potent Indeno[1,2-b]indole-Type Inhibitor of Protein Kinase CK2 Revealed by Complex Structures with the Catalytic Subunit CK2α and Its Paralog CK2α′

 , ,
, ,  , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
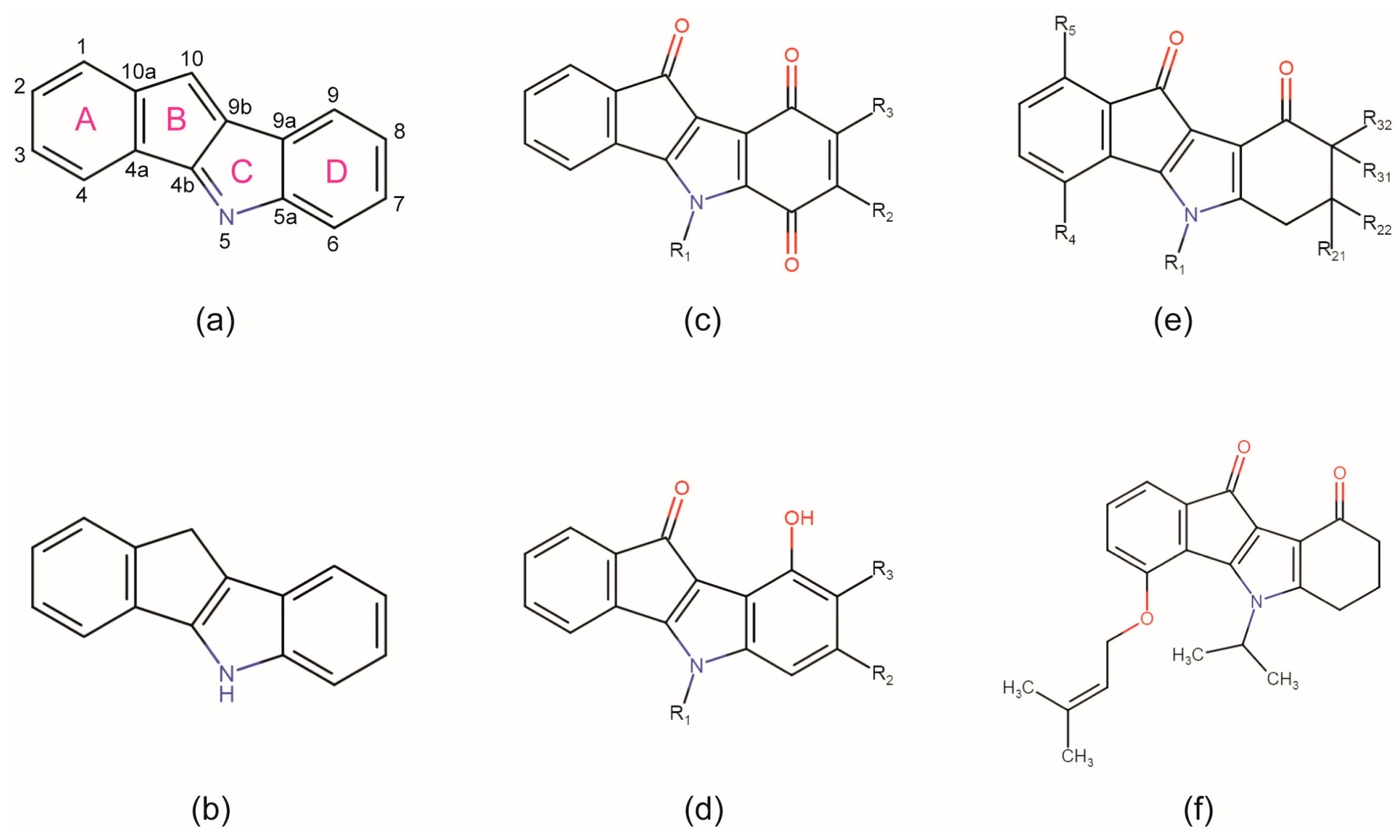
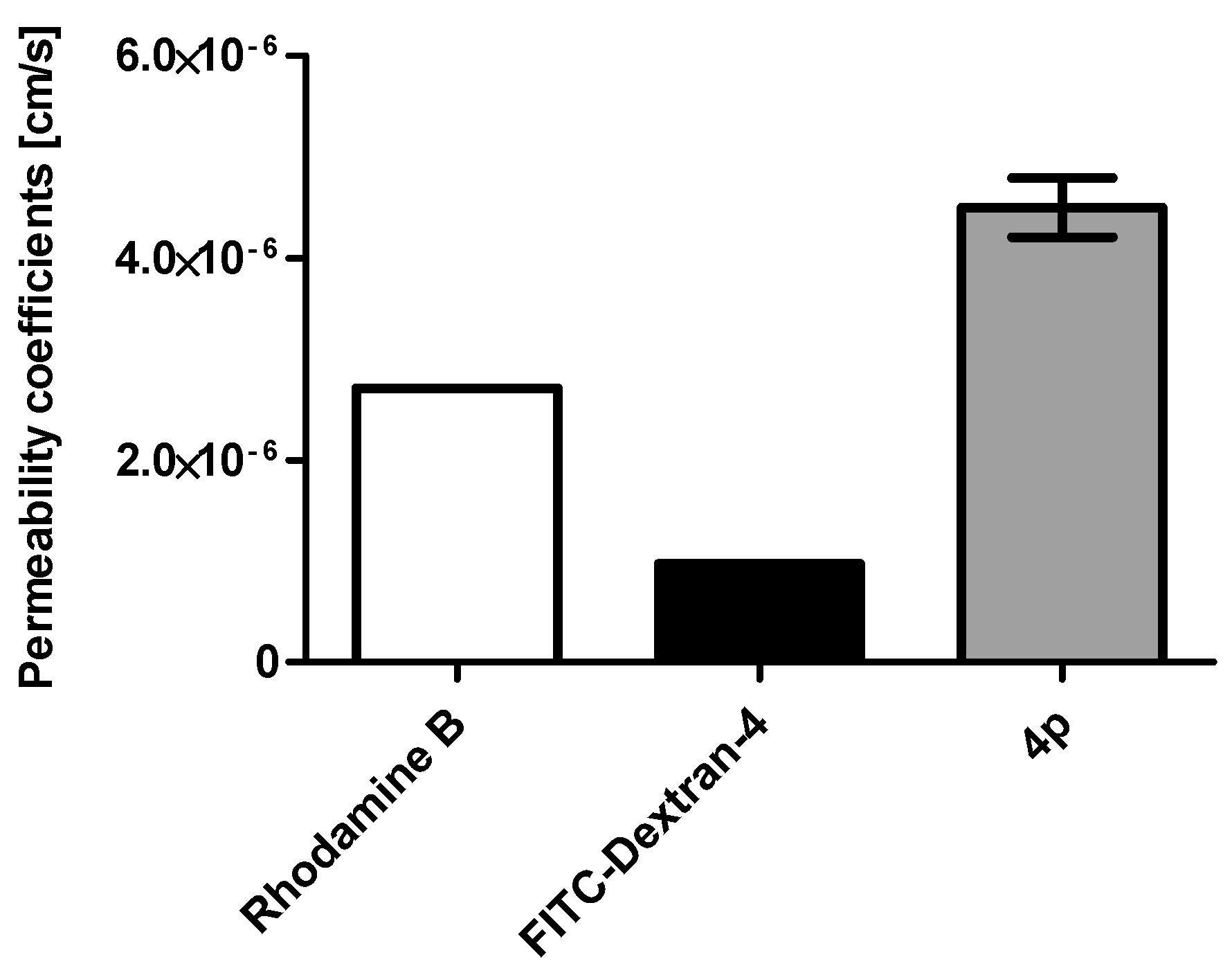
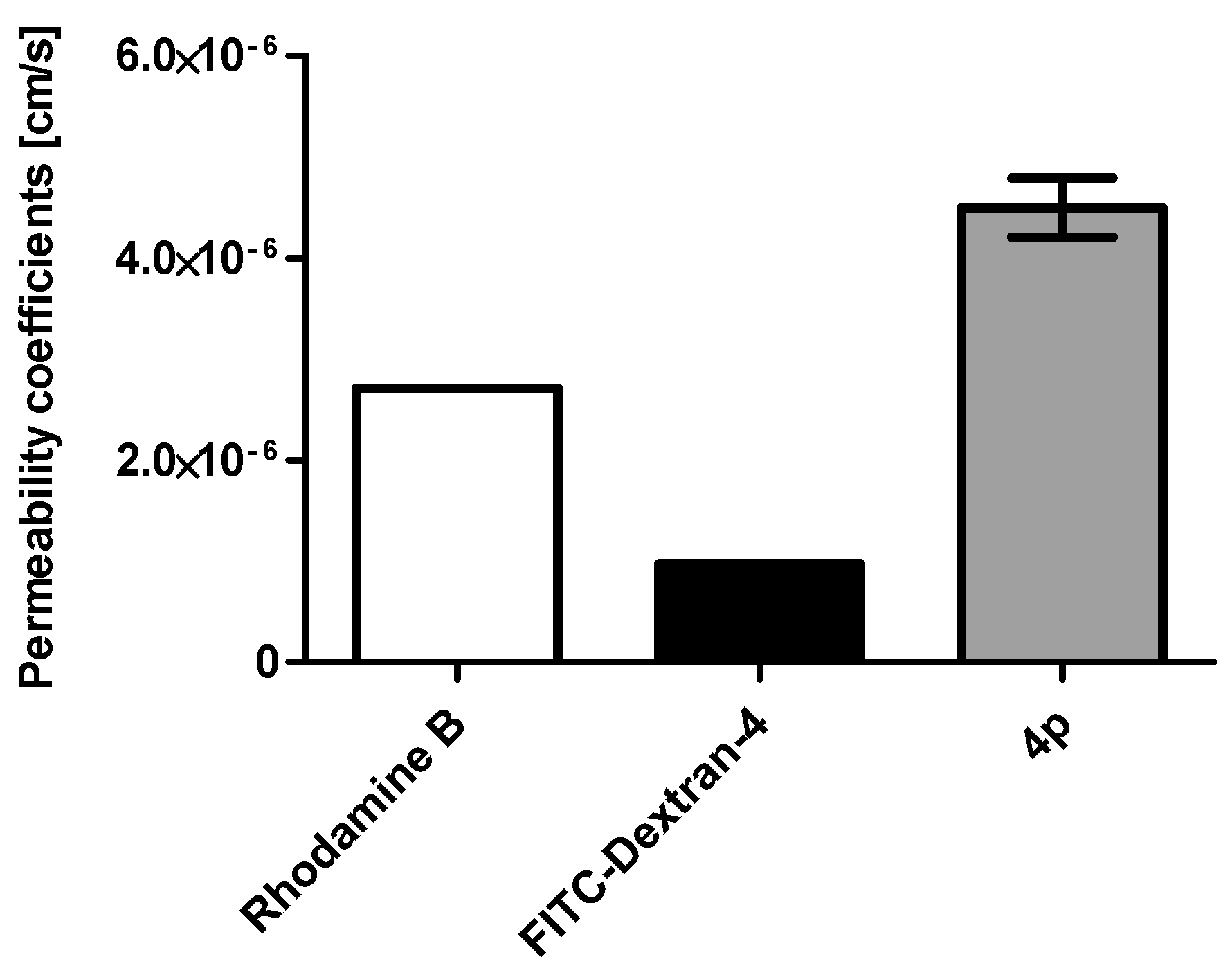
2.1. Membrane Permeability of the Indeno[1,2-b] Indole Compound 4p
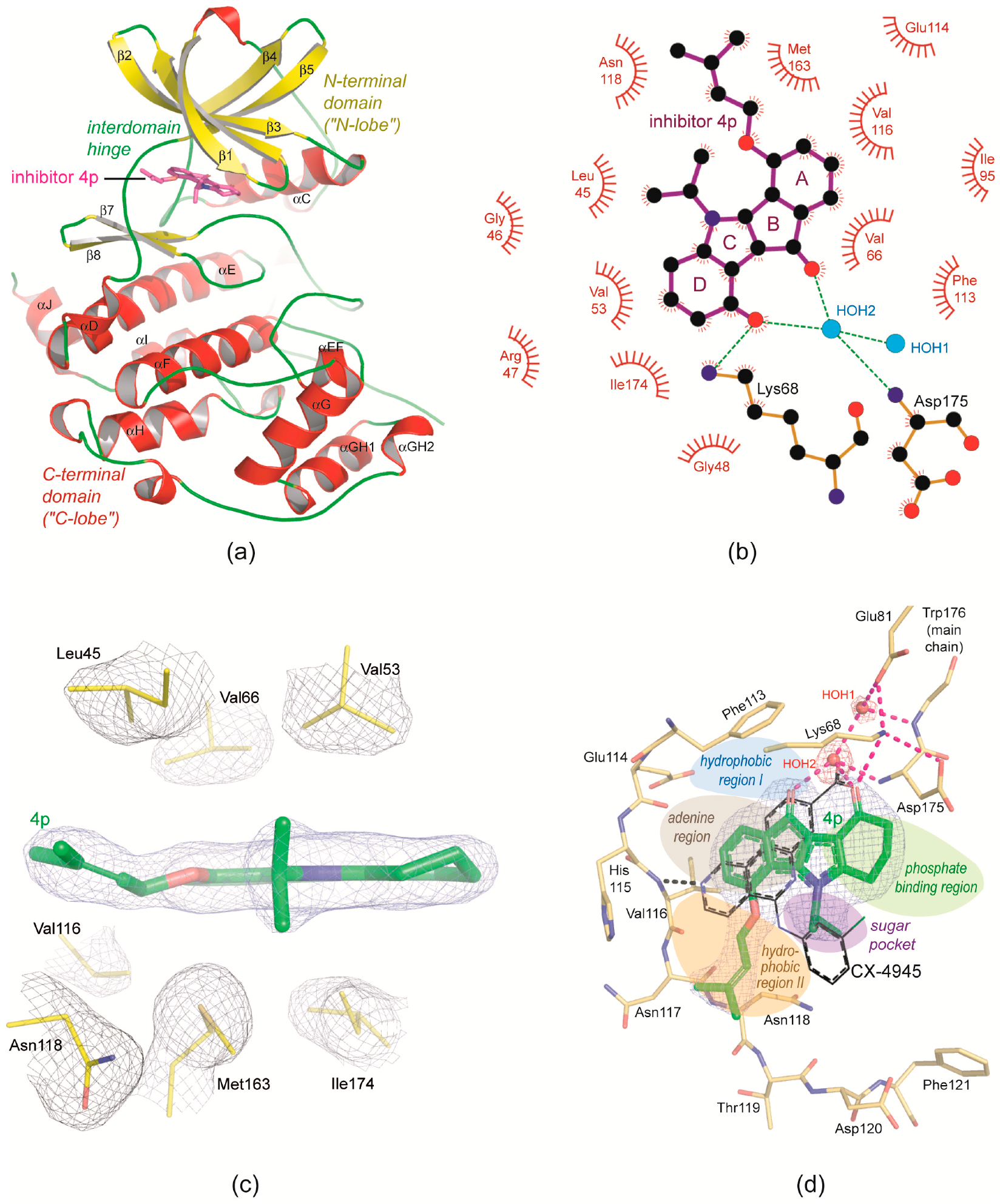
2.2. Overview of CK2α/CK2α′ Co-Crystal Structures with the Inhibitor 4p
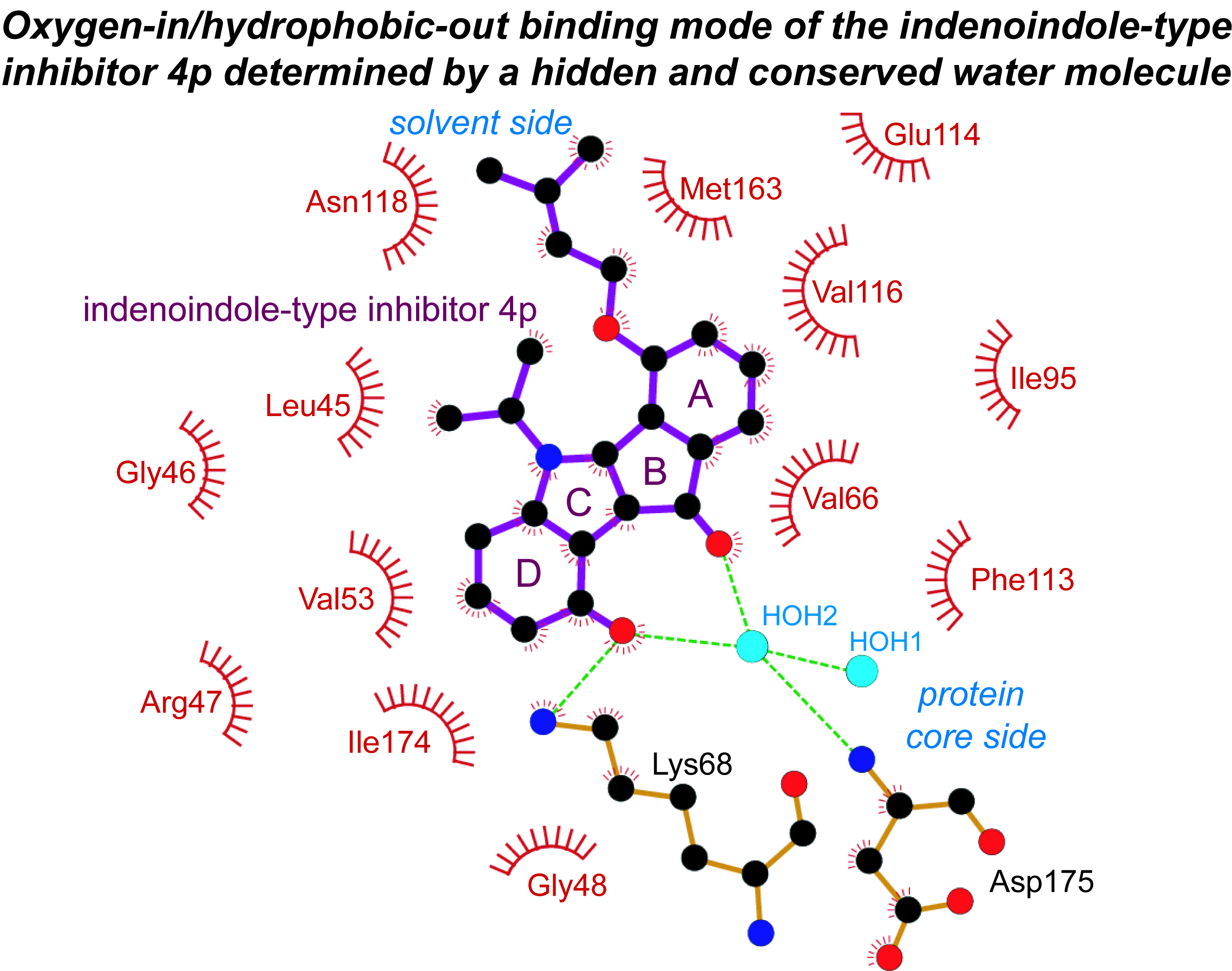
2.3. Principle Binding Mode of 4p to the ATP-Site of CK2α/CK2α′
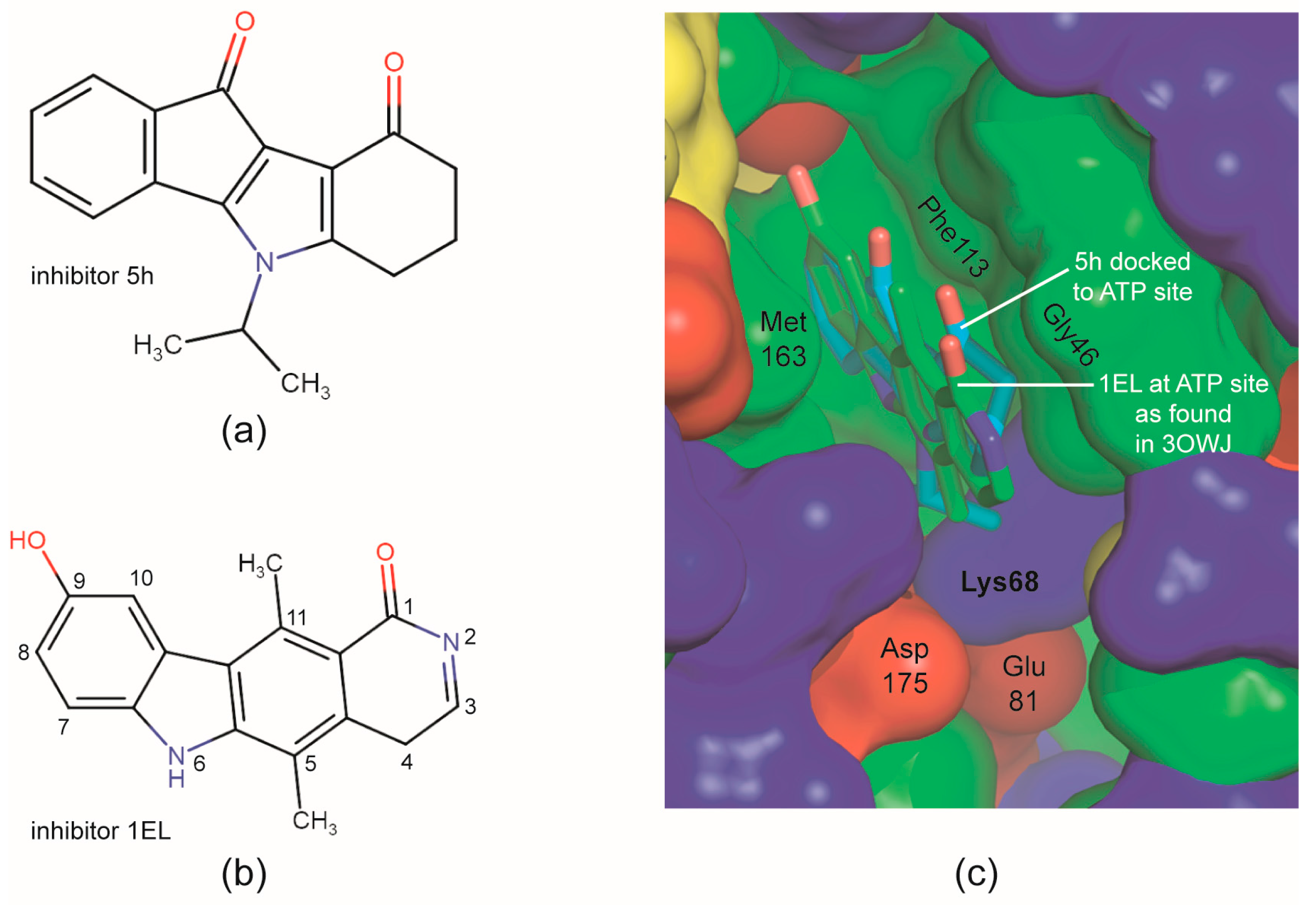
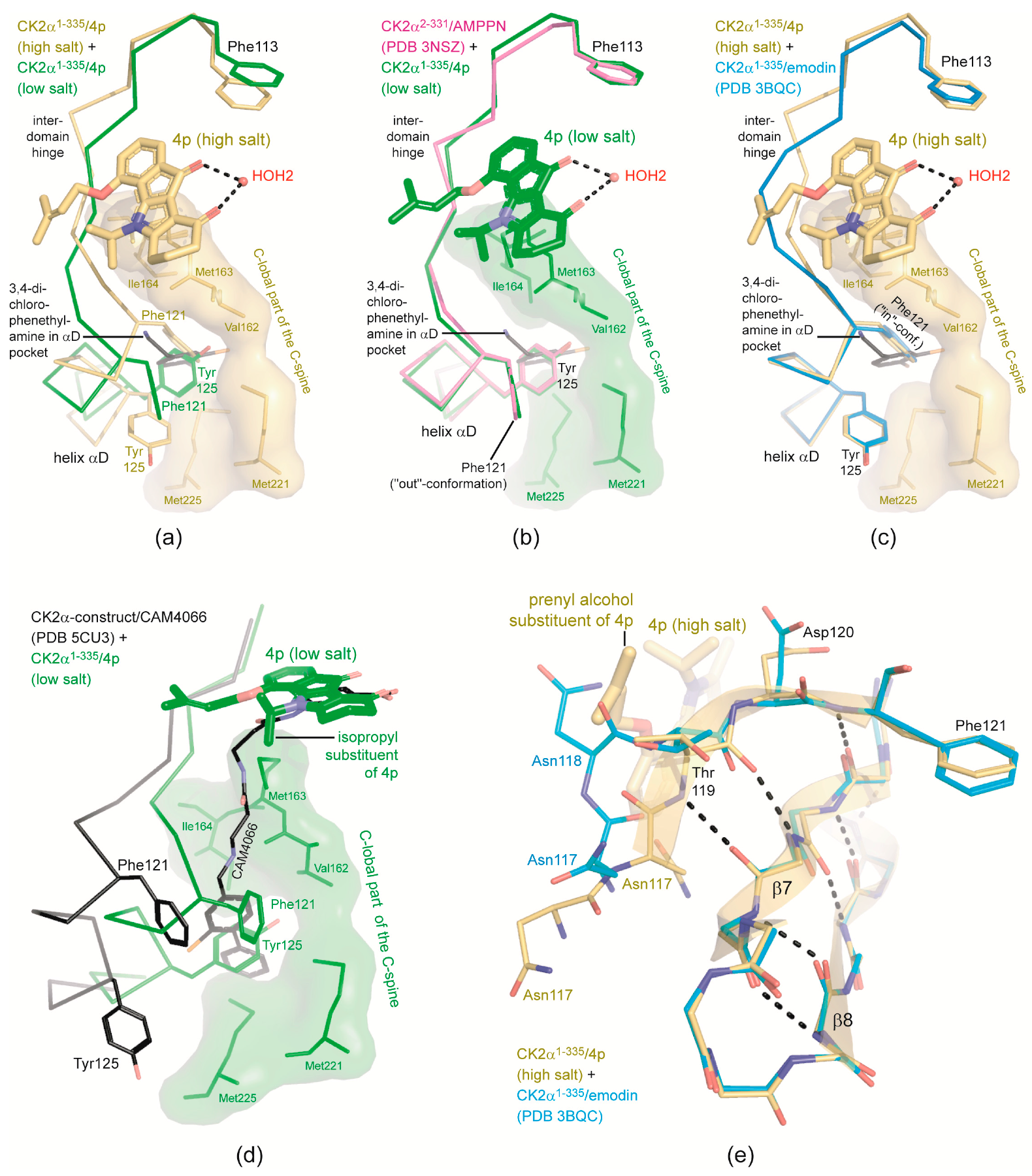
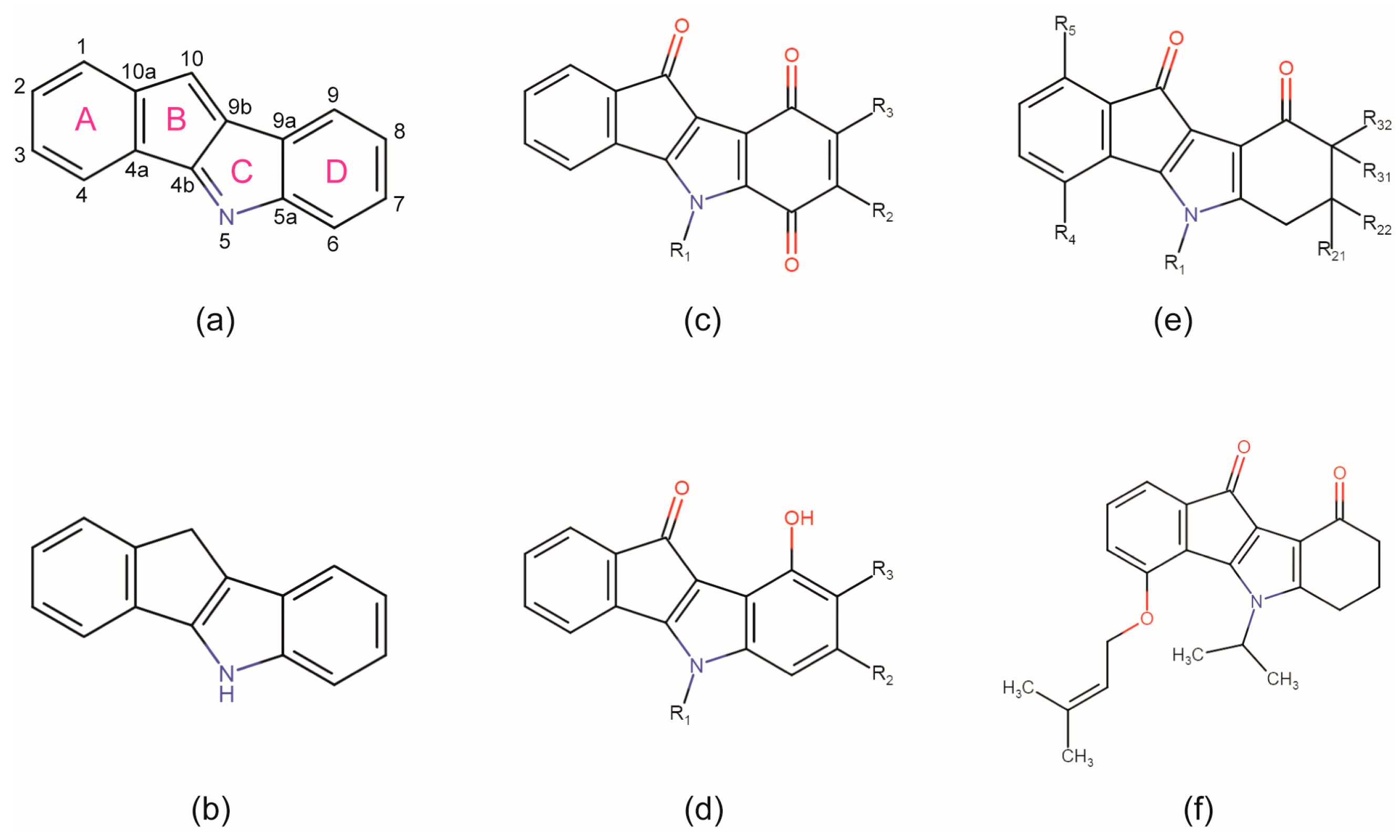
2.4. Is the CK2 Binding Mode of 4p Representative for Indeno[1,2-b]indole-Type CK2 Inhibitors?
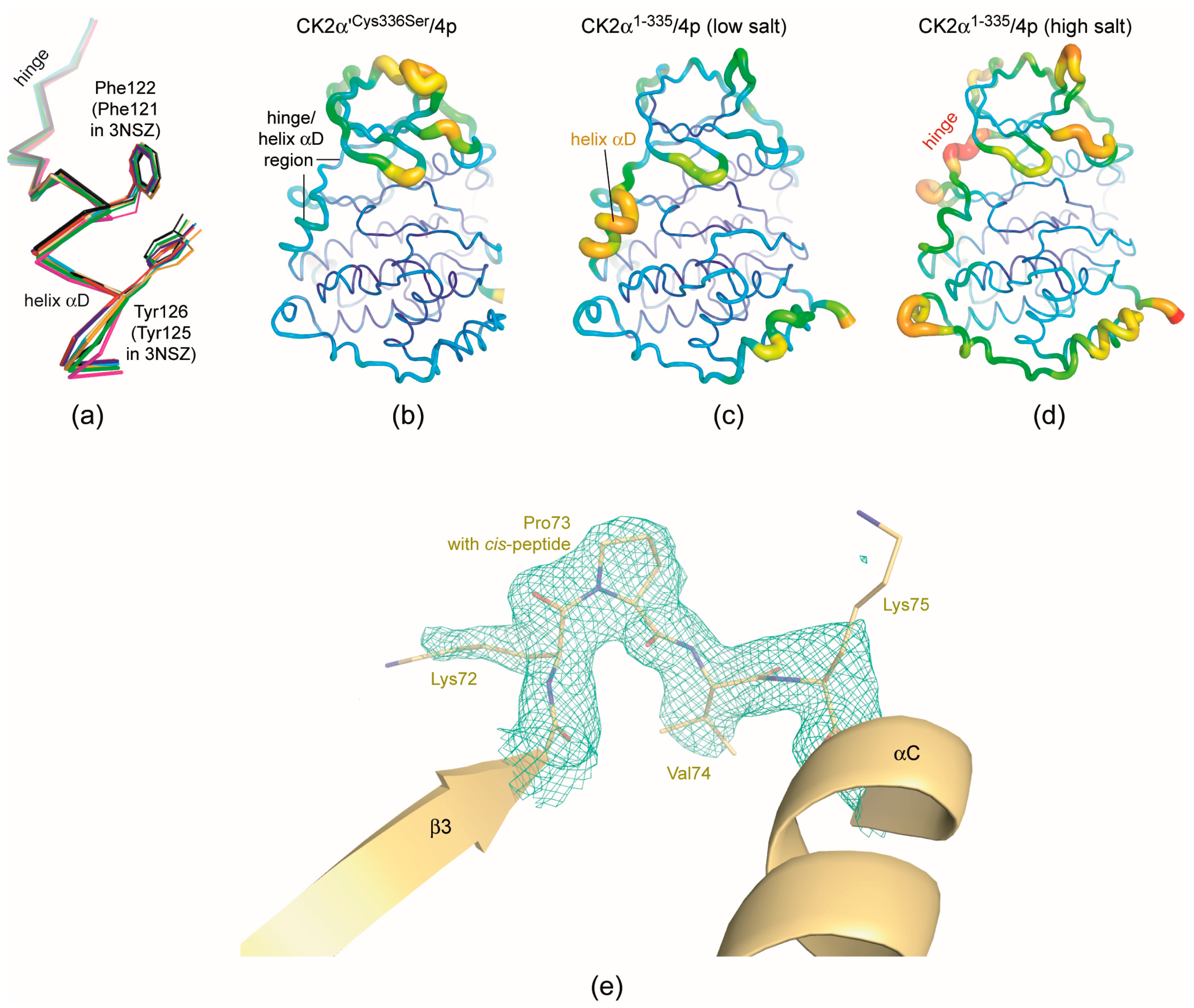
2.5. 4p Is Not Selective with Respect to the Interdomain Hinge/Helix αD region Conformation
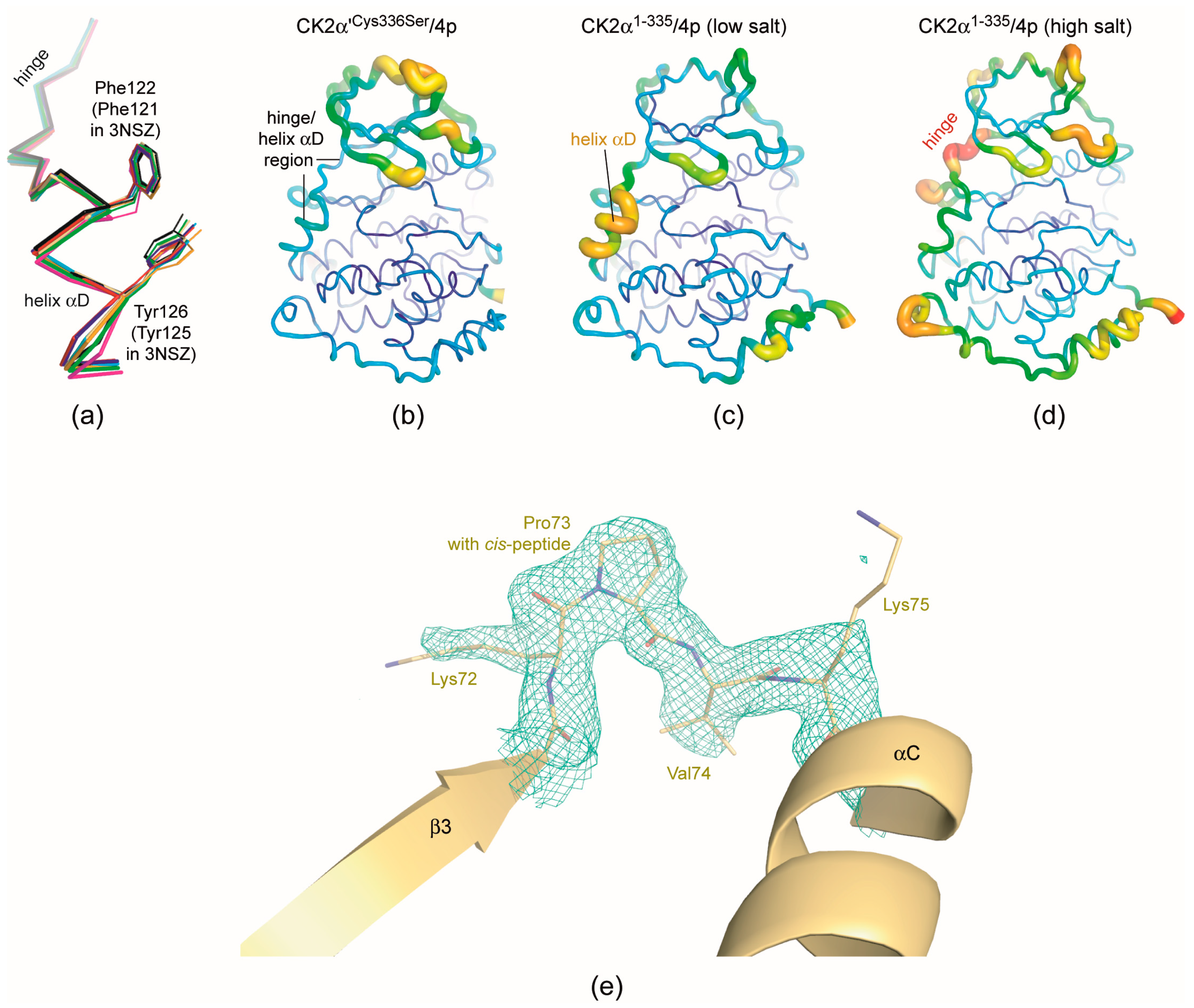
2.6. Structural Characteristics of the CK2α′Cys336Ser/4p Complex
- (i)
- (ii)
- As mentioned in the last section structural plasticity or even “hypervariability” of the hinge/helix αD region—casually accompanied by the occurrence of the αD pocket [19]—is a significant feature of human CK2α, but it was never observed so far in structures of maize or yeast CK2α and human CK2α′ where the hinge/helix αD region was always found in the open conformation without any exception. Correspondingly, crystals grown under high-salt conditions were never described for those CK2α homologs. The CK2α′Cys336Ser/4p complex of this study confirms these experiences: neither do the two CK2α′Cys336Ser protomers deviate from the open hinge/helix αD conformation (Figure 6a) nor did we observe any crystallization hit under high-salt conditions.The lack of any conformational ambiguity in the hinge/helix αD region of the CK2α′Cys336Ser/4p complex is also perceptible from the final atomic B-factors which reflect the mobilities of the atoms in the crystalline state: they are low in the whole hinge/helix αD area of the CK2α′Cys336Ser/4p complex (Figure 6b) while in both CK2α1−335/4p complex structures high mobility sections exist, namely either at the helix αD (low-salt structure; Figure 6c) or at the hinge (high-salt structure; Figure 6d). For maize CK2α the fixation to the open hinge/helix αD conformation was plausibly explained with restraints imposed by a proline residue at the C-terminal end of helix αD instead of Gln126 in human CK2α [70]. In the case of human CK2α′, however, no equivalent exchange exists. Rather, the sequences of the two human paralogs in this region are so similar that no particular enzyme-inherent restraints in favour of the open conformation are evident. Insofar, it is an open question whether in future CK2α′ structures the open hinge/helix αD conformation will prevail as well. For inhibitor development, it is even more interesting if CK2α′ conformations with an αD pocket accessible for small molecule exists at all. If not, inhibitors addressing the αD pocket should be selective for human CK2α over CK2α′.
- (iii)
- Finally, the CK2α′Cys336Ser/4p complex structure provides a further case of a cis-proline (Pro73) residue in the β3/αC loop (Figure 6e). As in previous instances of this phenomenon—a CK2α1−335 complex structure with a CK2β-competitive cyclic peptide (PDB 4IB5) [74] and a CK2α′Asp39Gly/Cys336Ser complex structure with the flavonol-derived inhibitor FLC21 (PDB 5M56) [38]—this peptide switch occurs only in one of two (5M56) or three (4IB5) protomers in the asymmetric unit, namely in chain A while in chain B the Lys72/Pro73 peptide has the normal trans-configuration. The Lys/Pro dipeptide in the β3/αC loop is absolutely conserved in the sequences of CK2α homologs, but it is completely unknown so far under which conditions a cis-peptide bond can be trapped within this dipeptide and whether a functional relevance is associated with this feature.
3. Materials and Methods
3.1. CK2 Inhibitor
3.2. Caco-2 Cell Permeability Assay
3.3. Protein
3.4. Crystallization
3.5. X-ray Diffraction Data Collection and Processing
3.6. Structure Solution, Refinement, Validation, Deposition and Illustration
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Niefind, K.; Yde, C.; Ermakova, I.; Issinger, O. Evolved to be active: Sulfate ions define substrate recognition sites of CK2α and emphasise its exceptional role within the CMGC family of eukaryotic protein kinases. J. Mol. Biol. 2007, 370, 427–438. [Google Scholar] [CrossRef] [PubMed]
- St-Denis, N.A.; Derksen, D.R.; Litchfield, D.W. Evidence for regulation of mitotic progression through temporal phosphorylation and dephosphorylation of CK2α. Mol. Cell. Biol. 2009, 29, 2068–2081. [Google Scholar] [CrossRef] [PubMed]
- St-Denis, N.A.; Litchfield, D.W. Protein kinase CK2 in health and disease: From birth to death: The role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell. Mol. Life Sci. 2009, 66, 1817–1829. [Google Scholar] [CrossRef] [PubMed]
- Loizou, J.I.; El-Khamisy, S.F.; Zlatanou, A.; Moore, D.J.; Chan, D.W.; Qin, J.; Sarno, S.; Meggio, F.; Pinna, L.A.; Caldecott, K.W. The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell 2004, 117, 17–28. [Google Scholar] [CrossRef]
- Münstermann, U.; Fritz, G.; Seitz, G.; Lu, Y.P.; Schneider, H.R.; Issinger, O.G. Casein kinase II is elevated in solid human tumours and rapidly proliferating non-neoplastic tissue. Eur. J. Biochem. 1990, 189, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Romieu-Mourez, R.; Landesman-Bollag, E.; Seldin, D.C.; Sonenshein, G.E. Protein kinase CK2 promotes aberrant activation of nuclear factor-kappaB, transformed phenotype, and survival of breast cancer cells. Cancer Res. 2002, 62, 6770–6778. [Google Scholar] [PubMed]
- Seldin, D.C.; Landesman-Bollag, E. The oncogenic potenial of CK2. In Protein Kinase CK2; Pinna, L.A., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 293–304. [Google Scholar]
- Zheng, Y.; McFarland, B.C.; Drygin, D.; Yu, H.; Bellis, S.L.; Kim, H.; Bredel, M.; Benveniste, E.N. Targeting protein kinase CK2 suppresses prosurvival signaling pathways and growth of glioblastoma. Clin. Cancer Res. 2013, 19, 6484–6494. [Google Scholar] [CrossRef] [PubMed]
- Nelson, N.; Szekeres, K.; Iclozan, C.; Rivera, I.O.; McGill, A.; Johnson, G.; Nwogu, O.; Ghansah, T. Apigenin: Selective CK2 inhibitor increases Ikaros expression and improves T cell homeostasis and function in murine pancreatic cancer. PLoS ONE 2017, 12, e0170197. [Google Scholar] [CrossRef] [PubMed]
- Quotti Tubi, L.; Canovas Nunes, S.; Brancalion, A.; Doriguzzi Breatta, E.; Manni, S.; Mandato, E.; Zaffino, F.; Macaccaro, P.; Carrino, M.; Gianesin, K.; et al. Protein kinase CK2 regulates AKT, NF-κB and STAT3 activation, stem cell viability and proliferation in acute myeloid leukemia. Leukemia 2017, 31, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Issinger, O.G. Protein kinase CK2 in human diseases. Curr. Med. Chem. 2008, 15, 1870–1886. [Google Scholar] [CrossRef] [PubMed]
- Okur, V.; Cho, M.T.; Henderson, L.; Retterer, K.; Schneider, M.; Sattler, S.; Niyazov, D.; Azage, M.; Smith, S.; Picker, J.; et al. De novo mutations in CSNK2A1 are associated with neurodevelopmental abnormalities and dysmorphic features. Hum. Genet. 2016, 135, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, A.F.; Morrema, T.H.; Gerritsen, W.H.; van Haastert, E.S.; Snkhchyan, H.; Hilhorst, R.; Rozemuller, A.J.; Scheltens, P.; van der Vies, S.M.; Hoozemans, J.J. Increased occurrence of protein kinase CK2 in astrocytes in Alzheimer’s disease pathology. J. Neuroinflamm. 2016, 13, 4. [Google Scholar] [CrossRef] [PubMed]
- Al Quobaili, F.; Montenarh, M. CK2 and the regulation of the carbohydrate metabolism. Metabolism 2012, 61, 1512–1517. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; Papinutto, E.; Franchin, C.; Bain, J.; Elliott, M.; Meggio, F.; Kazimierczuk, Z.; Orzeszko, A.; Zanotti, G.; Battistutta, R.; et al. ATP site-directed inhibitors of protein kinase CK2: An update. Curr. Top. Med. Chem. 2011, 11, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; Salvi, M.; Battistutta, R.; Zanotti, G.; Pinna, L.A. Features and potentials of ATP-site directed CK2 inhibitors. Biochim. Biophys. Acta 2005, 1754, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Moucadel, V.; Prudent, R.; Sautel, C.F.; Teillet, F.; Barette, C.; Lafanechere, L.; Receveur-Brechot, V.; Cochet, C. Antitumoral activity of allosteric inhibitors of protein kinase CK2. Oncotarget 2011, 2, 997–1010. [Google Scholar] [CrossRef] [PubMed]
- Viht, K.; Saaver, S.; Vahter, J.; Enkvist, E.; Lavogina, D.; Sinijärv, H.; Raidaru, G.; Guerra, B.; Issinger, O.G.; Uri, A. Acetoxymethyl ester of tetrabromobenzimidazole-peptoid conjugate for inhibition of protein kinase CK2 in living cells. Bioconjug. Chem. 2015, 26, 2325–2335. [Google Scholar] [CrossRef] [PubMed]
- Brear, P.; De Fusco, C.; Georgiou, K.H.; Francis-Newton, N.J.; Stubbs, C.J.; Sore, H.F.; Venkitaraman, A.R.; Abell, C.; Spring, D.R.; Hyvönen, M. Specific inhibition of CK2α from an anchor outside the active site. Chem. Sci. 2016, 7, 6839–6845. [Google Scholar] [CrossRef] [PubMed]
- De Fusco, C.; Brear, P.; Iegre, J.; Georgiou, K.H.; Sore, H.F.; Hyvönen, M.; Spring, D.R. A fragment-based approach leading to the discovery of a novel binding site and the selective CK2 inhibitor CAM4066. Bioorg. Med. Chem. 2017, 25, 3471–3482. [Google Scholar] [CrossRef] [PubMed]
- Enkvist, E.; Viht, K.; Bischoff, N.; Vahter, J.; Saaver, S.; Raidaru, G.; Issinger, O.G.; Niefind, K.; Uri, A. A subnanomolar fluorescent probe for protein kinase CK2 interaction studies. Org. Biomol. Chem. 2012, 10, 8645–8653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvi, M.; Sarno, S.; Marin, O.; Meggio, F.; Itarte, E.; Pinna, L.A. Discrimination between the activity of protein kinase CK2 holoenzyme and its catalytic subunits. FEBS Lett. 2006, 580, 3948–3952. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Guerra, B.; Ermakowa, I.; Issinger, O.-G. Crystal structure of human protein kinase CK2: Insights into basic properties of the CK2 holoenzyme. EMBO J. 2001, 20, 5320–5331. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Issinger, O.G. Primary and secondary interactions between CK2α and CK2β lead to ring-like structures in the crystals of the CK2 holoenzyme. Mol. Cell. Biochem. 2005, 274, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Valero, E.; De Bonis, S.; Filhol, O.; Wade, R.H.; Langowski, J.; Chambaz, E.M.; Cochet, C. Quaternary structure of casein kinase 2. Characterization of multiple oligomeric states and relation with its catalytic activity. J. Biol. Chem. 1995, 270, 8345–8352. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.; Poore, T.; Bandhakavi, S.; McCann, R.O.; Hanna, D.E.; Glover, C.V. A global view of CK2 function and regulation. Mol. Cell. Biochem. 2005, 274, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Hübner, G.M.; Larsen, J.N.; Guerra, B.; Niefind, K.; Vrecl, M.; Issinger, O.G. Evidence for aggregation of protein kinase CK2 in the cell: A novel strategy for studying CK2 holoenzyme interaction by BRET(2). Mol. Cell. Biochem. 2014, 397, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Pyerin, W.; Ackermann, K. The genes encoding human protein kinase CK2 and their functional links. Prog. Nucleic Acid Res. Mol. Biol. 2003, 74, 239–273. [Google Scholar] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; Olsson, I.; Edlund, K.; Lundberg, E.; Navani, S.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, D.W.; Bosc, D.G.; Canton, D.A.; Saulnier, R.B.; Vilk, G.; Zhang, C. Functional specialization of CK2 isoforms and characterization of isoform-specific binding partners. Mol. Cell. Biochem. 2001, 227, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, N.; Olsen, B.; Raaf, J.; Bretner, M.; Issinger, O.G.; Niefind, K. Structure of the human protein kinase CK2 catalytic subunit CK2α’ and interaction thermodynamics with the regulatory subunit CK2β. J. Mol. Biol. 2011, 407, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B.; Rasmussen, T.; Niefind, K.; Issinger, O. Biochemical characterization of CK2α and α’ paralogues and their derived holoenzymes: Evidence for the existence of a heterotrimeric CK2α’-holoenzyme forming trimeric complexes. Mol. Cell. Biochem. 2008, 316, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B.; Boldyreff, B.; Niefind, K.; Issinger, O. Purification and characterization of the CK2α′-based holoenzyme, an isozyme of CK2α: A comparative analysis. Protein Expr. Purif. 2006, 47, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Bosc, D.G.; Graham, K.C.; Saulnier, R.B.; Zhang, C.; Prober, D.; Gietz, R.D.; Litchfield, D.W. Identification and characterization of CKIP-1, a novel pleckstrin homology domain-containing protein that interacts with protein kinase CK2. J. Biol. Chem. 2000, 275, 14295–14306. [Google Scholar] [CrossRef] [PubMed]
- Hériché, J.K.; Lebrin, F.; Rabilloud, T.; Leroy, D.; Chambaz, E.M.; Goldberg, Y. Regulation of protein phosphatase 2A by direct interaction with casein kinase 2alpha. Science 1997, 276, 952–955. [Google Scholar] [CrossRef] [PubMed]
- Lou, D.Y.; Dominguez, I.; Toselli, P.; Landesman-Bollag, E.; O’Brien, C.; Seldin, D.C. The alpha catalytic subunit of protein kinase CK2 is required for mouse embryonic development. Mol. Cell. Biol. 2008, 28, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Toselli, P.A.; Russell, L.D.; Seldin, D.C. Globozoospermia in mice lacking the casein kinase II alpha’ catalytic subunit. Nat. Genet. 1999, 23, 118–121. [Google Scholar] [PubMed]
- Niefind, K.; Bischoff, N.; Golub, A.G.; Bdzhola, V.G.; Balanda, A.O.; Prykhod’ko, A. O.; Yarmoluk, S.M. Structural hypervariability of the two human protein kinase CK2 catalytic subunit paralogs revealed by complex structures with a flavonol- and a thieno[2,3-d]pyrimidine-based inhibitor. Pharmaceuticals 2017, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Nakaniwa, T.; Kinoshita, T.; Sekiguchi, Y.; Tada, T.; Nakanishi, I.; Kitaura, K.; Suzuki, Y.; Ohno, H.; Hirasawa, A.; Tsujimoto, G. Structure of human protein kinase CK2α2 with a potent indazole-derivative inhibitor. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2009, 65, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R.; De Moliner, E.; Sarno, S.; Zanotti, G.; Pinna, L.A. Structural features underlying selective inhibition of protein kinase CK2 by ATP site-directed tetrabromo-2-benzotriazole. Protein Sci. 2001, 10, 2200–2206. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Battistutta, R. Structural bases of protein kinase CK2 function and inhibition. In Protein Kinase CK2; Pinna, L.A., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 1–75. [Google Scholar]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R.; Mazzorana, M.; Cendron, L.; Bortolato, A.; Sarno, S.; Kazimierczuk, Z.; Zanotti, G.; Moro, S.; Pinna, L.A. The ATP-binding site of protein kinase CK2 holds a positive electrostatic area and conserved water molecules. ChemBioChem 2007, 8, 1804–1809. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.D.; Sheth, P.R.; Basso, A.D.; Paliwal, S.; Gray, K.; Fischmann, T.O.; Le, H.V. Structural basis of CX-4945 binding to human protein kinase CK2. FEBS Lett. 2011, 585, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shah, K.; Yang, F.; Witucki, L.; Shokat, K.M. A molecular gate which controls unnatural ATP analogue recognition by the tyrosine kinase v-Src. Bioorg. Med. Chem. 1998, 6, 1219–1226. [Google Scholar] [CrossRef]
- Raaf, J.; Klopffleisch, K.; Issinger, O.; Niefind, K. The catalytic subunit of human protein kinase CK2 structurally deviates from its maize homologue in complex with the nucleotide competitive inhibitor emodin. J. Mol. Biol. 2008, 377, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.M.; Vasiliou, V.; Zhu, H.; Duh, J.L.; Tabor, M.W.; Puga, A.; Nebert, D.W.; Sainsbury, M.; Shertzer, H.G. Regulation of [Ah] gene battery enzymes and glutathione levels by 5,10-dihydroindeno[1,2-b]indole in mouse hepatoma cell lines. Carcinogenesis 1994, 15, 2347–2352. [Google Scholar] [CrossRef] [PubMed]
- Rongved, P.; Kirsch, G.; Bouaziz, Z.; Jose, J.; Le Borgne, M. Indenoindoles and cyclopentacarbazoles as bioactive compounds: Synthesis and biological applications. Eur. J. Med. Chem. 2013, 69, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Bal, C.; Baldeyrou, B.; Moz, F.; Lansiaux, A.; Colson, P.; Kraus-Berthier, L.; Léonce, S.; Pierré, A.; Boussard, M.F.; Rousseau, A.; Wierzbicki, M.; Bailly, C. Novel antitumor indenoindole derivatives targeting DNA and topoisomerase II. Biochem. Pharmacol. 2004, 68, 1911–1922. [Google Scholar] [CrossRef] [PubMed]
- Hundsdörfer, C.; Hemmerling, H.J.; Götz, C.; Totzke, F.; Bednarski, P.; Le Borgne, M.; Jose, J. Indeno[1,2-b]indole derivatives as a novel class of potent human protein kinase CK2 inhibitors. Bioorg. Med. Chem. 2012, 20, 2282–2289. [Google Scholar] [CrossRef] [PubMed]
- Hundsdörfer, C.; Hemmerling, H.J.; Hamberger, J.; Le Borgne, M.; Bednarski, P.; Götz, C.; Totzke, F.; Jose, J. Novel indeno[1,2-b]indoloquinones as inhibitors of the human protein kinase CK2 with antiproliferative activity towards a broad panel of cancer cell lines. Biochem. Biophys. Res. Commun. 2012, 424, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Gozzi, J.G.; Bouaziz, Z.; Winter, E.; Daflon-Yunes, N.; Aichele, D.; Nacereddine, A.; Marminon, C.; Valdameri, G.; Zeinyeh, W.; Bollacke, A.; et al. Converting potent indeno[1,2-b]indole inhibitors of protein kinase CK2 into selective inhibitors of the breast cancer resistance protein ABCG2. J. Med. Chem. 2015, 58, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Gozzi, G.J.; Bouaziz, Z.; Winter, E.; Daflon-Yunes, N.; Honorat, M.; Guragossian, N.; Marminon, C.; Valdameri, G.; Bollacke, A.; Guillon, J.; et al. Phenolic indeno[1,2-b]indoles as ABCG2-selective potent and non-toxic inhibitors stimulating basal ATPase activity. Drug Des. Devel. Ther. 2015, 9, 3481–3495. [Google Scholar] [PubMed]
- Doyle, L.; Ross, D.D. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 2003, 22, 7340–7358. [Google Scholar] [CrossRef] [PubMed]
- Alchab, F.; Sibille, E.; Ettouati, L.; Bana, E.; Bouaziz, Z.; Mularoni, A.; Monniot, E.; Bagrel, D.; Jose, J.; Le Borgne, M.; et al. Screening of indeno[1,2-b]indoloquinones by MALDI-MS: A new set of potential CDC25 phosphatase inhibitors brought to light. J. Enzyme Inhib. Med. Chem. 2016, 31 (Suppl. 3), 25–32. [Google Scholar] [CrossRef] [PubMed]
- Haidar, S.; Bouaziz, Z.; Marminon, C.; Laitinen, T.; Poso, A.; Le Borgne, M.; Jose, J. Development of pharmacophore model for indeno[1,2-b]indoles as human protein kinase CK2 inhibitors and database mining. Pharmaceuticals 2017, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Alchab, F.; Ettouati, L.; Bouaziz, Z.; Bollacke, A.; Delcros, J.G.; Gertzen, C.G.; Gohlke, H.; Pinaud, N.; Marchivie, M.; Guillon, J.; et al. Synthesis, biological evaluation and molecular modeling of substituted indeno[1,2-b]indoles as inhibitors of human protein kinase CK2. Pharmaceuticals 2015, 8, 279–302. [Google Scholar] [CrossRef] [PubMed]
- Hubatsch, I.; Ragnarsson, E.G.; Artursson, P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat. Protoc. 2007, 2, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Artursson, P.; Karlsson, J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem. Biophys. Res. Commun. 1991, 175, 880–885. [Google Scholar] [CrossRef]
- Ermakova, I.; Boldyreff, B.; Issinger, O.; Niefind, K. Crystal structure of a C-terminal deletion mutant of human protein kinase CK2 catalytic subunit. J. Mol. Biol. 2003, 330, 925–934. [Google Scholar] [CrossRef]
- Guerra, B.; Bischoff, N.; Bdzhola, V.G.; Yarmoluk, S.M.; Issinger, O.G.; Golub, A.G.; Niefind, K. A note of caution on the role of halogen bonds for protein kinase/inhibitor recognition suggested by high- and low-salt CK2α complex structures. ACS Chem. Biol. 2015, 10, 1654–1660. [Google Scholar] [CrossRef] [PubMed]
- Traxler, P.; Furet, P. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol. Ther. 1999, 82, 195–206. [Google Scholar] [CrossRef]
- Guerra, B.; Rasmussen, T.D.; Schnitzler, A.; Jensen, H.H.; Boldyreff, B.S.; Miyata, Y.; Marcussen, N.; Niefind, K.; Issinger, O.G. Protein kinase CK2 inhibition is associated with the destabilization of HIF-1α in human cancer cells. Cancer Lett. 2015, 356, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Pütter, M.; Guerra, B.; Issinger, O.G.; Schomburg, D. GTP plus water mimic ATP in the active site of protein kinase CK2. Nat. Struct. Biol. 1999, 6, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Prudent, R.; Moucadel, V.; Nguyen, C.H.; Barette, C.; Schmidt, F.; Florent, J.C.; Lafanechère, L.; Sautel, C.F.; Duchemin-Pelletier, E.; Spreux, E.; Filhol, O.; et al. Antitumor activity of pyridocarbazole and benzopyridoindole derivatives that inhibit protein kinase CK2. Cancer Res. 2010, 70, 9865–9874. [Google Scholar] [CrossRef] [PubMed]
- Klopffleisch, K.; Issinger, O.; Niefind, K. Low-density crystal packing of human protein kinase CK2 catalytic subunit in complex with resorufin or other ligands: A tool to study the unique hinge-region plasticity of the enzyme without packing bias. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Issinger, O.G. Conformational plasticity of the catalytic subunit of protein kinase CK2 and its consequences for regulation and drug design. Biochim. Biophys. Acta 2010, 1804, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, N.; Raaf, J.; Olsen, B.; Bretner, M.; Issinger, O.; Niefind, K. Enzymatic activity with an incomplete catalytic spine: Insights from a comparative structural analysis of human CK2α and its paralogous isoform CK2α’. Mol. Cell. Biochem. 2011, 356, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R.; Lolli, G. Structural and functional determinants of protein kinase CK2α: Facts and open questions. Mol. Cell. Biochem. 2011, 356, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Yde, C.W.; Ermakova, I.; Issinger, O.G.; Niefind, K. Inclining the purine base binding plane in protein kinase CK2 by exchanging the flanking side-chains generates a preference for ATP as a cosubstrate. J. Mol. Biol. 2005, 347, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Kornev, A.P. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem. Sci. 2011, 36, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Schomburg, D. Amino-acid similarity coefficients for protein modeling and sequence alignment derived from main-chain folding angles. J. Mol. Biol. 1991, 219, 481–497. [Google Scholar]
- Raaf, J.; Guerra, B.; Neundorf, I.; Bopp, B.; Issinger, O.G.; Jose, J.; Pietsch, M.; Niefind, K. First structure of protein kinase CK2 catalytic subunit with an effective CK2β-competitive ligand. ACS Chem. Biol. 2013, 8, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Tóth, Z.A.; Raatikainen, O.; Naaranlahti, T.; Auriola, S. Isolation and determination of alizarin in cell cultures of Rubia tinctorum and emodin in Dermocybe sanguinea using solid-phase extraction and high-performance liquid chromatography. J. Chromatogr. A 1993, 630, 423–428. [Google Scholar] [CrossRef]
- Guerra, B.; Hochscherf, J.; Jensen, N.B.; Issinger, O.G. Identification of a novel potent, selective and cell permeable inhibitor of protein kinase CK2 from the NIH/NCI Diversity Set Library. Mol. Cell. Biochem. 2015, 406, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; McCoy, A.J.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, version 1.7; Schrödinger, LLC: New York, NY, USA, 2013.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure No. | 1 | 2 | 3 | |
|---|---|---|---|---|
| PDB Code | 5OMY | 5ONI | 5OOI | |
| Crystallized Complex | CK2α1−335 + 4p | CK2αCys336Ser′ + 4p | ||
| Crystallization | ||||
| Vapour diffusion reservoir composition | 4.2 M NaCl, 0.1 M sodium citrate, pH 5.5 | 25% (w/v) PEG5000, 0.2 M ammonium sulfate, 0.1 M MES, pH 6.5 | 25% (w/v) PEG3350, 0.2 M ammonium acetate, 0.1 M HEPES, pH 7.5 | |
| Sitting drop composition before equilibration | 1 μL reservoir + 1 μL enzyme/4p mixture (90 μL 5 mg/mL enzyme, 0.5 M NaCl, 25 mM Tris/HCl, pH 8.5, mixed and pre-equilibrated with 10 μL 10 mM 4p in DMSO) | 1 μL reservoir + 1 μL enzyme/4p mixture (90 μL 5 mg/mL enzyme, 0.5 M NaCl, 25 mM Tris/HCl, pH 8.5, mixed and pre-equilibrated with 10 μL 10 mM 4p in DMSO) | ||
| X-ray Diffraction Data Collection | ||||
| Wavelength [Å] | 0.97625 | 0.9660 | 1.0000 | |
| Synchrotron (beamline) | SLS (X06DA) | ESRF (ID30A-1) | PETRA III at DESY (P13) | |
| Space group | P43212 | P43212 | P212121 | |
| Unit cell | a, b, c [Å] | 72.70, 72.70, 132.89 | 128.45, 128.45, 124.11 | 46.49, 112.13, 143.69 |
| α, β, γ [°] | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | |
| Protomers per asym. unit | 1 | 2 | 2 | |
| Resolution [Å] (highest resolution shell) | 63.78–1.95 (2.02–1.95) 1 | 57.04–2.00 (2.072–2.00) 1 | 60.49–2.00 (2.07–2.00) 1 | |
| Rsym [%] | 9.2 (228.1) 1 | 9.3 (65.7) 1 | 17.7 (119.1) 1 | |
| CC1/2 | 0.999 (0.640) 1 | 0.996 (0.685) 1 | 0.995 (0.673) 1 | |
| Signal-to-noise ratio (I/σI) | 20.77 (1.52) 1 | 14.20 (0.93) 1 | 7.39 (1.41) 1 | |
| No. of unique reflections | 26747 (2639) 1 | 70221 (6881) 1 | 51742 (4979) 1 | |
| Completeness [%] | 99.94 (100.0) 1 | 99.51 (98.84) 1 | 99.69 (97.59) 1 | |
| Multiplicity | 24.9 (26.1)1 | 6.2 (6.3) 1 | 6.7 (6.7) 1 | |
| Wilson B-factor [Å2] | 43.54 | 44.40 | 24.98 | |
| Structure Refinement | ||||
| No. of reflections for Rwork/Rfree | 25417/1316 | 68727/1388 | 50784/1041 | |
| Rwork/Rfree [%] | 19.25/22.08 | 17.51/20.42 | 17.37/22.10 | |
| Number of non-H-atoms | 2966 | 6055 | 6050 | |
| Protein | 2812 | 5645 | 5524 | |
| Ligand/Ion | 32 | 113 | 83 | |
| Water | 122 | 297 | 443 | |
| Average B-factor [Å2] | 56.42 | 55.51 | 34.15 | |
| Protein | 56.63 | 55.10 | 33.80 | |
| Ligand/Ion | 64.83 | 79.16 | 34.33 | |
| water | 49.51 | 54.20 | 38.55 | |
| RMS deviations | ||||
| Bond lengths [Å] | 0.003 | 0.007 | 0.011 | |
| Bond angles [°] | 0.570 | 0.81 | 1.13 | |
| Ramachandran plot | ||||
| favoured (%) | 96.36 | 96.99 | 97.38 | |
| allowed (%) | 3.64 | 3.01 | 2.46 | |
| outliers (%) | 0.30 | 0.00 | 0.15 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hochscherf, J.; Lindenblatt, D.; Witulski, B.; Birus, R.; Aichele, D.; Marminon, C.; Bouaziz, Z.; Le Borgne, M.; Jose, J.; Niefind, K. Unexpected Binding Mode of a Potent Indeno[1,2-b]indole-Type Inhibitor of Protein Kinase CK2 Revealed by Complex Structures with the Catalytic Subunit CK2α and Its Paralog CK2α′. Pharmaceuticals 2017, 10, 98. https://doi.org/10.3390/ph10040098
Hochscherf J, Lindenblatt D, Witulski B, Birus R, Aichele D, Marminon C, Bouaziz Z, Le Borgne M, Jose J, Niefind K. Unexpected Binding Mode of a Potent Indeno[1,2-b]indole-Type Inhibitor of Protein Kinase CK2 Revealed by Complex Structures with the Catalytic Subunit CK2α and Its Paralog CK2α′. Pharmaceuticals. 2017; 10(4):98. https://doi.org/10.3390/ph10040098
Chicago/Turabian StyleHochscherf, Jennifer, Dirk Lindenblatt, Benedict Witulski, Robin Birus, Dagmar Aichele, Christelle Marminon, Zouhair Bouaziz, Marc Le Borgne, Joachim Jose, and Karsten Niefind. 2017. "Unexpected Binding Mode of a Potent Indeno[1,2-b]indole-Type Inhibitor of Protein Kinase CK2 Revealed by Complex Structures with the Catalytic Subunit CK2α and Its Paralog CK2α′" Pharmaceuticals 10, no. 4: 98. https://doi.org/10.3390/ph10040098