The Molecular Mechanisms of Anesthetic Action: Updates and Cutting Edge Developments from the Field of Molecular Modeling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: From Lipid to Protein Theories of Anesthetic Action
1.1. The Meyer-Overton and Lipid Theories of Anesthetic Action
1.2. Exceptions to the Meyer-Overton Correlation
1.3. Direct Interactions of Anesthetics with Proteins
1.4. Altered Anesthetic Modulation of Ion Channel Electrophysiology via a Direct Effect
1.5. Studies of the Physicochemical Characteristics of an Anesthetic Binding Site
1.6. Reasons to Study Molecular Mechanisms of Anesthetic Action
2. Modeling A Putative Site of Anesthetic Action within Ligand-gated Ion Channels (LGICs): From Protein Schematic to Full-Scale Atomic Models
2.1. Physicochemical Basis of Anesthetic-Protein Interactions
2.2 Defining the Structural Problem

2.3. Predicting Transmembrane Secondary Structure
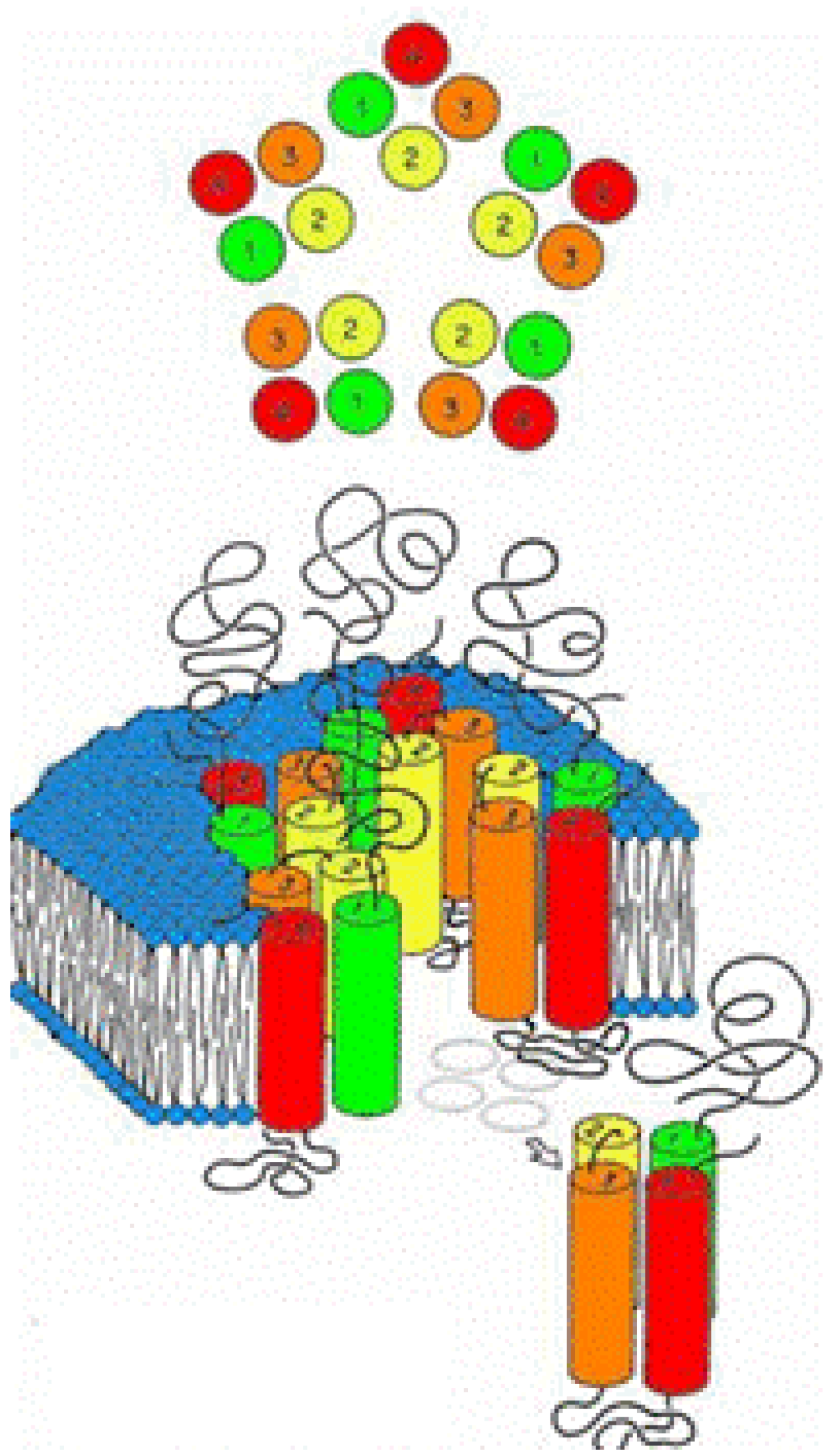
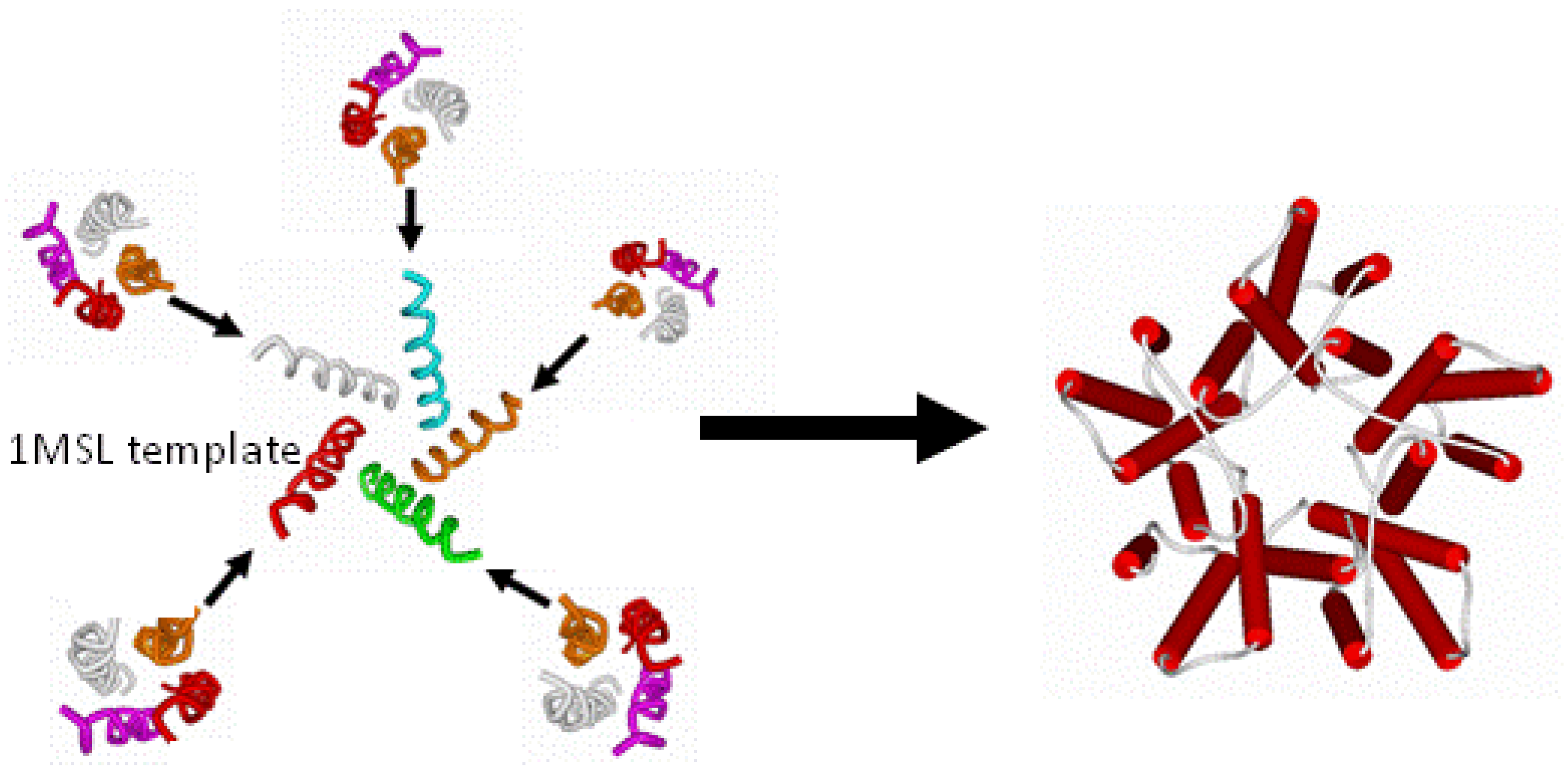
2.4. Building the Transmembrane Four-Helix Bundle and the Convergence of Residues Relevant to Anesthetic Binding

2.5 Building the Entire Transmembrane Domain

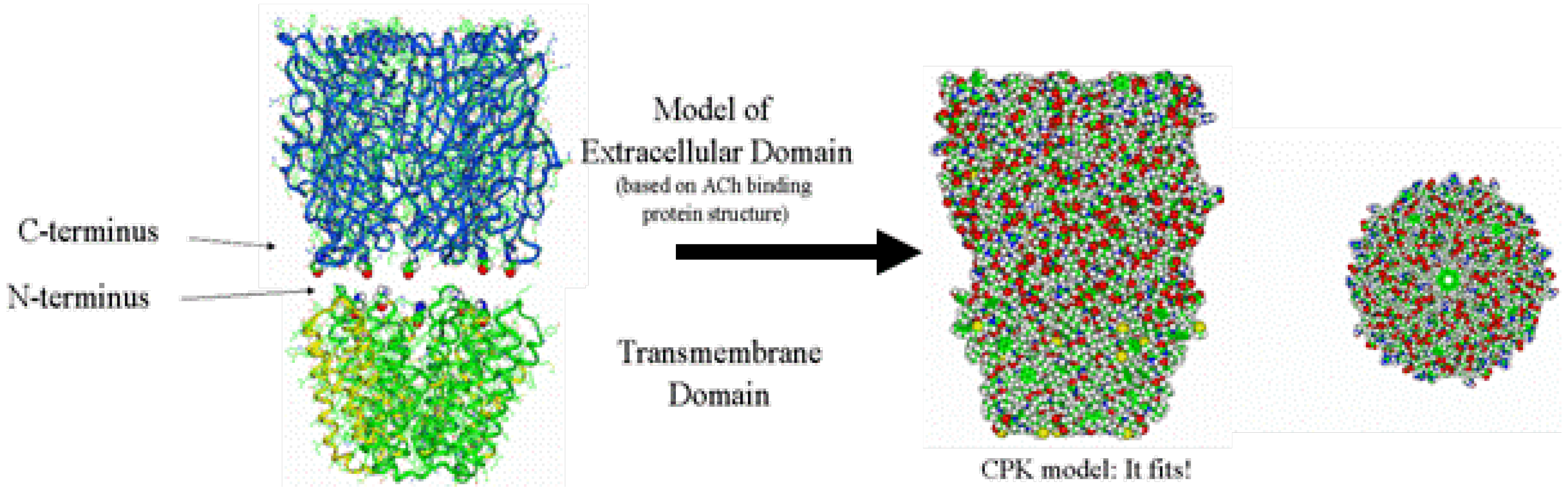
2.6. Building the Ligand-Binding Domain (LBD)
2.7. Aligning and Merging the TMD to the LBD


3. A Newer Alternative Model Based on Updated Prokaryotic Templates Changes the Putative Location of the Anesthetic Binding Site

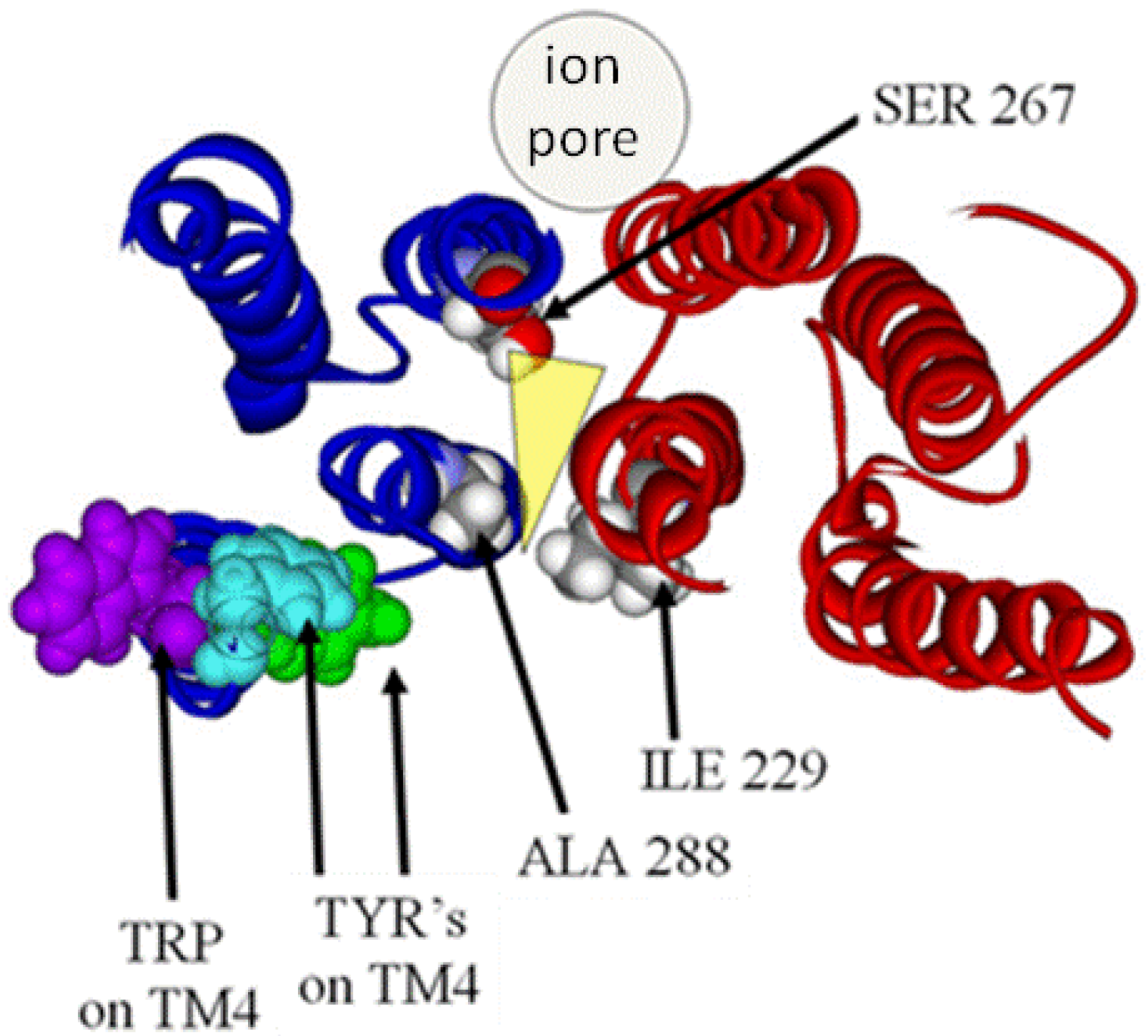
4. Results and Analysis of the LGIC Model: The Quandary of the Location of the Anesthetic Binding Site
5. How the LGIC Moves: The Results of Normal Mode Analyses
5.1. In Vacuo Simulations
5.2. Simulations of an LGIC in the Fully Hydrated Lipid Bilayer

5.3. Implications

6. Conclusion and Future Directions
Acknowledgements
References
- Meyer, H. Zur Theorie der alkoholnarkose I. Mit welch Eigenshaft der Anasthetika bedingt ihre narkotische Wirkung? Arch. Exp. Path. Pharmakol. (Naunyn Schmiedebergs) 1899, 42, 109–137. [Google Scholar] [CrossRef]
- Taheri, S.; Halsey, M.J.; Liu, J.; Eger, E.I., 2nd; Koblin, D.D.; Laster, M.J. What solvent best represents the site of action of inhaled anesthetics in humans, rats, and dogs? Anesth. Analg. 1991, 72, 627–634. [Google Scholar] [PubMed]
- Johansson, J.S.; Zou, H. Partitioning of four modern volatile general anesthetics into solvents that model buried amino acid side-chains. Biophys. Chem. 1999, 79, 107–116. [Google Scholar]
- Franks, N.P.; Lieb, W.R. Is membrane expansion relevant to anaesthesia? Nature 1981, 292, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Bertaccini, E.; Loew, G.H. Molecular dynamics simulation of anesthetic-phospholipid bilayer interactions. J. Biomol. Struct. Dyn. 1995, 12, 725–754. [Google Scholar]
- Cantor, R.S. The lateral pressure profile in membranes: a physical mechanism of general anesthesia. Biochemistry 1997, 36, 2339–2344. [Google Scholar]
- Koblin, D.D.; Chortkoff, B.S.; Laster, M.J.; Eger, E.I., 2nd; Halsey, M.J.; Ionescu, P. Polyhalogenated and perfluorinated compounds that disobey the Meyer-Overton hypothesis. Anesth Analg. 1994, 79, 1043–1048. [Google Scholar] [PubMed]
- Koblin, D.D.; Chortkoff, B.S.; Laster, M.J.; Eger, E.I.; Halsey, M.J.; Ionescu, P. Nonanesthetic polyhalogenated alkanes and deviations from the Meyer-Overton hypothesis. Prog. Anesth. Mech. 1995, 3, 451–456. [Google Scholar]
- Moss, G.W.; Lieb, W.R.; Franks, N.P. Anesthetic inhibition of firefly luciferase, a protein model for general anesthesia, does not exhibit pressure reversal. Biophys. J. 1991, 60, 1309–1314. [Google Scholar] [CrossRef] [PubMed]
- Franks, N.P.; Jenkins, A.; Conti, E.; Lieb, W.R.; Brick, P. Structural basis for the inhibition of firefly luciferase by a general anesthetic. Biophys. J. 1998, 75, 2205–2211. [Google Scholar]
- Mihic, S.J.; Ye, Q.; Wick, M.J.; Koltchine, V.V.; Krasowski, M.D.; Finn, S.E.; Mascia, M.P.; Valenzuela, C.F.; Hanson, K.K.; Greenblatt, E.P.; Harris, R.A.; Harrison, N.L. Sites of alcohol and volatile anaesthetic action on GABA(A) and glycine receptors. Nature 1997, 389, 385–389. [Google Scholar]
- Mascia, M.P.; Trudell, J.R.; Harris, R.A. Specific binding sites for alcohols and anesthetics on ligand-gated ion channels. Proc. Natl. Acad. Sci. USA 2000, 97, 9305–9310. [Google Scholar]
- Jurd, R.; Arras, M.; Lambert, S.; Drexler, B.; Siegwart, R.; Crestani, F.; Zaugg, M.; Vogt, K.E.; Ledermann, B.; Antkowiak, B.; Rudolph, U. General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor beta3 subunit. FASEB. J. 2003, 17, 250–252. [Google Scholar] [PubMed]
- Sandorfy, C. Weak intermolecular associations and anesthesia. Anesthesiology 2004, 101, 1225–1227. [Google Scholar]
- Abraham, M.H.; Lieb, W.R.; Franks, N.P. Role of hydrogen bonding in general anesthesia. J. Pharm. Sci. 1991, 80, 719–724. [Google Scholar]
- Trudell, J.R.; Koblin, D.D.; Eger, E.I., 2nd. A molecular description of how noble gases and nitrogen bind to a model site of anesthetic action. Anesth. Analg. 1998, 87, 411–418. [Google Scholar]
- Bertaccini, E.J.; Trudell, J.R.; Franks, N.P. The common chemical motifs within anesthetic binding sites. Anesth. Analg. 2007, 104, 318–324. [Google Scholar]
- Trudell, J.R.; Bertaccini, E. Comparative modeling of a GABAA alpha1 receptor using three crystal structures as templates. J. Mol. Graph Model 2004, 23, 39–49. [Google Scholar]
- Liu, R.; Loll, P.J.; Eckenhoff, R.G. Structural basis for high-affinity volatile anesthetic binding in a natural 4-helix bundle protein. FASEB J. 2005, 19, 567–576. [Google Scholar]
- Vedula, L.S.; Brannigan, G.; Economou, N.J.; Xi, J.; Hall, M.A.; Liu, R.; Rossi, M.J.; Dailey, W.P.; Grasty, K.C.; Klein, M.L.; Eckenhoff, R.G.; Loll, P.J. A unitary anesthetic binding site at high resolution. J. Biol. Chem. 2009, 284, 24176–24184. [Google Scholar]
- Gursky, O.; Fontano, E.; Bhyravbhatla, B.; Caspar, D.L. Stereospecific dihaloalkane binding in a pH-sensitive cavity in cubic insulin crystals. Proc Natl. Acad. Sci. USA 1994, 91, 12388–12392. [Google Scholar]
- Sachsenheimer, W.; Pai, E.F.; Schulz, G.E.; Schirmer, R.H. Halothane binds in the adenine-specific niche of crystalline adenylate kinase. FEBS Lett. 1977, 79, 310–312. [Google Scholar]
- Ishizawa, Y.; Sharp, R.; Liebman, P.A.; Eckenhoff, R.G. Halothane binding to a G protein coupled receptor in retinal membranes by photoaffinity labeling. Biochemistry 2000, 39, 8497–8502. [Google Scholar]
- Johansson, J.S.; Scharf, D.; Davies, L.A.; Reddy, K.S.; Eckenhoff, R.G. A designed four-alpha-helix bundle that binds the volatile general anesthetic halothane with high affinity. Biophys. J. 2000, 78, 982–993. [Google Scholar]
- Das, J.; Addona, G.H.; Sandberg, W.S.; Husain, S.S.; Stehle, T.; Miller, K.W. Identification of a general anesthetic binding site in the diacylglycerol-binding domain of protein kinase Cdelta. J. Biol. Chem. 2004, 279, 37964–37972. [Google Scholar]
- Ziebell, M.R.; Nirthanan, S.; Husain, S.S.; Miller, K.W.; Cohen, J.B. Identification of binding sites in the nicotinic acetylcholine receptor for [3H] azietomidate, a photoactivatable general anesthetic. J. Biol. Chem. 2004, 279, 17640–17649. [Google Scholar]
- Franks, N.P.; Lieb, W.R. Mechanisms of general anesthesia. Environ. Health Perspect 1990, 87, 199–205. [Google Scholar]
- Sewell, J.C.; Halsey, M.J. Shape similarity indices are the best predictors of substituted fluoroethane and ether anaesthesia. Eur. J. Med. Chem. 1997, 32, 731–737. [Google Scholar]
- Sewell, J.C.; Sear, J.W. Determinants of Volatile General Anesthetic Potency: A Preliminary Three-Dimensional Pharmacophore for Halogenated Anesthetics. Anesth. Analg. 2006, 102, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Unwin, N. Nicotinic acetylcholine receptor at 9 A resolution. J. Mol. Biol. 1993, 229, 1101–1124. [Google Scholar]
- Trudell, J.R.; Bertaccini, E. Evaluation of forcefields for molecular mechanics/dynamics calculations involving halogenated anesthetics. Toxicol. Lett. 1998, 100, 413–419. [Google Scholar]
- Trudell, J.R.; Bertaccini, E.; Eger, E.I.; Harrison, N.L.; Mihic, S.J.; Harris, R.A. The interface of molecular modeling and molecular genetics: A search for sites of anesthetic action. Prog. Anes. Mech. 2000, 6, 172–178. [Google Scholar]
- Bertaccini, E.; Trudell, J.R. Molecular modeling of ligand-gated ion channels: progress and challenges. Int. Rev. Neurobiol. 2001, 48, 141–166. [Google Scholar]
- Jenkins, A.; Greenblatt, E.P.; Faulkner, H.J.; Bertaccini, E.; Light, A.; Lin, A.; Andreasen, A.; Viner, A.; Trudell, J.R.; Harrison, N.L. Evidence for a common binding cavity for three general anesthetics within the GABAA receptor. J. Neurosci. 2001, 21, RC136. [Google Scholar]
- Yamakura, T.; Bertaccini, E.; Trudell, J.R.; Harris, R.A. Anesthetics and ion channels: molecular models and sites of action. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 23–51. [Google Scholar]
- Bertaccini, E.; Trudell, J.R. Predicting the transmembrane secondary structure of ligand-gated ion channels. Protein Eng. 2002, 15, 443–454. [Google Scholar]
- Trudell, J.R.; Bertaccini, E. Molecular modelling of specific and non-specific anaesthetic interactions. Br. J. Anaesth. 2002, 89, 32–40. [Google Scholar]
- Bertaccini, E.J.; Shapiro, J.; Brutlag, D.L.; Trudell, J.R. Homology modeling of a human glycine alpha 1 receptor reveals a plausible anesthetic binding site. J. Chem. Inf. Model 2005, 45, 128–135. [Google Scholar]
- Jeanmougin, F.; Thompson, J.D.; Gouy, M.; Higgins, D.G.; Gibson, T.J. Multiple sequence alignment with Clustal X. Trends Biochem. Sci. 1998, 23, 403–405. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar]
- Olszewski, K.; Yan, L.; Edwards, D.J. SeqFold. Fully automated fold recognition and modeling software. Evaluation and application. Theor. Chem. Acc. 1999, 101, 57–61. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar]
- Trudell, J. Unique assignment of inter-subunit association in GABA(A) alpha 1 beta 3 gamma 2 receptors determined by molecular modeling. Biochim. Biophys. Acta 2002, 1565, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Bocquet, N.; Nury, H.; Baaden, M.; Le Poupon, C.; Changeux, J.P.; Delarue, M.; Corringer, P.J. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature 2009, 457, 111–114. [Google Scholar]
- Hilf, R.J.; Dutzler, R. Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature 2009, 457, 115–118. [Google Scholar]
- Hilf, R.J.; Dutzler, R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature 2008, 452, 375–379. [Google Scholar]
- Bertaccini, E.; Trudell, J. Putative anesthetic binding sites within a GlyRa1 receptor model based on a prokaryotic template. Anesthesiology 2008, A644. [Google Scholar]
- Bertaccini, E.; Trudell, J.; Lindahl, E.; Murail, S. Anesthetic Binding Sites in a GlyRa1 Model Based on Open State Prokaryotic Ion Channel Templates. Anesthesiology 2009, A1399. [Google Scholar]
- Jensen, M.L.; Schousboe, A.; Ahring, P.K. Charge selectivity of the Cys-loop family of ligand-gated ion channels. J. Neurochem. 2005, 92, 217–225. [Google Scholar]
- Unwin, N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J. Mol. Biol. 2005, 346, 967–989. [Google Scholar]
- Akabas, M.H.; Karlin, A. Identification of acetylcholine receptor channel-lining residues in the M1 segment of the alpha-subunit. Biochemistry 1995, 34, 12496–12500. [Google Scholar]
- Akabas, M.H.; Kaufmann, C.; Archdeacon, P.; Karlin, A. Identification of acetylcholine receptor channel-lining residues in the entire M2 segment of the alpha subunit. Neuron 1994, 13, 919–927. [Google Scholar]
- Blanton, M.P.; Cohen, J.B. Identifying the lipid-protein interface of the Torpedo nicotinic acetylcholine receptor: secondary structure implications. Biochemistry 1994, 33, 2859–2872. [Google Scholar]
- Lobo, I.A.; Harris, R.A.; Trudell, J.R. Cross-linking of sites involved with alcohol action between transmembrane segments 1 and 3 of the glycine receptor following activation. J. Neurochem. 2008, 104, 1649–1662. [Google Scholar]
- Jenkins, A.; Andreasen, A.; Trudell, J.R.; Harrison, N.L. Tryptophan scanning mutagenesis in TM4 of the GABA(A) receptor alpha1 subunit: implications for modulation by inhaled anesthetics and ion channel structure. Neuropharmacology 2002, 43, 669–678. [Google Scholar]
- Richardson, J.E.; Garcia, P.S.; O'Toole, K.K.; Derry, J.M.; Bell, S.V.; Jenkins, A. A conserved tyrosine in the beta2 subunit M4 segment is a determinant of gamma-aminobutyric acid type A receptor sensitivity to propofol. Anesthesiology 2007, 107, 412–418. [Google Scholar]
- McCracken, L.; McCracken, M.; Gong, D.; Trudell, J.; Harris, R. Linking of Glycine Receptor Transmembrane Segments Three and Four Allows Assignment of Intrasubunit-Facing Residues. ACS Chem. Neurosci. 2010, 1, 14–18. [Google Scholar]
- Cheng, M.H.; Cascio, M.; Coalson, R.D. Homology modeling and molecular dynamics simulations of the alpha1 glycine receptor reveals different states of the channel. Proteins 2007, 68, 581–593. [Google Scholar]
- Stewart, D.; Desai, R.; Cheng, Q.; Liu, A.; Forman, S.A. Tryptophan Mutations at Azi-Etomidate Photo-Incorporation Sites on α1 or β2 Subunits Enhance GABAA Receptor Gating and Reduce Etomidate Modulation. Mol. Pharmacol. 2008, 6, 1687–1695. [Google Scholar]
- Bali, M.; Jansen, M.; Akabas, M.H. GABA-induced intersubunit conformational movement in the GABAA receptor alpha1M1-beta2M3 transmembrane subunit interface: experimental basis for homology modeling of an intravenous anesthetic binding site. J. Neurosci. 2009, 29, 3083–3092. [Google Scholar]
- Jansen, M.; Akabas, M.H. State-dependent cross-linking of the M2 and M3 segments: functional basis for the alignment of GABAA and acetylcholine receptor M3 segments. J. Neurosci. 2006, 26, 4492–4499. [Google Scholar]
- Li, G.D.; Chiara, D.C.; Sawyer, G.W.; Husain, S.S.; Olsen, R.W.; Cohen, J.B. Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J. Neurosci. 2006, 26, 11599–11605. [Google Scholar]
- Bertaccini, E.J.; Trudell, J.R.; Lindahl, E. Normal Mode Analysis Reveals the Channel Gating Motion within a Ligand Gated Ion Channel Model. In 7th International Conference on Basic and Systematic Mechanisms of Anesthesia; Elsevier: Nara, Japan, 2005; Volume 1283, pp. 160–163. [Google Scholar]
- Bertaccini, E.; Trudell, J.; Lindahl, E. Understanding Effects Of Anesthetics On Ligand-Gated Ion Channels (LGIC) in Lipid Membranes. Anesthesiology 2008, A852. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bertaccini, E.J. The Molecular Mechanisms of Anesthetic Action: Updates and Cutting Edge Developments from the Field of Molecular Modeling. Pharmaceuticals 2010, 3, 2178-2196. https://doi.org/10.3390/ph3072178
Bertaccini EJ. The Molecular Mechanisms of Anesthetic Action: Updates and Cutting Edge Developments from the Field of Molecular Modeling. Pharmaceuticals. 2010; 3(7):2178-2196. https://doi.org/10.3390/ph3072178
Chicago/Turabian StyleBertaccini, Edward J. 2010. "The Molecular Mechanisms of Anesthetic Action: Updates and Cutting Edge Developments from the Field of Molecular Modeling" Pharmaceuticals 3, no. 7: 2178-2196. https://doi.org/10.3390/ph3072178