
Exploring the Prominent and Concealed Inhibitory Features for Cytoplasmic Isoforms of Hsp90 Using QSAR Analysis


 ,
,
Abstract
:1. Introduction
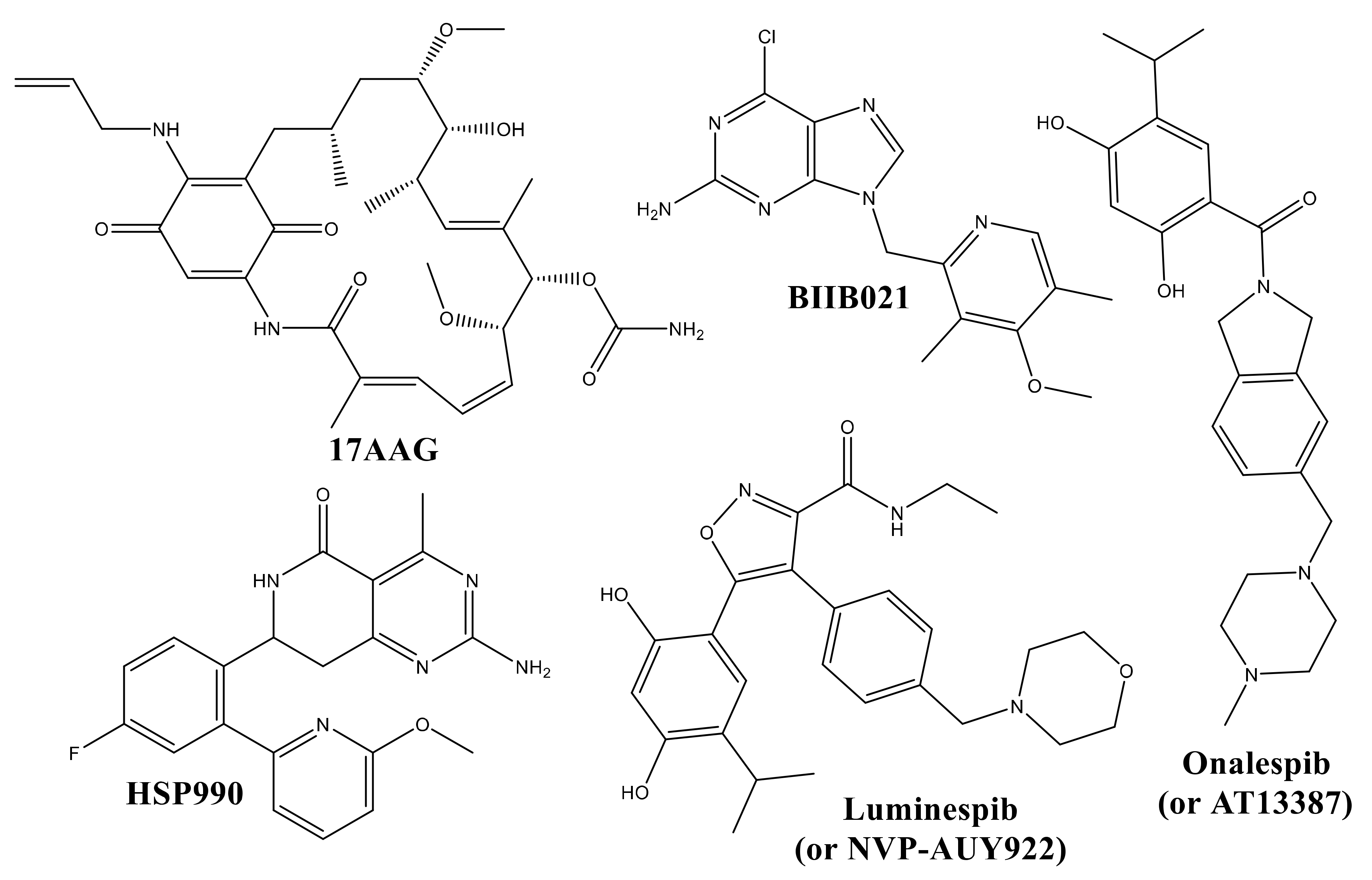
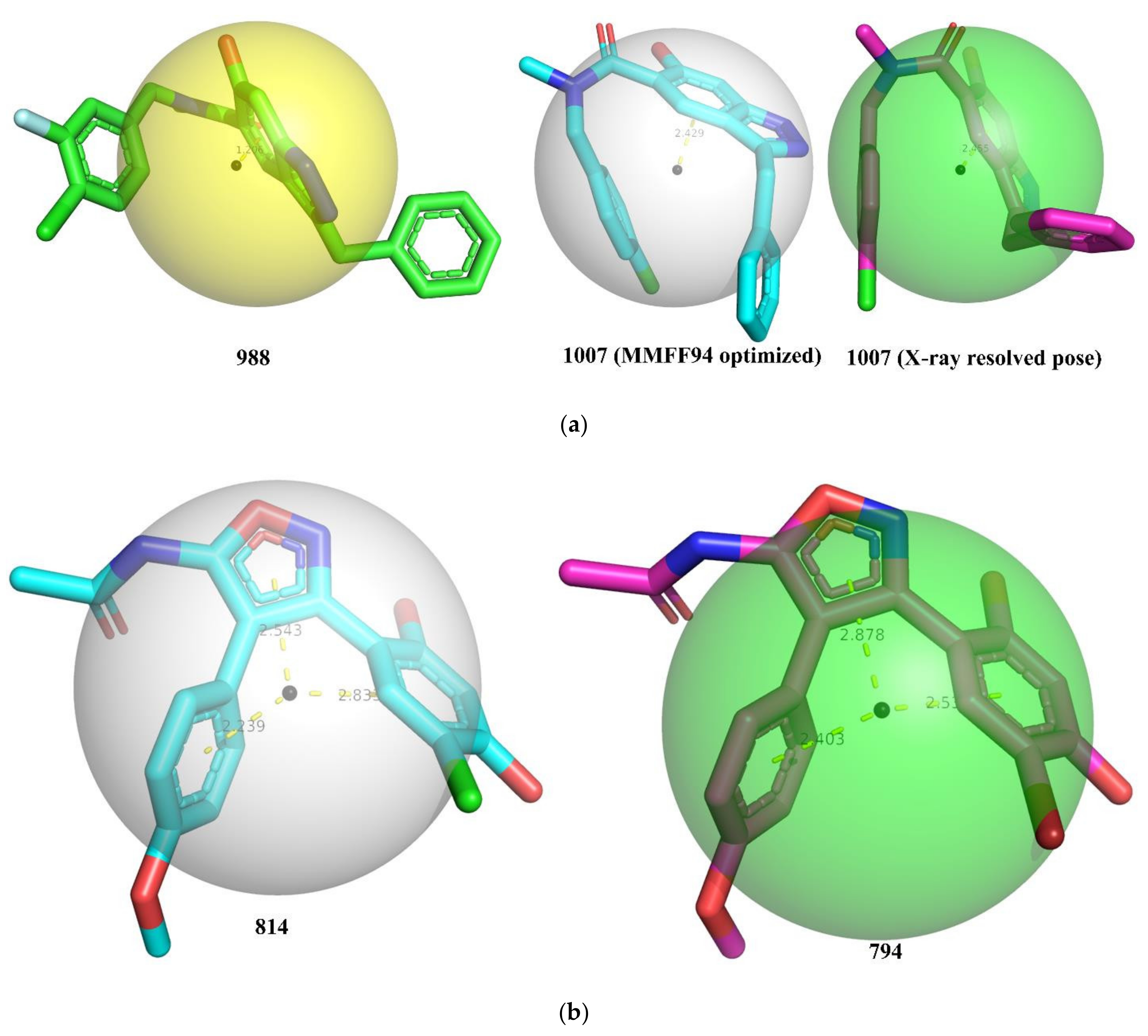
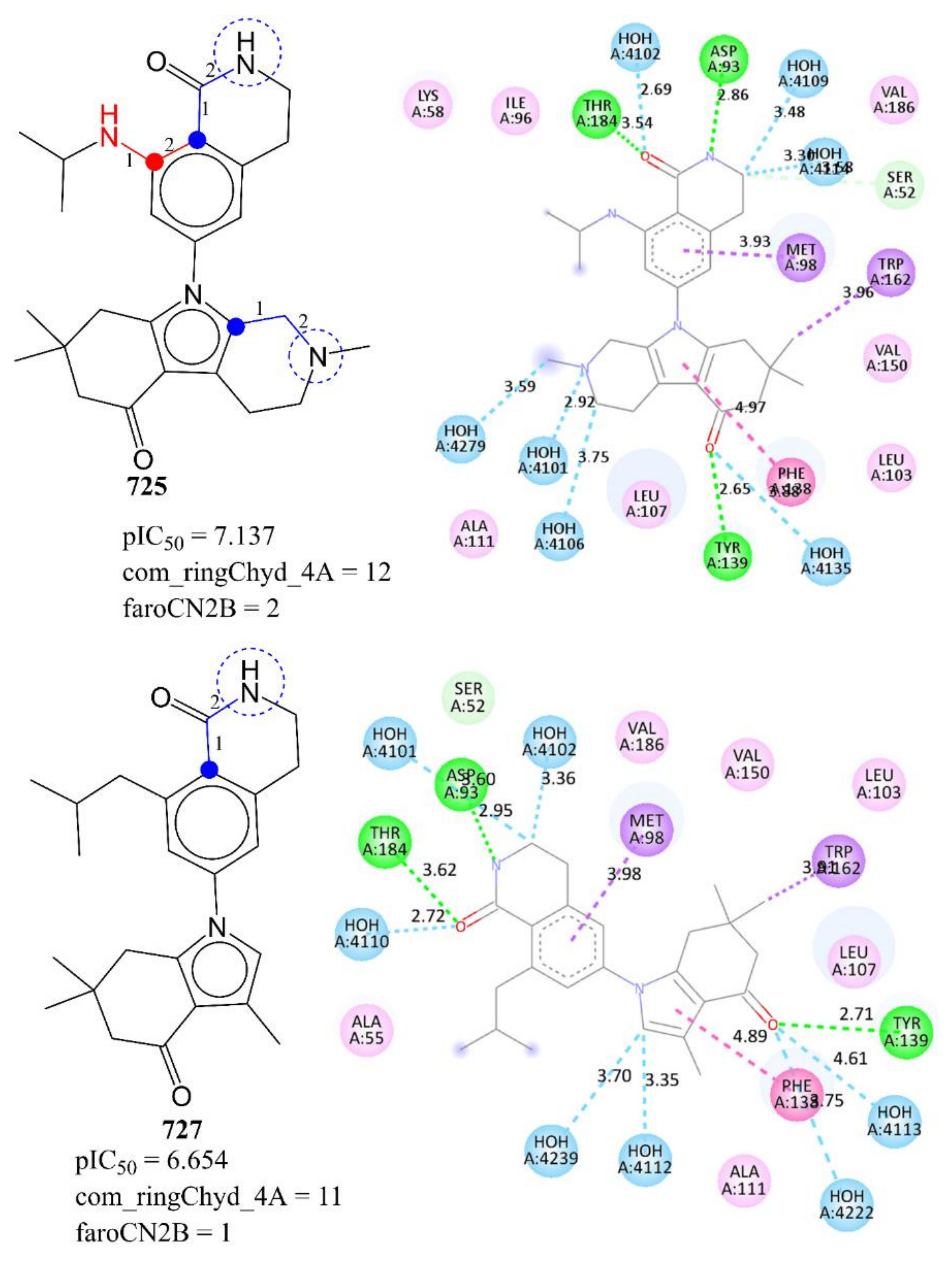
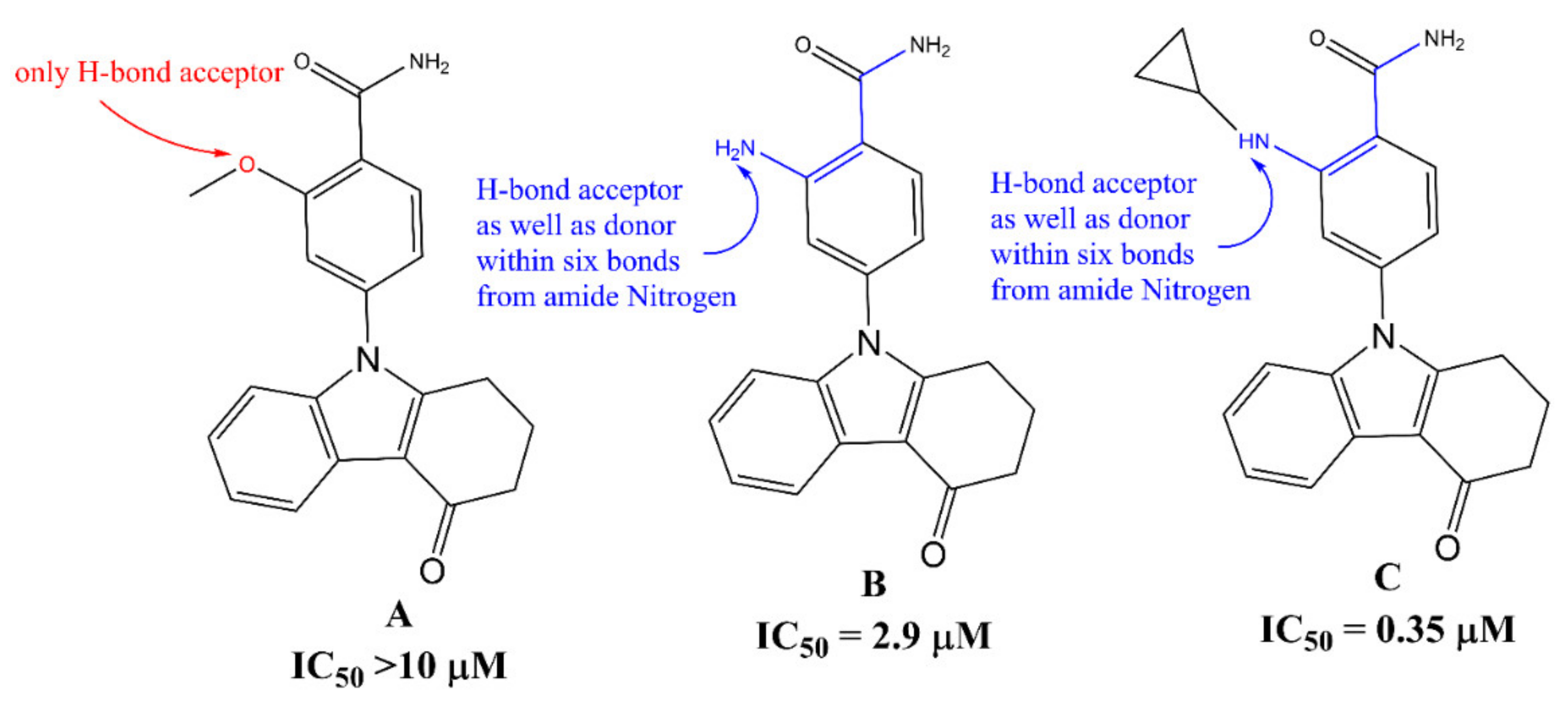
2. Results
3. Discussion
Mechanistic Interpretation of QSAR Model
4. Materials and Methods
4.1. Data Collection and Its Curation
4.2. Calculation of Molecular Descriptors and Objective Feature Selection (OFS)
4.3. Splitting the Dataset into Training and External Sets and SFS (Subjective Feature Selection)
4.4. Building Regression Model and Its Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SMILES | Simplified molecular-input line-entry system |
| GA | Genetic algorithm |
| MLR | Multiple linear regression |
| QSAR | Quantitative structure−activity relationship |
| WHO | World Health Organization |
| ADMET | Absorption, distribution, metabolism, excretion, and toxicity |
| OLS | Ordinary least square |
| QSARINS | QSAR Insubria |
| OECD | Organization for Economic Cooperation and Development |
References
- Ho, N.; Li, A.; Li, S.; Zhang, H. Heat Shock Protein 90 and Role of Its Chemical Inhibitors in Treatment of Hematologic Malignancies. Pharmaceuticals 2012, 5, 779–801. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; You, Q.-D.; Xu, X.-L. Heat Shock Protein 90 Inhibitors: An Update on Achievements, Challenges, and Future Directions. J. Med. Chem. 2019, 63, 1798–1822. [Google Scholar] [CrossRef]
- Bhat, R.; Tummalapalli, S.R.; Rotella, D.P. Progress in the Discovery and Development of Heat Shock Protein 90 (Hsp90) Inhibitors. J. Med. Chem. 2014, 57, 8718–8728. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Moroni, E.; Colombo, G.; Blagg, B.S.J. Identification of a New Scaffold for Hsp90 C-Terminal Inhibition. ACS Med. Chem. Lett. 2013, 5, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, T.; Schwartz, S.J.; Sun, D. New developments in Hsp90 inhibitors as anti-cancer therapeutics: Mechanisms, clinical perspective and more potential. Drug Resist. Updates 2009, 12, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Hoter, A.; El-Sabban, M.; Naim, H. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef] [Green Version]
- Zuehlke, A.D.; Moses, M.A.; Neckers, L. Heat shock protein 90: Its inhibition and function. Philos. Trans. R. Soc. B Biol. Sci. 2017, 373, 20160527. [Google Scholar] [CrossRef] [Green Version]
- Biamonte, M.A.; Van de Water, R.; Arndt, J.W.; Scannevin, R.H.; Perret, D.; Lee, W.-C. Heat Shock Protein 90: Inhibitors in Clinical Trials. J. Med. Chem. 2009, 53, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.M.; Masand, N.; Gupta, S.P.; Blagg, B.S.J. QSAR Studies to Predict Activity of HSP90 Inhibitors. Curr. Top. Med. Chem. 2021, 21, 2272–2291. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, K.; Taldone, T.; Modi, S.; Chiosis, G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim. Biophys. Acta BBA Mol. Cell Res. 2012, 1823, 742–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramatica, P. Principles of QSAR Modeling. Int. J. Quant. Struct.-Prop. Relatsh. 2020, 5, 61–97. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [Green Version]
- Gramatica, P. On the development and validation of QSAR models. Methods Mol. Biol. 2013, 930, 499–526. [Google Scholar] [CrossRef]
- Gramatica, P.; Cassani, S.; Roy, P.P.; Kovarich, S.; Yap, C.W.; Papa, E. QSAR Modeling is not Push a Button and Find a Correlation: A Case Study of Toxicity of (Benzo-)triazoles on Algae. Mol. Inform. 2012, 31, 817–835. [Google Scholar] [CrossRef]
- Li, J.; Gramatica, P. The importance of molecular structures, endpoints’ values, and predictivity parameters in QSAR research: QSAR analysis of a series of estrogen receptor binders. Mol. Divers. 2010, 14, 687–696. [Google Scholar] [CrossRef]
- Muratov, E.N.; Bajorath, J.; Sheridan, R.P.; Tetko, I.V.; Filimonov, D.; Poroikov, V.; Oprea, T.I.; Baskin, I.I.; Varnek, A.; Roitberg, A.; et al. QSAR without borders. Chem. Soc. Rev. 2020, 49, 3525–3564. [Google Scholar] [CrossRef]
- Fujita, T.; Winkler, D.A. Understanding the Roles of the “Two QSARs”. J. Chem. Inf. Model. 2016, 56, 269–274. [Google Scholar] [CrossRef]
- Zaki, M.E.A.; Al-Hussain, S.A.; Masand, V.H.; Sabnani, M.K.; Samad, A. Mechanistic and Predictive QSAR Analysis of Diverse Molecules to Capture Salient and Hidden Pharmacophores for Anti-Thrombotic Activity. Int. J. Mol. Sci. 2021, 22, 8352. [Google Scholar] [CrossRef]
- Zhao, H.; Moroni, E.; Yan, B.; Colombo, G.; Blagg, B.S.J. 3D-QSAR-Assisted Design, Synthesis, and Evaluation of Novobiocin Analogues. ACS Med. Chem. Lett. 2012, 4, 57–62. [Google Scholar] [CrossRef]
- Barta, T.E.; Veal, J.M.; Rice, J.W.; Partridge, J.M.; Fadden, R.P.; Ma, W.; Jenks, M.; Geng, L.; Hanson, G.J.; Huang, K.H.; et al. Discovery of benzamide tetrahydro-4H-carbazol-4-ones as novel small molecule inhibitors of Hsp90. Bioorg. Med. Chem. Lett. 2008, 18, 3517–3521. [Google Scholar] [CrossRef]
- Bussenius, J.; Blazey, C.M.; Aay, N.; Anand, N.K.; Arcalas, A.; Baik, T.; Bowles, O.J.; Buhr, C.A.; Costanzo, S.; Curtis, J.K.; et al. Discovery of XL888: A novel tropane-derived small molecule inhibitor of HSP90. Bioorg. Med. Chem. Lett. 2012, 22, 5396–5404. [Google Scholar] [CrossRef]
- Abbasi, M.; Sadeghi-Aliabadi, H.; Amanlou, M. Prediction of new Hsp90 inhibitors based on 3,4-isoxazolediamide scaffold using QSAR study, molecular docking and molecular dynamic simulation. DARU J. Pharm. Sci. 2017, 25, 17. [Google Scholar] [CrossRef]
- Gramatica, P. External Evaluation of QSAR Models, in Addition to Cross-Validation Verification of Predictive Capability on Totally New Chemicals. Mol. Inform. 2014, 33, 311–314. [Google Scholar] [CrossRef]
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
- Chirico, N.; Gramatica, P. Real external predictivity of QSAR models. Part 2. New intercomparable thresholds for different validation criteria and the need for scatter plot inspection. J. Chem. Inf. Model. 2012, 52, 2044–2058. [Google Scholar] [CrossRef]
- Chirico, N.; Gramatica, P. Real external predictivity of QSAR models: How to evaluate it? Comparison of different validation criteria and proposal of using the concordance correlation coefficient. J. Chem. Inf. Model. 2011, 51, 2320–2335. [Google Scholar] [CrossRef]
- Gramatica, P.; Pilutti, P.; Papa, E. Approaches for externally validated QSAR modelling of Nitrated Polycyclic Aromatic Hydrocarbon mutagenicity. SAR QSAR Environ. Res. 2007, 18, 169–178. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR models validation internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Martin, T.M.; Harten, P.; Young, D.M.; Muratov, E.N.; Golbraikh, A.; Zhu, H.; Tropsha, A. Does rational selection of training and test sets improve the outcome of QSAR modeling? J. Chem. Inf. Model. 2012, 52, 2570–2578. [Google Scholar] [CrossRef]
- Tropsha, A.; Gramatica, P.; Gombar, V.K. The Importance of Being Earnest Validation is the Absolute Essential for Successful Application and Interpretation of QSPR Models. QSAR Comb. Sci. 2003, 22, 69–77. [Google Scholar] [CrossRef]
- Golbraikh, A.; Shen, M.; Xiao, Z.; Xiao, Y.D.; Lee, K.H.; Tropsha, A. Rational selection of training and test sets for the development of validated QSAR models. J. Comput.-Aided Mol. Des. 2003, 17, 241–253. [Google Scholar] [CrossRef]
- Polishchuk, P. Interpretation of Quantitative Structure–Activity Relationship Models: Past, Present, and Future. J. Chem. Inf. Model. 2017, 57, 2618–2639. [Google Scholar] [CrossRef]
- Jackson, S.E. Hsp90: Structure and Function. In Molecular Chaperones; Springer: Berlin/Heidelberg, Germany, 2012; pp. 155–240. [Google Scholar]
- Vallée, F.; Carrez, C.; Pilorge, F.; Dupuy, A.; Parent, A.; Bertin, L.; Thompson, F.; Ferrari, P.; Fassy, F.; Lamberton, A.; et al. Tricyclic Series of Heat Shock Protein 90 (Hsp90) Inhibitors Part I: Discovery of Tricyclic Imidazo[4,5-c]pyridines as Potent Inhibitors of the Hsp90 Molecular Chaperone. J. Med. Chem. 2011, 54, 7206–7219. [Google Scholar] [CrossRef]
- Baruchello, R.; Simoni, D.; Grisolia, G.; Barbato, G.; Marchetti, P.; Rondanin, R.; Mangiola, S.; Giannini, G.; Brunetti, T.; Alloatti, D.; et al. Novel 3,4-Isoxazolediamides as Potent Inhibitors of Chaperone Heat Shock Protein 90. J. Med. Chem. 2011, 54, 8592–8604. [Google Scholar] [CrossRef]
- Davies, N.G.M.; Browne, H.; Davis, B.; Drysdale, M.J.; Foloppe, N.; Geoffrey, S.; Gibbons, B.; Hart, T.; Hubbard, R.; Jensen, M.R.; et al. Targeting conserved water molecules: Design of 4-aryl-5-cyanopyrrolo[2,3-d]pyrimidine Hsp90 inhibitors using fragment-based screening and structure-based optimization. Bioorg. Med. Chem. 2012, 20, 6770–6789. [Google Scholar] [CrossRef]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, but verify: On the importance of chemical structure curation in cheminformatics and QSAR modeling research. J. Chem. Inf. Model. 2010, 50, 1189–1204. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Masand, V.H.; Rastija, V. PyDescriptor: A new PyMOL plugin for calculating thousands of easily understandable molecular descriptors. Chemom. Intell. Lab. Syst. 2017, 169, 12–18. [Google Scholar] [CrossRef]
- Zaki, M.E.A.; Al-Hussain, S.A.; Masand, V.H.; Akasapu, S.; Bajaj, S.O.; El-Sayed, N.N.E.; Ghosh, A.; Lewaa, I. Identification of Anti-SARS-CoV-2 Compounds from Food Using QSAR-Based Virtual Screening, Molecular Docking, and Molecular Dynamics Simulation Analysis. Pharmaceuticals 2021, 14, 357. [Google Scholar] [CrossRef]
- Kar, S.; Roy, K.; Leszczynski, J. Applicability Domain: A Step Toward Confident Predictions and Decidability for QSAR Modeling. In Computational Toxicology; Humana Press: New York, NY, USA, 2018; pp. 141–169. [Google Scholar]
- Gramatica, P.; Kovarich, S.; Roy, P.P. Reply to the comment of S. Rayne on “QSAR model reproducibility and applicability: A case study of rate constants of hydroxyl radical reaction models applied to polybrominated diphenyl ethers and (benzo-)triazoles”. J. Comput. Chem. 2013, 34, 1796. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
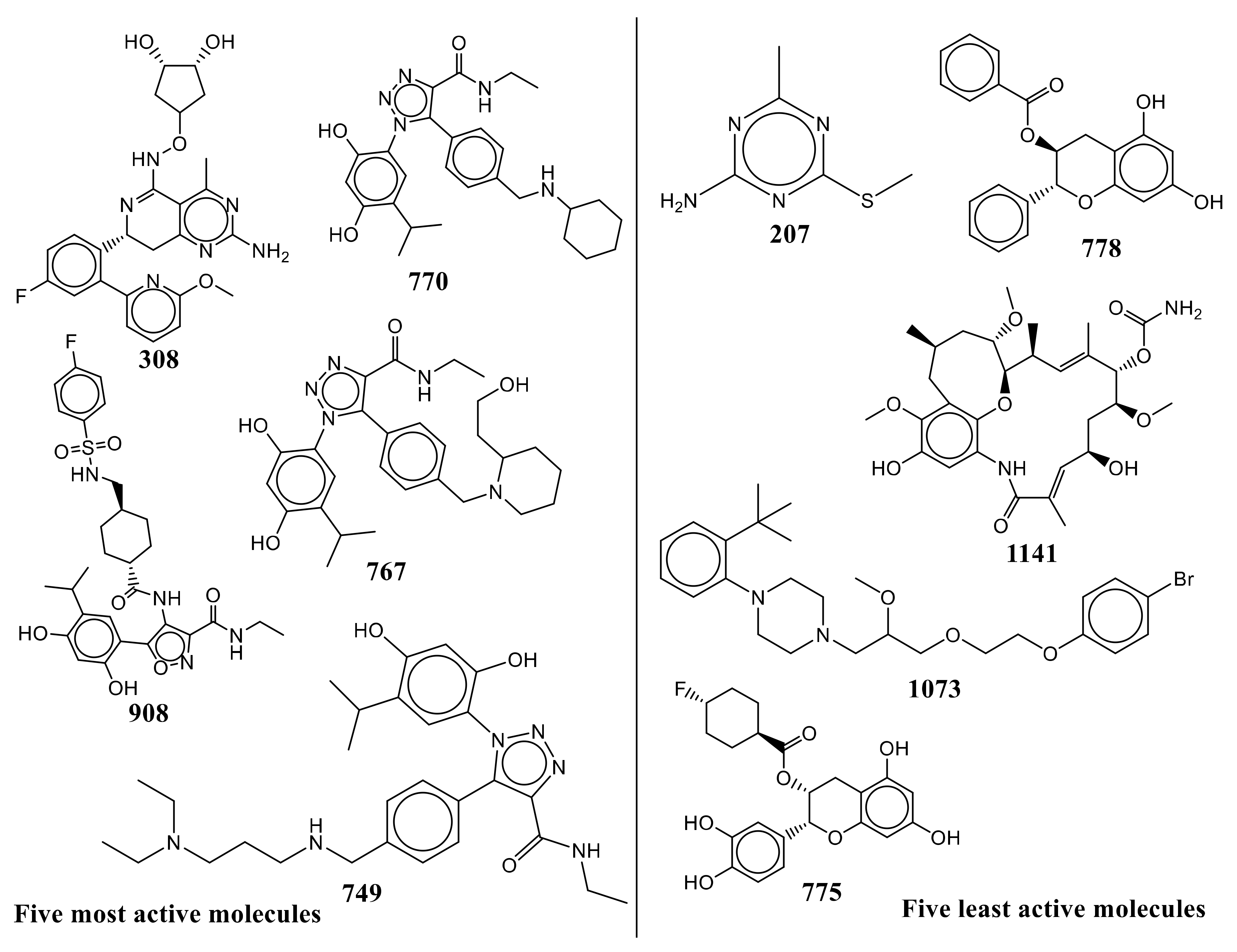
| S.N. | Ligand SMILES | IC50 (nM) | pIC50 (M) |
|---|---|---|---|
| 308 | COc1cccc(n1)-c1cc(F)ccc1[C@H]1Cc2nc(N)nc(C)c2C(NOC2C[C@H](O)[C@H](O)C2)=N1 | 5 | 8.301 |
| 908 | CCNC(=O)c1noc(c1NC(=O)[C@H]1CC[C@H](CNS(=O)(=O)c2ccc(F)cc2)CC1)-c1cc(C(C)C)c(O)cc1O | 5.4 | 8.268 |
| 770 | CCNC(=O)c1nnn(c1-c1ccc(CNC2CCCCC2)cc1)-c1cc(C(C)C)c(O)cc1O | 6.8 | 8.167 |
| 767 | CCNC(=O)c1nnn(c1-c1ccc(CN2CCCCC2CCO)cc1)-c1cc(C(C)C)c(O)cc1O | 10 | 8 |
| 749 | CCNC(=O)c1nnn(c1-c1ccc(CNCCCN(CC)CC)cc1)-c1cc(C(C)C)c(O)cc1O | 12 | 7.921 |
| 775 | Oc1cc(O)c2C[C@@H](OC(=O)[C@H]3CC[C@H](F)CC3)[C@H](Oc2c1)c1ccc(O)c(O)c1 | 69,000 | 4.161 |
| 1073 | COC(COCCOc1ccc(Br)cc1)CN1CCN(CC1)c1ccccc1C(C)(C)C | 70,430 | 4.152 |
| 1141 | CO[C@H]1C[C@H](C)Cc2c(OC)c(O)cc3NC(=O)\C(C)=C\[C@H](O)C[C@H](OC)[C@@H](OC(N)=O)\C(C)=C\[C@H](C)[C@H]1Oc23 | 96,000 | 4.018 |
| 778 | Oc1cc(O)c2C[C@H](OC(=O)c3ccccc3)[C@H](Oc2c1)c1ccccc1 | 120,000 | 3.921 |
| 207 | CSc1nc(C)nc(N)n1 | 350,000 | 3.456 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaki, M.E.A.; Al-Hussain, S.A.; Bukhari, S.N.A.; Masand, V.H.; Rathore, M.M.; Thakur, S.D.; Patil, V.M. Exploring the Prominent and Concealed Inhibitory Features for Cytoplasmic Isoforms of Hsp90 Using QSAR Analysis. Pharmaceuticals 2022, 15, 303. https://doi.org/10.3390/ph15030303
Zaki MEA, Al-Hussain SA, Bukhari SNA, Masand VH, Rathore MM, Thakur SD, Patil VM. Exploring the Prominent and Concealed Inhibitory Features for Cytoplasmic Isoforms of Hsp90 Using QSAR Analysis. Pharmaceuticals. 2022; 15(3):303. https://doi.org/10.3390/ph15030303
Chicago/Turabian StyleZaki, Magdi E. A., Sami A. Al-Hussain, Syed Nasir Abbas Bukhari, Vijay H. Masand, Mithilesh M. Rathore, Sumer D. Thakur, and Vaishali M. Patil. 2022. "Exploring the Prominent and Concealed Inhibitory Features for Cytoplasmic Isoforms of Hsp90 Using QSAR Analysis" Pharmaceuticals 15, no. 3: 303. https://doi.org/10.3390/ph15030303