
Meat-Borne-Parasite: A Nanopore-Based Meta-Barcoding Work-Flow for Parasitic Microbiodiversity Assessment in the Wild Fauna of French Guiana
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
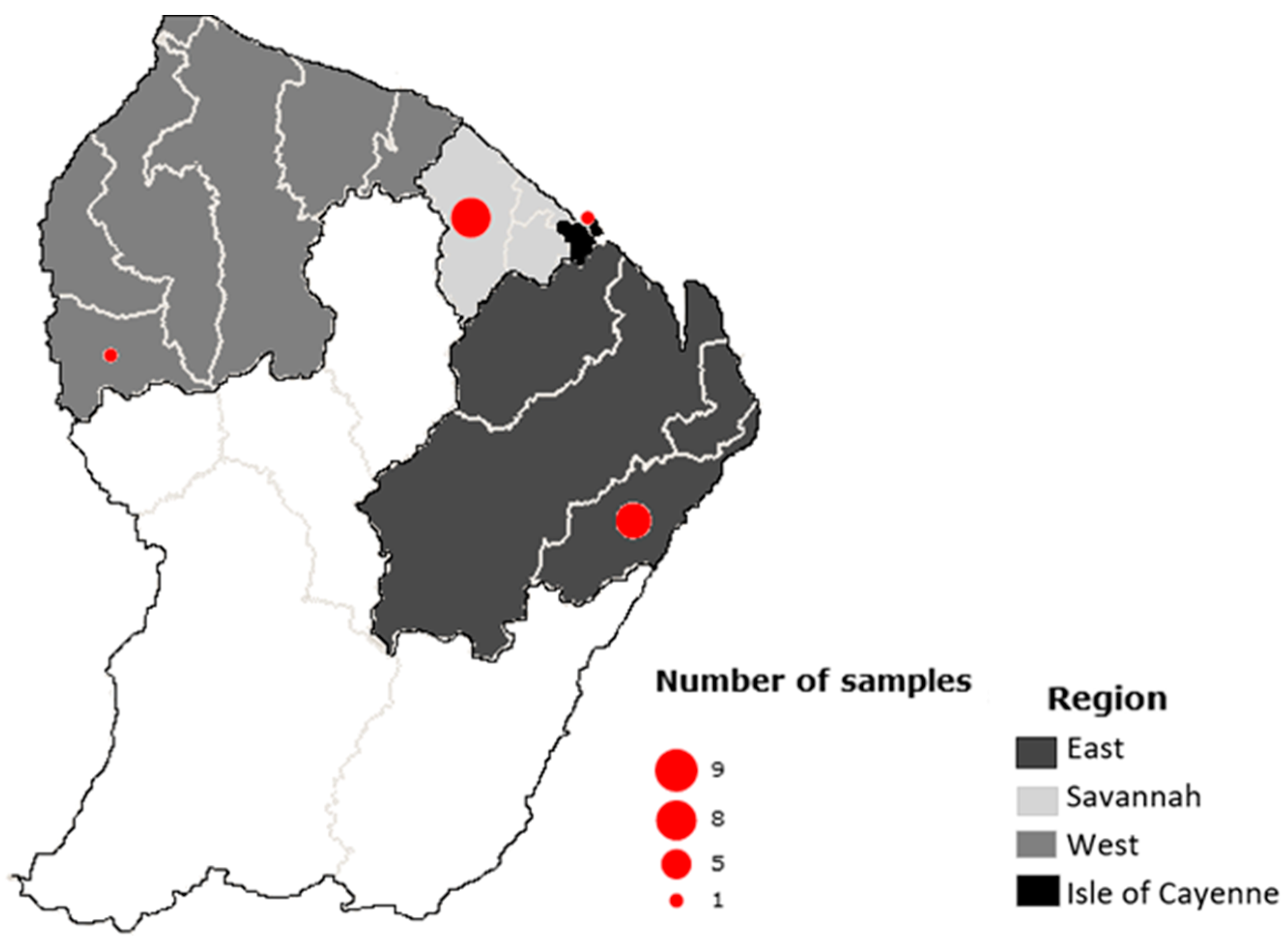
2.1. Sampling and Samples
2.2. Molecular Analysis
2.2.1. DNA Extraction
2.2.2. DNA Amplification
2.2.3. Nanopore Library Preparation and Sequencing
2.2.4. Illumina Library Preparation and Sequencing
2.3. Bioinformatics Processing
2.3.1. Meta-Barcoding Pipeline for Nanopore Data Analysis
2.3.2. Meta-Barcoding Pipeline for Illumina Data Analysis
2.3.3. Comparison of Nanopore and Illumina Meta-Barcoding Results
3. Results
3.1. Sampling and Apicomplexa Positive Samples Identification
3.2. Matched Samples Sequencing with Nanopore and Illumina Platforms
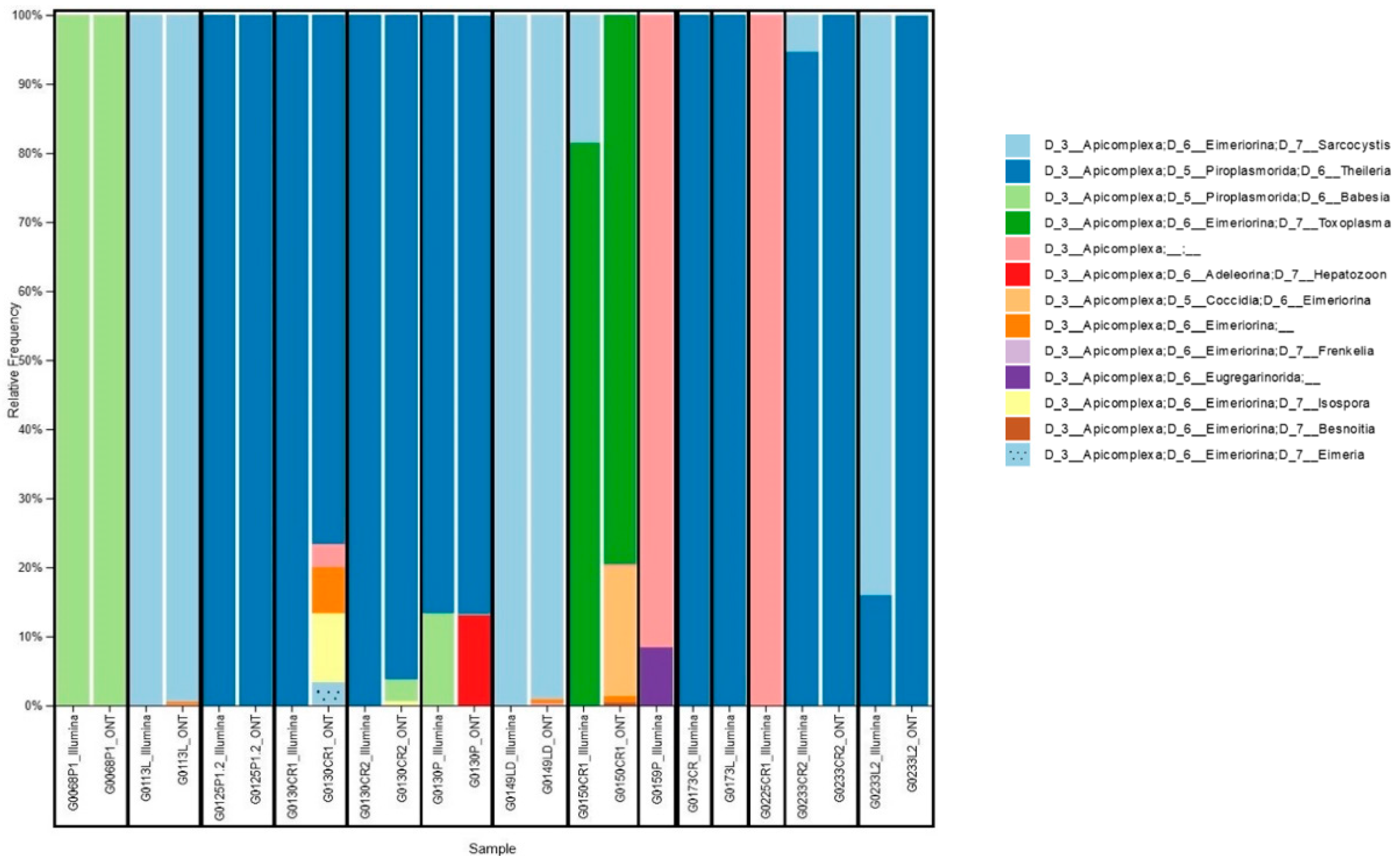
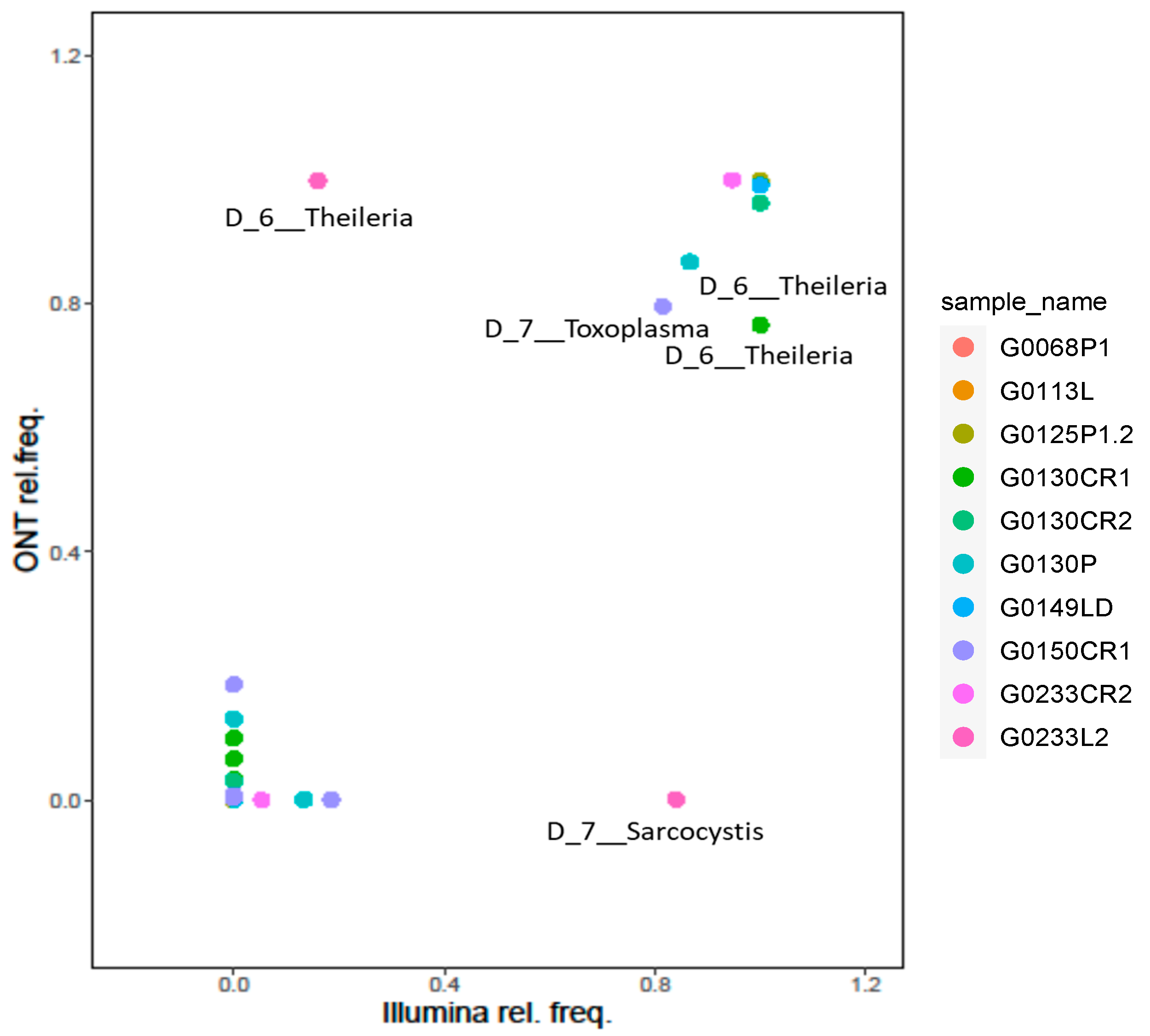
3.3. Taxonomy Assignment and Platforms Comparison
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pathmanathan, R.; Kan, S.-P. Three cases of human Sarcocystis infection with a review of human muscular sarcocystosis in Malaysia. Trop. Geogr. Med. 1992, 44, 102–108. [Google Scholar] [PubMed]
- Winter, M.; Abate, S.D.; Pasqualetti, M.I.; Fariña, F.A.; Ercole, M.E.; Pardini, L.; Moré, G.; Venturini, M.C.; Perera, N.; Corominas, M.J. Toxoplasma gondii and Trichinella infections in wild boars (Sus scrofa) from Northeastern Patagonia, Argentina. Prev. Vet. Med. 2019, 168, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Demar, M.; Ajzenberg, D.; Serrurier, B.; Dardé, M.-L.; Carme, B. Atypical Toxoplasma gondii strain from a free-living jaguar (Panthera onca) in French Guiana. Am. J. Trop. Med. Hyg. 2008, 78, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Dubey, J.P.; Van Wilpe, E.; Calero-Bernal, R.; Verma, S.K.; Fayer, R. Sarcocystis heydorni, n. sp. (Apicomplexa: Sarcocystidae) with cattle (Bos taurus) and human (Homo sapiens) cycle. Parasitol. Res. 2015, 114, 4143–4147. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, A.; Murer, L.; dos Santos, H.F.; Ludwig, A.; Sangioni, L.A.; Vogel, F.S. Molecular detection of Apicomplexa protozoa in tissues from Alouatta guariba clamitans. Pesqui. Veterinária Bras. 2021, 41, e06717. [Google Scholar] [CrossRef]
- Fayer, R. Sarcocystis spp. in Human Infections. Clin. Microbiol. Rev. 2004, 17, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Beck, H.-P.; Blake, D.; Dardé, M.-L.; Felger, I.; Pedraza-Díaz, S.; Regidor-Cerrillo, J.; Gómez-Bautista, M.; Ortega-Mora, L.M.; Putignani, L.; Shiels, B. Molecular approaches to diversity of populations of apicomplexan parasites. Int. J. Parasitol. 2009, 39, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Howells, R.; Carvalho, A.; Mello, M.; Rangel, N. Morphological and histochemical observations on Sarcocystis from the nine-banded armadillo, Dasypus novemcinctus. Ann. Trop. Med. Parasitol. 1975, 69, 463–474. [Google Scholar] [CrossRef]
- do Rêgo, W.M.F.; Costa, J.G.L.; Baraviera, R.C.d.A.; Pinto, L.V.; Bessa, G.d.L.; Lopes, R.E.N.; da Silveira, J.A.G.; Vitor, R.W.A. Sarcocystidae em aves silvestres do sudeste do Brasil. Rev. Bras. Parasitol. Veterinária 2021, 30, e028520. [Google Scholar]
- Laghoe, L. Toxoplasma gondii infection in free-living animals in French Guiana. 2019; Unpublished data. [Google Scholar]
- Brasseur, P.; Agnamey, P.; Emmanuel, E.; Pape, J.W.; Vaillant, M.; Raccurt, C.P. Cryptosporidium contamination of surface and water supplies in Haiti. Arch. Environ. Occup. Health 2011, 66, 12–17. [Google Scholar] [CrossRef]
- Demar, M.; Hommel, D.; Djossou, F.; Peneau, C.; Boukhari, R.; Louvel, D.; Bourbigot, A.-M.; Nasser, V.; Ajzenberg, D.; Darde, M.-L. Acute toxoplasmoses in immunocompetent patients hospitalized in an intensive care unit in French Guiana. Clin. Microbiol. Infect. 2012, 18, E221–E231. [Google Scholar] [CrossRef]
- Demar, M.; Ajzenberg, D.; Maubon, D.; Djossou, F.; Panchoe, D.; Punwasi, W.; Valery, N.; Peneau, C.; Daigre, J.-L.; Aznar, C. Fatal outbreak of human toxoplasmosis along the Maroni River: Epidemiological, clinical, and parasitological aspects. Clin. Infect. Dis. 2007, 45, e88–e95. [Google Scholar] [CrossRef]
- Carme, B.; Bissuel, F.; Ajzenberg, D.; Bouyne, R.; Aznar, C.; Demar, M.; Bichat, S.; Louvel, D.; Bourbigot, A.; Peneau, C. Severe acquired toxoplasmosis in immunocompetent adult patients in French Guiana. J. Clin. Microbiol. 2002, 40, 4037–4044. [Google Scholar] [CrossRef]
- Dahlgren, S.S.; Gjerde, B. Genetic characterisation of six Sarcocystis species from reindeer (Rangifer tarandus tarandus) in Norway based on the small subunit rRNA gene. Vet. Parasitol. 2007, 146, 204–213. [Google Scholar] [CrossRef]
- Hůrková, L.; Modrý, D. PCR detection of Neospora caninum, Toxoplasma gondii and Encephalitozoon cuniculi in brains of wild carnivores. Vet. Parasitol. 2006, 137, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Moré, G.; Basso, W.; Bacigalupe, D.; Venturini, M.C.; Venturini, L. Diagnosis of Sarcocystis cruzi, Neospora caninum, and Toxoplasma gondii infections in cattle. Parasitol. Res. 2008, 102, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012, 251364. [Google Scholar] [CrossRef]
- Santos, A.; van Aerle, R.; Barrientos, L.; Martinez-Urtaza, J. Computational methods for 16S metabarcoding studies using Nanopore sequencing data. Comput. Struct. Biotechnol. J. 2020, 18, 296–305. [Google Scholar] [CrossRef]
- DeMone, C.; McClure, J.T.; Greenwood, S.J.; Fung, R.; Hwang, M.-H.; Feng, Z.; Shapiro, K. A metabarcoding approach for detecting protozoan pathogens in wild oysters from Prince Edward Island, Canada. Int. J. Food Microbiol. 2021, 360, 109315. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, N.V.; Zemlak, T.S.; Hanner, R.H.; Hebert, P.D. Universal primer cocktails for fish DNA barcoding. Mol. Ecol. Notes 2007, 7, 544–548. [Google Scholar] [CrossRef]
- Hadziavdic, K.; Lekang, K.; Lanzen, A.; Jonassen, I.; Thompson, E.M.; Troedsson, C. Characterization of the 18S rRNA gene for designing universal eukaryote specific primers. PLoS ONE 2014, 9, e87624. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Jardillier, L.; Deschamps, P.; Moreira, D.; Restoux, G.; Bertolino, P.; López-García, P. Complex communities of small protists and unexpected occurrence of typical marine lineages in shallow freshwater systems. Environ. Microbiol. 2015, 17, 3610–3627. [Google Scholar] [CrossRef]
- De Coster, W.; D’hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag: New York, NY, USA, 2019; ISBN 978-3-319-24277-4. Available online: https://ggplot2.tidyverse.org (accessed on 5 January 2023).
- Wang, M.; Fu, A.; Hu, B.; Tong, Y.; Liu, R.; Liu, Z.; Gu, J.; Xiang, B.; Liu, J.; Jiang, W. Nanopore targeted sequencing for the accurate and comprehensive detection of SARS-CoV-2 and other respiratory viruses. Small 2020, 16, 2002169. [Google Scholar] [CrossRef]
- Maestri, S.; Grosso, V.; Alfano, M.; Lavezzari, D.; Piubelli, C.; Bisoffi, Z.; Rossato, M.; Delledonne, M. STArS (STrain-Amplicon-Seq), a targeted nanopore sequencing workflow for SARS-CoV-2 diagnostics and genotyping. Biol. Methods Protoc. 2022, 7, bpac020. [Google Scholar] [CrossRef]
- Cuscó, A.; Viñes, J.; D’Andreano, S.; Riva, F.; Casellas, J.; Sánchez, A.; Francino, O. Using MinIONTM to characterize dog skin microbiota through full-length 16S rRNA gene sequencing approach. bioRxiv 2017, 167015. [Google Scholar] [CrossRef]
- Daugaliyeva, A.; Daugaliyeva, S.; Ashanin, A.; Kanatbayev, S.; Beltramo, C.; Peletto, S. Study of cattle microbiota in different regions of Kazakhstan using 16S metabarcoding analysis. Sci. Rep. 2022, 12, 16410. [Google Scholar] [CrossRef]
- Ho, J.K.; Puniamoorthy, J.; Srivathsan, A.; Meier, R. MinION sequencing of seafood in Singapore reveals creatively labelled flatfishes, confused roe, pig DNA in squid balls, and phantom crustaceans. Food Control 2020, 112, 107144. [Google Scholar] [CrossRef]
- Quéméré, E.; Aucourd, M.; Troispoux, V.; Brosse, S.; Murienne, J.; Covain, R.; Le Bail, P.; Olivier, J.; Tysklind, N.; Galan, M. Unraveling the dietary diversity of Neotropical top predators using scat DNA metabarcoding: A case study on the elusive Giant Otter. Environ. DNA 2021, 3, 889–900. [Google Scholar] [CrossRef]
- Maestri, S.; Cosentino, E.; Paterno, M.; Freitag, H.; Garces, J.M.; Marcolungo, L.; Alfano, M.; Njunjić, I.; Schilthuizen, M.; Slik, F. A rapid and accurate MinION-based workflow for tracking species biodiversity in the field. Genes 2019, 10, 468. [Google Scholar] [CrossRef] [PubMed]
- Marcolungo, L.; Passera, A.; Maestri, S.; Segala, E.; Alfano, M.; Gaffuri, F.; Marturano, G.; Casati, P.; Bianco, P.A.; Delledonne, M. Real-time on-site diagnosis of quarantine pathogens in plant tissues by nanopore-based sequencing. Pathogens 2022, 11, 199. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Meng, Z.; Xu, X.; Wang, L.; Zhao, K.; Zhu, X.; Qiao, Q.; Ge, Y.; Mao, L.; Cui, L. Systematic benchmarking of nanopore Q20+ kit in SARS-CoV-2 whole genome sequencing. Front. Microbiol. 2022, 13, 4059. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.E.; Dabernig-Heinz, J.; Lipp, M.; Cabal, A.; Simantzik, J.; Kohl, M.; Scheiber, M.; Lichtenegger, S.; Ehricht, R.; Leitner, E. Real-time nanopore Q20+ sequencing enables extremely fast and accurate core genome MLST typing and democratizes access to high-resolution bacterial pathogen surveillance. J. Clin. Microbiol. 2023, 61, e01631-22. [Google Scholar] [CrossRef] [PubMed]
- Acharya, K.; Khanal, S.; Pantha, K.; Amatya, N.; Davenport, R.J.; Werner, D. A comparative assessment of conventional and molecular methods, including MinION nanopore sequencing, for surveying water quality. Sci. Rep. 2019, 9, 15726. [Google Scholar] [CrossRef] [PubMed]
- Egeter, B.; Veríssimo, J.; Lopes-Lima, M.; Chaves, C.; Pinto, J.; Riccardi, N.; Beja, P.; Fonseca, N.A. Speeding up the detection of invasive bivalve species using environmental DNA: A Nanopore and Illumina sequencing comparison. Mol. Ecol. Resour. 2022, 22, 2232–2247. [Google Scholar] [CrossRef] [PubMed]
- Heikema, A.P.; Horst-Kreft, D.; Boers, S.A.; Jansen, R.; Hiltemann, S.D.; de Koning, W.; Kraaij, R.; de Ridder, M.A.; van Houten, C.B.; Bont, L.J. Comparison of illumina versus nanopore 16S rRNA gene sequencing of the human nasal microbiota. Genes 2020, 11, 1105. [Google Scholar] [CrossRef]
- Srivathsan, A.; Loh, R.K.; Ong, E.J.; Lee, L.; Ang, Y.; Kutty, S.N.; Meier, R. Network analysis with either Illumina or MinION reveals that detecting vertebrate species requires metabarcoding of iDNA from a diverse fly community. Mol. Ecol. 2022, 32, 6418–6435. [Google Scholar] [CrossRef]
- Iadarola, B.; Xumerle, L.; Lavezzari, D.; Paterno, M.; Marcolungo, L.; Beltrami, C.; Fortunati, E.; Mei, D.; Vetro, A.; Guerrini, R. Shedding light on dark genes: Enhanced targeted resequencing by optimizing the combination of enrichment technology and DNA fragment length. Sci. Rep. 2020, 10, 9424. [Google Scholar] [CrossRef] [PubMed]
- Maestri, S.; Maturo, M.G.; Cosentino, E.; Marcolungo, L.; Iadarola, B.; Fortunati, E.; Rossato, M.; Delledonne, M. A long-read sequencing approach for direct haplotype phasing in clinical settings. Int. J. Mol. Sci. 2020, 21, 9177. [Google Scholar] [CrossRef] [PubMed]
- Dubey, J. Re-examination of resistance of Toxoplasma gondii tachyzoites and bradyzoites to pepsin and trypsin digestion. Parasitology 1998, 116, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Soares, H.S.; Marcili, A.; Barbieri, A.R.; Minervino, A.H.; Moreira, T.R.; Gennari, S.M.; Labruna, M.B. Novel piroplasmid and Hepatozoon organisms infecting the wildlife of two regions of the Brazilian Amazon. Int. J. Parasitol. Parasites Wildl. 2017, 6, 115–121. [Google Scholar] [CrossRef]
- Bilhassi, T.B.; Oliveira, H.N.; Ibelli, A.M.; Giglioti, R.; Regitano, L.C.; Oliveira-Sequeira, T.C.; Bressani, F.A.; Malago, W., Jr.; Resende, F.D.; Oliveira, M.C. Quantitative study of Babesia bovis infection in beef cattle from São Paulo state, Brazil. Ticks Tick-Borne Dis. 2014, 5, 234–238. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Order | Family | Species | Common Name | Individual Distribution |
|---|---|---|---|---|
| Rodentia | Agoutidae | Cuniculus paca | Lowland paca, Paca | 1 |
| Rodentia | Dasyproctidae | Dasyprocta leporina | Red-rumped agouti | 1 |
| Cingulata | Dasypodidae | Dasypus sp. nov. | Nine-banded armadillo | 7 |
| Rodentia | Caviidae | Hydrochoerus hydrochaeris | Capybara | 3 |
| Cetartiodactyla | Cervidae | Mazama americana | Red brocket deer | 2 |
| Perissodactyla | Tapiridae | Tapirus terrestris | Tapir | 1 |
| Total | 15 |
| Illumina Reads | Nanopore Reads | |
|---|---|---|
| Minimum | 71,565 | 18 |
| Median | 78,510 | 5821 |
| Mean | 78,495.5 | 11,127.4 |
| Maximum | 91,032 | 42,449 |
| Total | 1,569,910 | 233,676 |
| Host Identification Code | Host Species |
|---|---|
| G0068 | Hydrochoerus hydrochaeris |
| G0113 | Cuniculus paca |
| G0125 | Mazama americana |
| G0130 | Tapirus terrestris |
| G0149 | Dasypus sp. nov. |
| G0173 | Dasyprocta leporina |
| G0225 | Dasypus sp. nov. |
| G0233 | Mazama americana |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matoute, A.; Maestri, S.; Saout, M.; Laghoe, L.; Simon, S.; Blanquart, H.; Hernandez Martinez, M.A.; Pierre Demar, M. Meat-Borne-Parasite: A Nanopore-Based Meta-Barcoding Work-Flow for Parasitic Microbiodiversity Assessment in the Wild Fauna of French Guiana. Curr. Issues Mol. Biol. 2024, 46, 3810-3821. https://doi.org/10.3390/cimb46050237
Matoute A, Maestri S, Saout M, Laghoe L, Simon S, Blanquart H, Hernandez Martinez MA, Pierre Demar M. Meat-Borne-Parasite: A Nanopore-Based Meta-Barcoding Work-Flow for Parasitic Microbiodiversity Assessment in the Wild Fauna of French Guiana. Current Issues in Molecular Biology. 2024; 46(5):3810-3821. https://doi.org/10.3390/cimb46050237
Chicago/Turabian StyleMatoute, Adria, Simone Maestri, Mona Saout, Laure Laghoe, Stéphane Simon, Hélène Blanquart, Miguel Angel Hernandez Martinez, and Magalie Pierre Demar. 2024. "Meat-Borne-Parasite: A Nanopore-Based Meta-Barcoding Work-Flow for Parasitic Microbiodiversity Assessment in the Wild Fauna of French Guiana" Current Issues in Molecular Biology 46, no. 5: 3810-3821. https://doi.org/10.3390/cimb46050237