
Optimization of 2-Aminoquinazolin-4-(3H)-one Derivatives as Potent Inhibitors of SARS-CoV-2: Improved Synthesis and Pharmacokinetic Properties

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
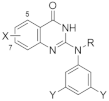
2.1. Synthesis of 2-Aminoquinazolin-4-(3H)-one Derivatives
2.2. In Vitro Activity against SARS-CoV-2
2.3. Mode of Action Study
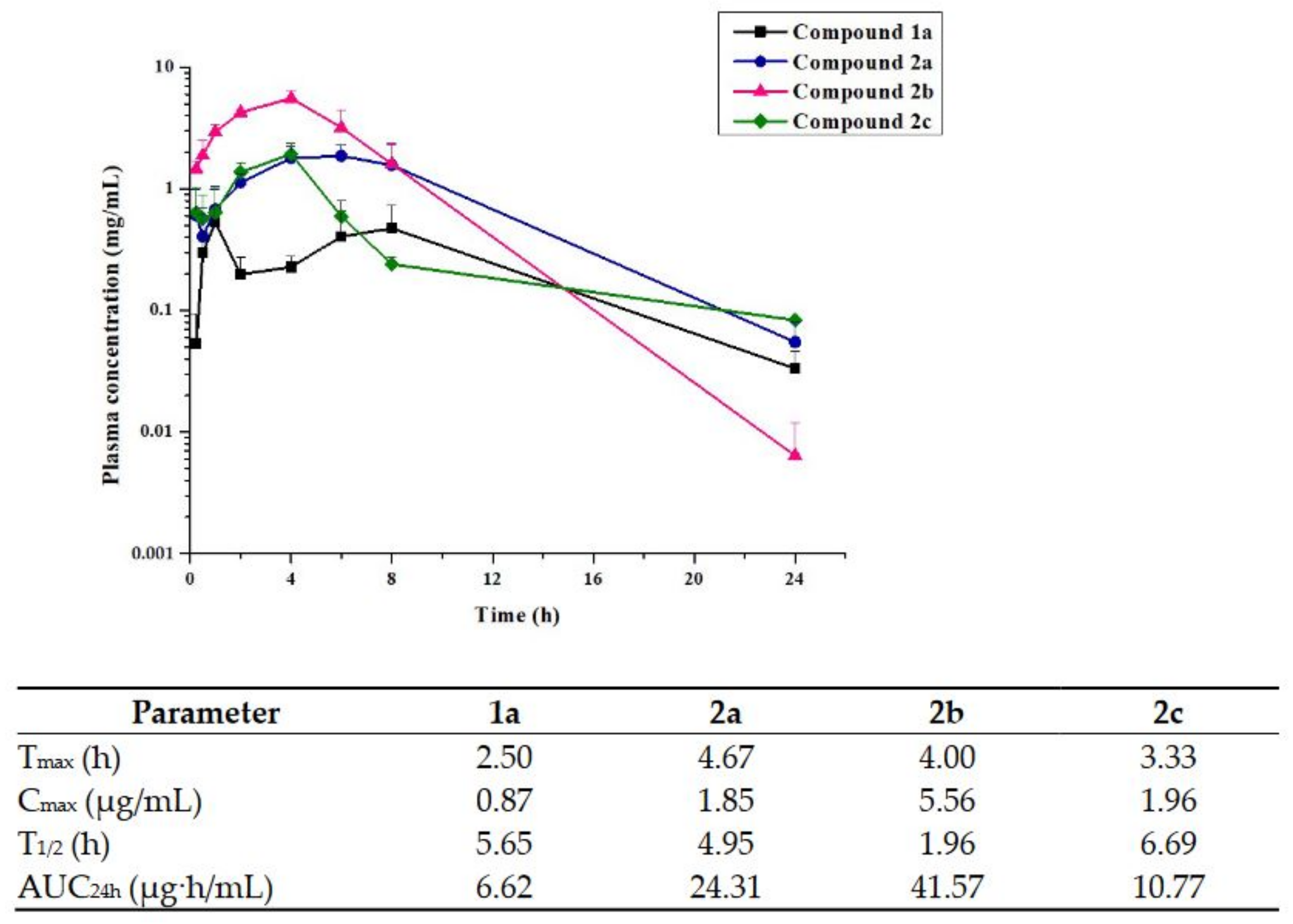
2.4. Pharmacokinetics
2.5. hERG Affinity, Microsomal Stability, and Cytotoxicity
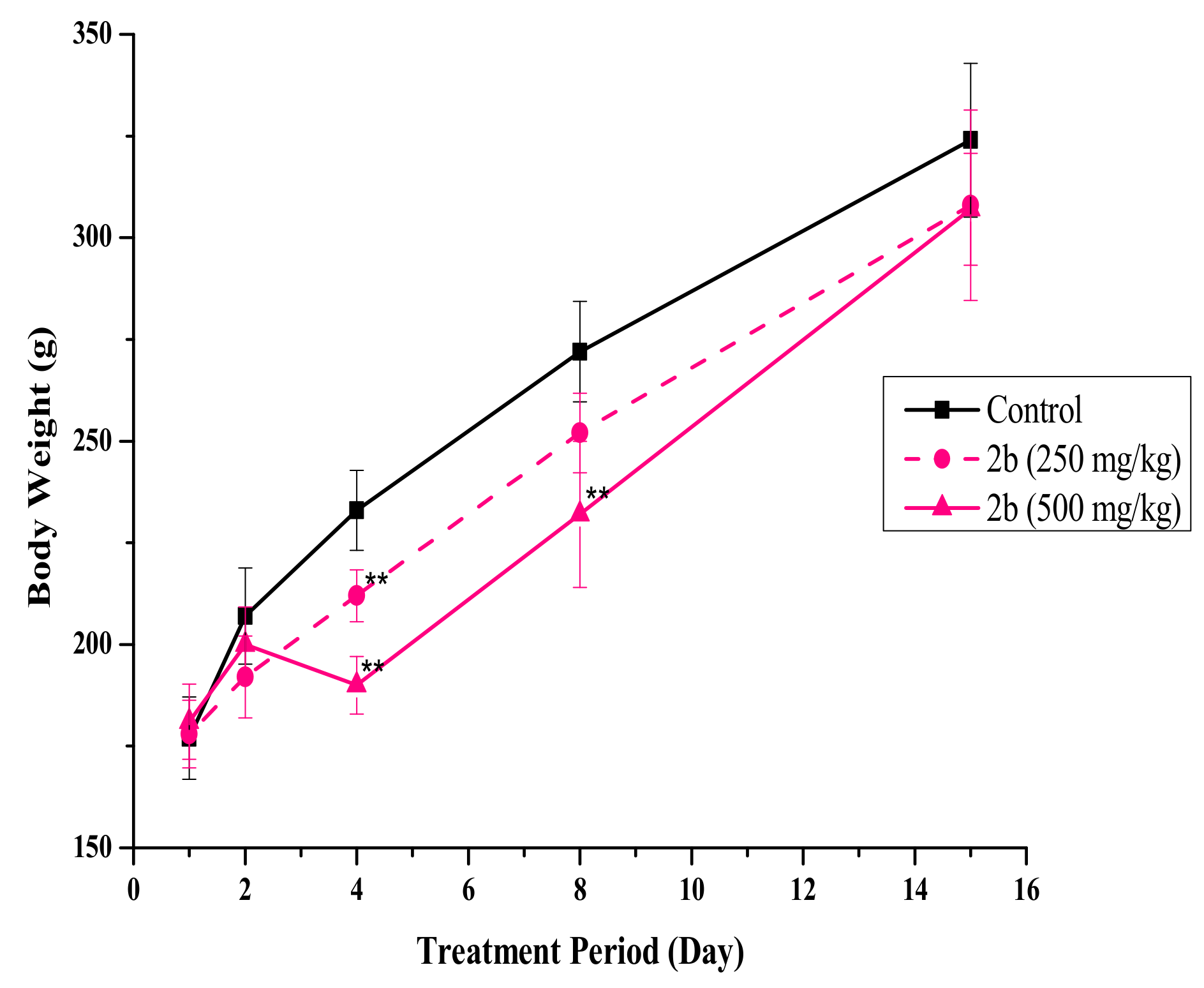
2.6. In Vivo Single-Dose Toxicity
3. Materials and Methods
3.1. Chemistry
3.1.1. 7-Chloro-2-((3,5-dichlorophenyl)amino)quinazolin-4(3H)-one (1a)
3.1.2. 7-Chloro-2-((3,5-difluorophenyl)amino)quinazolin-4(3H)-one (1b)
3.1.3. 2-((3,5-Dichlorophenyl)amino)-5-methoxyquinazolin-4(3H)-one (1c)
3.1.4. N-(3,5-Dichlorophenyl)cyanamide (4a)
3.1.5. N-(3,5-Dichlorophenyl)cyanamide (4b)
3.1.6. 2-((3,5-Dichlorophenyl)amino)-5-hydroxyquinazolin-4(3H)-one (5c)
3.1.7. N-(7-Dichloro-4-oxo-3,4-dihydroquinazolin-2-yl)-N-(3,5-dichlorophenyl)acetamide (2a)
3.1.8. N-(7-Chloro-4-oxo-3,4-dihydroquinazolin-2-yl)-N-(3,5-difluorophenyl)acetamide (2b)
3.1.9. N-(3,5-Dichlorophenyl)-N-(5-hydroxy-4-oxo-3,4-dihydroquinazolin-2-yl)acetamide (2c)
3.1.10. N-(7-Chloro-4-oxo-3,4-dihydroquinazolin-2-yl)-N-(3,5-dichlorophenyl)propionamide (6a)
3.1.11. Methyl (7-Chloro-4-oxo-3,4-dihydroquinazolin-2-yl)(3,5-dichlorophenyl)carbamate (6b)
3.1.12. 7-Chloro-2-((3,5-dichlorophenyl)(methyl)amino)quinazolin-4(3H)-one (6c)
3.1.13. N-(7-Chloro-4-oxo-3,4-dihydroquinazolin-2-yl)-N-(3,5-dichlorophenyl)-4-methylben-zenesulfonamide (6d)
3.1.14. 2-(Benzyloxy)-N-(7-chloro-4-oxo-3,4-dihydroquinazolin-2-yl)-N-(3,5-dichlorophenyl)-2-hydroxy-acetamide (7)
3.1.15. N-(7-Chloro-4-oxo-3,4-dihydroquinazolin-2-yl)-N-(3,5-dichlorophenyl)-2-hydroxy-acetamide (8)
3.2. Cell Line and Virus
3.3. Concentration–Response Curve Analysis by Immunofluorescence Assay
3.4. Pseudovirus-Based Entry Assay
3.5. hERG Channel Patch-Clamp Assay
3.6. Microsomal Stability Assay
3.7. Cell Viability Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; De Clercq, E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 2020, 19, 149–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.; Urquiza, J.; Ramírez, D.; Alonso, C.; Campillo, N.E.; et al. COVID-19: Drug Targets and Potential Treatments. J. Med. Chem. 2020, 63, 12359–12386. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Weekly Operational Update on COVID-19. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19 (accessed on 15 June 2022).
- Liu, D.X.; Liang, J.Q.; Fung, T.S. Human coronavirus-229E, -OC43, -NL63, and -HKU1 (Coronaviridae). In Encyclopedia of Virology; Academic Press: Cambridge, MA, USA, 2021; pp. 428–440. [Google Scholar]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Olivera, T.; Karim, S.A. New SARS-CoV-2 Variants—Clinical, Public Health, and Vaccine Implications. N. Engl. J. Med. 2021, 384, 1866–1868. [Google Scholar]
- Mahase, E. Delta variant: What is happening with transmission, hospital admissions, and restrictions. BMJ 2021, 373, n1513. [Google Scholar] [CrossRef]
- Mahase, E. Covid-19: Hospital admission 50–70% less likely with omicron than delta, but transmission a major concern. BMJ 2021, 375, n3151. [Google Scholar] [CrossRef]
- Gatti, M.; De Ponti, F. Drug Repurposing in the COVID-19 Era: Insights from Case Studies Showing Pharmaceutical Peculiarities. Pharmaceutics 2021, 13, 302. [Google Scholar] [CrossRef]
- The RECOVERY Collaborative Group. Effect of Hydroxychloroquine in Hospitalized Patients with Covid-19. N. Engl. J. Med. 2020, 383, 2030–2040. [Google Scholar] [CrossRef]
- Horby, P.W.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Emberson, J.; Palfreeman, A.; Raw, J.; Elmahi, E.; Prudon, B.; et al. Lopinavir–ritonavir in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2020, 396, 1345–1352. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Jayk Bernal, A.; Gomes da Silva, M.M.; Musungaie, D.B.; Kovalchuk, E.; Gonzalez, A.; Delos Reyes, V.; Martín-Quirós, A.; Caraco, Y.; Williams-Diaz, A.; Brown, M.L.; et al. Molnupiravir for Oral Treatment of COVID-19 in Nonhospitalized Patients. N. Engl. J. Med. 2022, 386, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Whitley, R. Molnupiravir—A Step toward Orally Bioavailable Therapies for Covid-19. N. Engl. J. Med. 2021, 386, 592–593. [Google Scholar] [CrossRef] [PubMed]
- Painter, G.R.; Natchus, M.G.; Cohen, O.; Holman, W.; Painter, W.P. Developing a direct acting, orally available antiviral agent in a pandemic: The evolution of molnupiravir as a potential treatment for COVID-19. Curr. Opin. Virol. 2021, 50, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.A.; Eron, J.J.; Holman, W.; Cohen, M.S.; Fang, L.; Szewczyk, L.J.; Sheahan, T.P.; Baric, R.; Mollan, K.R.; Wolfe, C.R.; et al. A phase 2a clinical trial of molnupiravir in patients with COVID-19 shows accelerated SARS-CoV-2 RNA clearance and elimination of infectious virus. Sci. Transl. Med. 2022, 14, 1–11. [Google Scholar] [CrossRef]
- Gordon, C.J.; Tchesnokov, E.P.; Schinazi, R.F.; Götte, M. Molnupiravir promotes SARS-CoV-2 mutagenesis via the RNA template. J. Biol. Chem. 2021, 297, 100770. [Google Scholar] [CrossRef]
- Janion, C. On the different response of Salmonella Typhimurium HISG46 and TA1530 to mutagenic acion of base analogues. Acta Biochim. Pol. 1979, 26, 171–177. [Google Scholar]
- Press Release Archive in Pfizer. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-announces-additional-phase-23-study-results (accessed on 1 April 2022).
- Lee, J.Y.; Shin, Y.S.; Jeon, S.; Lee, S.I.; Noh, S.; Cho, J.E.; Jang, M.S.; Kim, S.; Song, J.H.; Kim, H.R.; et al. Design, synthesis and biological evaluation of 2-aminoquinazolin-4(3H)-one derivatives as potential SARS-CoV-2 and MERS-CoV treatments. Bioorg. Med. Chem. Lett. 2021, 39, 127885. [Google Scholar] [CrossRef]
- Lee, J.Y.; Shin, Y.S.; Jeon, S.; Lee, S.I.; Cho, J.; Myung, S.; Jang, M.S.; Kim, S.; Song, J.H.; Kim, H.R.; et al. Synthesis and biological evaluation of 2-benzylaminoquinazolin-4(3H)-one derivatives as a potential treatment for SARS-CoV-2. Bull. Korea Chem. Soc. 2022, 43, 412–416. [Google Scholar] [CrossRef]
- Liao, Z.Y.; Yeh, W.H.; Liao, P.Y.; Liu, Y.T.; Chen, Y.C.; Chen, Y.H.; Hsieh, T.H.; Lin, C.C.; Lu, M.H.; Chen, Y.S.; et al. Regioselective synthesis and biological evaluation of: N -substituted 2-aminoquinazolin-4-ones. Org. Biomol. Chem. 2018, 16, 4482–4494. [Google Scholar] [CrossRef]
- Boyle, P.H.; Lockhart, R.J. Synthesis and properties of 7-alkoxyfurazano[3,4-d]pyrimidines and their use in the preparation of 4-alkoxypteridines and N3-substituted pterins. J. Org. Chem. 1985, 50, 5127–5132. [Google Scholar] [CrossRef]
- Shin, Y.S.; Lee, J.Y.; Noh, S.; Kwak, Y.; Jeon, S.; Kwon, S.; Jin, Y.-H.; Jang, M.S.; Kim, S.; Song, J.H.; et al. Discovery of cyclic sulfonamide derivatives as potent inhibitors of SARS-CoV-2. Bioorg. Med. Chem. Lett. 2021, 31, 127667. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.Y.; Jeon, S.; Jang, Y.; Gotina, L.; Won, J.; Ju, Y.H.; Kim, S.; Jang, M.W.; Won, W.; Park, M.G.; et al. Platycodin D, a natural component of Platycodon grandiflorum, prevents both lysosome- and TMPRSS2-driven SARS-CoV-2 infection by hindering membrane fusion. Exp. Mol. Med. 2021, 53, 956–972. [Google Scholar] [CrossRef] [PubMed]
- El-Haj, B.M.; Ahmed, S.B.M.; Garawi, M.A.; Ali, H.S. Linking aromatic hydroxy metabolic functionalization of drug molecules to structure and pharmacologic activity. Molecules 2018, 23, 2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Lee, H.; Lee, E.; Kang, S.K.; Nam, J.M.; Lee, M. Responsive nematic gels from the self-assembly of aqueous nanofibres. Nat. Commun. 2011, 2, 6–10. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Hong, Y.; Kleintop, T.A.; McConnell, O.J.; Huryn, D.M. Optimization of a higher throughput microsomal stability screening assay for profiling drug discovery candidates. J. Biomol. Screen. 2003, 8, 453–462. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Compound | R | X | Y | IC50 a (μM) | CC50 b (μM) | SI c | ClogP d |
| 1a | H | 7-Cl | Cl | 0.23 | >25 | 110 | 5.0 |
| 1b | H | 7-Cl | F | 0.24 | 18 | 74 | 3.9 |
| 2a | -COCH3 | 7-Cl | Cl | 0.33 | 9.39 | 26 | 2.4 |
| 2b | -COCH3 | 7-Cl | F | 0.29 | >25 | 87 | 1.3 |
| 2c | -COCH3 | 5-OH | Cl | 0.11 | >25 | 227 | 1.8 |
| 5c | H | 5-OH | Cl | 0.15 | >25 | 168 | 4.5 |
| 6a | -COCH2CH3 | 7-Cl | Cl | 0.21 | >25 | 117 | 5.1 |
| 6b | -CO2CH3 | 7-Cl | Cl | 7.05 | >25 | 4 | 5.1 |
| 6c | -CH3 | 7-Cl | Cl | 5.66 | >25 | 4 | 4.8 |
| 6d | p-Ts | 7-Cl | Cl | 0.57 | >25 | 44 | 6.5 |
| 8 | -COCH2OH | 7-Cl | Cl | 2.57 | >25 | 10 | 3.8 |
| Remdesivir | 3.47 | >50 | 14 | 3.2 | |||
| Compound | hERG K+ Channel IC50 (μM) | Microsomal Stability (%) a | Cytotoxicity (μM) b | ||||
|---|---|---|---|---|---|---|---|
| Rat | Human | HFL-1 | L929 | NIH 3T3 | CHO-K1 | ||
| 2a | 15.2 | 78 | 83 | 2.2 | 1.2 | 7.6 | 1.5 |
| 2b | 30.0 | >99 | >99 | 12.6 | 17.9 | 22.7 | 14.6 |
| 2c | >50 | 57 | 10 | 13.7 | 42.7 | 59.9 | 17.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, Y.S.; Lee, J.Y.; Jeon, S.; Cho, J.-E.; Myung, S.; Jang, M.S.; Kim, S.; Song, J.H.; Kim, H.R.; Park, H.-g.; et al. Optimization of 2-Aminoquinazolin-4-(3H)-one Derivatives as Potent Inhibitors of SARS-CoV-2: Improved Synthesis and Pharmacokinetic Properties. Pharmaceuticals 2022, 15, 831. https://doi.org/10.3390/ph15070831
Shin YS, Lee JY, Jeon S, Cho J-E, Myung S, Jang MS, Kim S, Song JH, Kim HR, Park H-g, et al. Optimization of 2-Aminoquinazolin-4-(3H)-one Derivatives as Potent Inhibitors of SARS-CoV-2: Improved Synthesis and Pharmacokinetic Properties. Pharmaceuticals. 2022; 15(7):831. https://doi.org/10.3390/ph15070831
Chicago/Turabian StyleShin, Young Sup, Jun Young Lee, Sangeun Jeon, Jung-Eun Cho, Subeen Myung, Min Seong Jang, Seungtaek Kim, Jong Hwan Song, Hyoung Rae Kim, Hyeung-geun Park, and et al. 2022. "Optimization of 2-Aminoquinazolin-4-(3H)-one Derivatives as Potent Inhibitors of SARS-CoV-2: Improved Synthesis and Pharmacokinetic Properties" Pharmaceuticals 15, no. 7: 831. https://doi.org/10.3390/ph15070831