
Search for Novel Potent Inhibitors of the SARS-CoV-2 Papain-like Enzyme: A Computational Biochemistry Approach

 ,
,  ,
,
Abstract
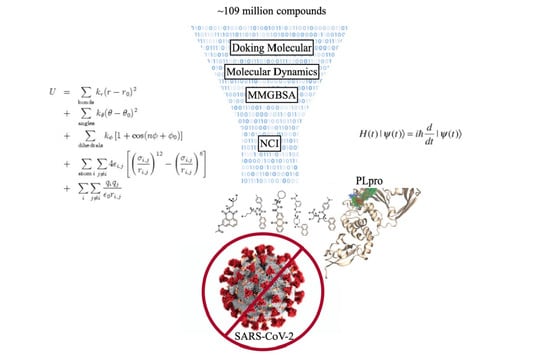
:
1. Introduction
2. Results and Discussion
2.1. Molecular Docking
2.2. Physicochemical Descriptors and ADME Properties
2.3. Molecular Dynamics Simulation and Protein/Ligand Interactions
2.4. Non-Covalent Interactions
2.5. Free Energy of Binding by MMGBSA
3. Materials and Methods
3.1. Molecular Docking
3.2. Ligand Efficiency (LE)
3.3. BEI and LLE
3.4. ADME-Tox Properties
3.5. Molecular Dynamics (MD) Simulation
3.6. Cluster Analysis
3.7. Free Energy Calculation
3.8. Non-Covalent Interactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meister, K.D.; Pandian, V.; Hillel, A.T.; Walsh, B.K.; Brodsky, M.B.; Balakrishnan, K.; Best, S.R.; Chinn, S.B.; Cramer, J.D.; Graboyes, E.M.; et al. Multidisciplinary Safety Recommendations after Tracheostomy during COVID-19 Pandemic: State of the Art Review. Otolaryngol. Head Neck Surg. 2021, 164, 984–1000. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T.; Weeraratne, D.; Snowdon, J.L.; Parida, L. Emergence of Drift Variants that May Affect COVID-19 Vaccine Development and Antibody Treatment. Pathogens 2020, 9, 324. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. Rapid displacement of SARS-CoV-2 variant B.1.1.7 by B.1.617.2 and P.1 in the United States. Nature 2021, 595, 17–18. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; Bassi, J.; De Marco, A.; Chen, A.; Walls, A.C.; Di Iulio, J.; Tortorici, M.A.; Navarro, M.-J.; Silacci-Fregni, C.; Saliba, C.; et al. SARS-CoV-2 immune evasion by the B.1.427/B.1.429 variant of concern. Science 2021, 7994, 6. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, S.; Tang, Y.; Zhang, S.; Xu, D.; Yue, S. Clinical Characteristics, Transmissibility, Pathogenicity, Susceptible Populations, and Re-infectivity of Prominent COVID-19 Variants. Aging Dis. 2022, 13, 402–422. [Google Scholar] [CrossRef]
- Zumla, A.; Chan, J.F.W.; Azhar, E.I.; Hui, D.S.C.; Yuen, K.Y. Coronaviruses-drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Cavasotto, C.N.; Lamas, M.S.; Maggini, J. Functional and druggability analysis of the SARS-CoV-2 proteome. Eur. J. Pharmacol. 2021, 890, 173705. [Google Scholar] [CrossRef]
- Ul Qamar, M.T.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020, 39, e106275. [Google Scholar] [CrossRef]
- Ratia, K.; Pegan, S.; Takayama, J.; Sleeman, K.; Coughlin, M.; Baliji, S.; Chaudhuri, R.; Fu, W.; Prabhakar, B.S.; Johnson, M.E.; et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. USA 2008, 105, 16119–16124. [Google Scholar] [CrossRef] [Green Version]
- Clasman, J.R.; Everett, R.K.; Srinivasan, K.; Mesecar, A.D. Decoupling Deisgylating and Deubiquitinating Activities of the MERS Virus Papain-Like Protease; Elsevier: Amsterdam, The Netherlands, 2020; Volume 174, ISBN 7654961189. [Google Scholar]
- Ratia, K.; Kilianski, A.; Baez-Santos, Y.M.; Baker, S.C.; Mesecar, A. Structural Basis for the Ubiquitin-Linkage Specificity and deISGylating Activity of SARS-CoV Papain-Like Protease. PLoS Pathog. 2014, 10, e1004113. [Google Scholar] [CrossRef] [Green Version]
- Békés, M.; van der Heden van Noort, G.J.; Ekkebus, R.; Ovaa, H.; Huang, T.T.; Lima, C.D. Recognition of Lys48-Linked Di-ubiquitin and Deubiquitinating Activities of the SARS Coronavirus Papain-like Protease. Mol. Cell 2016, 62, 572–585. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Qin, B.; Chen, P.; Zhu, K.; Hou, P.; Wojdyla, J.A.; Wang, M.; Cui, S. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B 2021, 11, 237–245. [Google Scholar] [CrossRef]
- Fu, Z.; Huang, B.; Tang, J.; Liu, S.; Liu, M.; Ye, Y.; Liu, Z.; Xiong, Y.; Zhu, W.; Cao, D.; et al. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Shan, H.; Liu, J.; Shen, J.; Dai, J.; Xu, G.; Lu, K.; Han, C.; Wang, Y.; Xu, X.; Tong, Y.; et al. Development of potent and selective inhibitors targeting the papain-like protease of SARS-CoV-2. Cell Chem. Biol. 2021, 28, 855–865.e9. [Google Scholar] [CrossRef]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Yañez, O.; Osorio, M.I.; Areche, C.; Vasquez-Espinal, A.; Bravo, J.; Sandoval-Aldana, A.; Pérez-Donoso, J.M.; González-Nilo, F.; Matos, M.J.; Osorio, E.; et al. Theobroma cacao L. compounds: Theoretical study and molecular modeling as inhibitors of main SARS-CoV-2 protease. Biomed. Pharmacother. 2021, 140, 111764. [Google Scholar] [CrossRef]
- Alamri, M.A.; Tahir ul Qamar, M.; Mirza, M.U.; Bhadane, R.; Alqahtani, S.M.; Muneer, I.; Froeyen, M.; Salo-Ahen, O.M.H. Pharmacoinformatics and molecular dynamics simulation studies reveal potential covalent and FDA-approved inhibitors of SARS-CoV-2 main protease 3CLpro. J. Biomol. Struct. Dyn. 2021, 39, 4936–4948. [Google Scholar] [CrossRef]
- Khan, A.; Ali, S.S.; Khan, M.T.; Saleem, S.; Ali, A.; Suleman, M.; Babar, Z.; Shafiq, A.; Khan, M.; Wei, D.Q. Combined drug repurposing and virtual screening strategies with molecular dynamics simulation identified potent inhibitors for SARS-CoV-2 main protease (3CLpro). J. Biomol. Struct. Dyn. 2021, 39, 4659–4670. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Řezáč, J.; Hobza, P. Advanced corrections of hydrogen bonding and dispersion for semiempirical quantum mechanical methods. J. Chem. Theory Comput. 2012, 8, 141–151. [Google Scholar] [CrossRef] [PubMed]
- James, J.P. Stewart MOPAC: A semiempirical molecular orbital program. J. Comput. Aided. Mol. Des. 1990, 4, 1–105. [Google Scholar]
- Abad-Zapatero, C. Ligand Efficiency Indices for Drug Discovery. Ligand Effic. Indices Drug Discov. 2013, 469–488. [Google Scholar] [CrossRef]
- Cavalluzzi, M.M.; Mangiatordi, G.F.; Nicolotti, O.; Lentini, G. Ligand efficiency metrics in drug discovery: The pros and cons from a practical perspective. Expert Opin. Drug Discov. 2017, 12, 1087–1104. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. Ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Peters, M.B.; Yang, Y.; Wang, B.; Füsti-Molnár, L.; Weaver, M.N.; Merz, K.M. Structural survey of zinc-containing proteins and development of the zinc AMBER force field (ZAFF). J. Chem. Theory Comput. 2010, 6, 2935–2947. [Google Scholar] [CrossRef] [Green Version]
- Seritan, S.; Bannwarth, C.; Fales, B.S.; Hohenstein, E.G.; Kokkila-Schumacher, S.I.L.; Luehr, N.; Snyder, J.W.; Song, C.; Titov, A.V.; Ufimtsev, I.S.; et al. TeraChem: Accelerating electronic structure and ab initio molecular dynamics with graphical processing units. J. Chem. Phys. 2020, 152, 224110. [Google Scholar] [CrossRef]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. generalized born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Osorio, M.; Cabrera, M.Á.; Gonzalez-Nilo, F.D.; Pérez-Donoso, J.M. The odd loop regions of XenA and XenB enzymes modulate their interaction with nitro-explosives and provide structural support for their regioselectivity. J. Chem. Inf. Model. 2019, 59, 3860–3870. [Google Scholar] [CrossRef]
- Fogha, J.; Diharce, J.; Obled, A.; Aci-Sèche, S.; Bonnet, P. Computational Analysis of Crystallization Additives for the Identification of New Allosteric Sites. ACS Omega 2020, 5, 2114–2122. [Google Scholar] [CrossRef] [Green Version]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting noncovalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Calculated Free Energy of Decomposition (kcal/mol) | |||||
|---|---|---|---|---|---|
| ΔGbind | ΔEvdW | ΔEelect | ΔGgas | ΔGsolv | |
| GRL0617 | −32.6 | −37.9 | −21.6 | −59.52 | 26.9 |
| 12C | −32.8 | −38.9 | −22.1 | −61.0 | 28.2 |
| D24 | −13.6 | −22.2 | −10.3 | −32.5 | 18.9 |
| D28 | −30.2 | −38.2 | −31.9 | −70.1 | 39.8 |
| D04 | −34.2 | −42.4 | −37,1 | −79.5 | 45.3 |
| D59 | −30.5 | −42.2 | −25.5 | −67.7 | 37.2 |
| D06 | −36.8 | −40.3 | −36.9 | −77.2 | 40.5 |
| D60 | −42.0 | −44.7 | −42.2 | −86.9 | 44.9 |
| D99 | −42.3 | −44.8 | −42.6 | −87.4 | 45.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osorio, M.I.; Yáñez, O.; Gallardo, M.; Zuñiga-Bustos, M.; Mulia-Rodríguez, J.; López-Rendón, R.; García-Beltrán, O.; González-Nilo, F.; Pérez-Donoso, J.M. Search for Novel Potent Inhibitors of the SARS-CoV-2 Papain-like Enzyme: A Computational Biochemistry Approach. Pharmaceuticals 2022, 15, 986. https://doi.org/10.3390/ph15080986
Osorio MI, Yáñez O, Gallardo M, Zuñiga-Bustos M, Mulia-Rodríguez J, López-Rendón R, García-Beltrán O, González-Nilo F, Pérez-Donoso JM. Search for Novel Potent Inhibitors of the SARS-CoV-2 Papain-like Enzyme: A Computational Biochemistry Approach. Pharmaceuticals. 2022; 15(8):986. https://doi.org/10.3390/ph15080986
Chicago/Turabian StyleOsorio, Manuel I., Osvaldo Yáñez, Mauricio Gallardo, Matías Zuñiga-Bustos, Jorge Mulia-Rodríguez, Roberto López-Rendón, Olimpo García-Beltrán, Fernando González-Nilo, and José M. Pérez-Donoso. 2022. "Search for Novel Potent Inhibitors of the SARS-CoV-2 Papain-like Enzyme: A Computational Biochemistry Approach" Pharmaceuticals 15, no. 8: 986. https://doi.org/10.3390/ph15080986