

SARS-CoV-2-Induced Type I Interferon Signaling Dysregulation in Olfactory Networks Implications for Alzheimer’s Disease

 ,
,  , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Concept Design
2.2. Dataset Selection
- Regarding study design:
- Reviews.
- Studies on children.
- In vitro or animal experiments, including gene expression data derived from such experiments.
- Study design unrelated to COVID-19 or focusing on a specific comorbidity.
- Sample size <5 per group.
- Regarding gene expression data:
- Gene expression data limited on SARS-CoV-2 entry factors.
- Gene expression data are limited on immune or other gene panels.
2.3. Leveraging Gene Set Enrichment Analysis (GSEA) Results from COVID-19 and Alzheimer’s Disease Datasets
2.4. Detection of Specific Overlapping Genes in Type I Interferon Signatures and Their Determination as Gene Network–the Minimal Dysregulated Network
2.5. Leveraging the Agora Multi-Omics Database to Identify Gene-Disease Associations with Alzheimer’s Disease
3. Results
3.1. Interferon Signaling Pathways Is Significantly Enriched along the Transolfactory Route in COVID-19 and in Response to Alzheimer’s Disease Pathology
3.2. Shared Genes between the Transolfactory Route and in Response to Alzheimer’s Disease Pathology Represent a Type I Interferon Network Containing IFITM and OAS Family Genes
3.3. Gene-Disease Associations between the Type I Interferon Signature and Alzheimer’s Disease Diagnosis
4. Discussion
4.1. Type I Interferon as a Mechanism for Cognitive Impairment in Long COVID
4.2. Type I Interferon Signaling as Common Ground between COVID-19 and Alzheimer’s Disease: A Hint towards Nucleic Acid Immunity and the cGAS-STING-IFITM3 Axis
4.3. Limitations, Strengths, and Outstanding Questions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de Erausquin, G.A.; Snyder, H.; Carrillo, M.; Hosseini, A.A.; Brugha, T.S.; Seshadri, S. The chronic neuropsychiatric sequelae of COVID-19: The need for a prospective study of viral impact on brain functioning. Alzheimer’s Dement. 2021, 17, 1056–1065. [Google Scholar] [CrossRef]
- Vavougios, G.D.; Nday, C.; Pelidou, S.H.; Gourgoulianis, K.I.; Stamoulis, G.; Doskas, T.; Zarogiannis, S.G. Outside-in induction of the IFITM3 trafficking system by infections, including SARS-CoV-2, in the pathobiology of Alzheimer’s disease. Brain Behav. Immun. Health 2021, 14, 100243. [Google Scholar] [CrossRef]
- Vavougios, G.D.; de Erausquin, G.A.; Snyder, H.M. Type I interferon signaling in SARS-CoV-2 associated neurocognitive disorder (SAND): Mapping host-virus interactions to an etiopathogenesis. Front. Neurol. 2022, 13, 1063298. [Google Scholar] [CrossRef]
- Tan, P.H.; Ji, J.; Hsing, C.H.; Tan, R.; Ji, R.R. Emerging Roles of Type-I Interferons in Neuroinflammation, Neurological Diseases, and Long-Haul COVID. Int. J. Mol. Sci. 2022, 23, 14394. [Google Scholar] [CrossRef]
- Suzzi, S.; Tsitsou-Kampeli, A.; Schwartz, M. The type I interferon antiviral response in the choroid plexus and the cognitive risk in COVID-19. Nat. Immunol. 2023, 24, 220–224. [Google Scholar] [CrossRef]
- Hosseini, S.; Michaelsen-Preusse, K.; Grigoryan, G.; Chhatbar, C.; Kalinke, U.; Korte, M. Type I Interferon Receptor Signaling in Astrocytes Regulates Hippocampal Synaptic Plasticity and Cognitive Function of the Healthy CNS. Cell Rep. 2020, 31, 107666. [Google Scholar] [CrossRef]
- Ekdahl, C.T.; Claasen, J.-H.; Bonde, S.; Kokaia, Z.; Lindvall, O. Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13632–13637. [Google Scholar] [CrossRef]
- Kim, H.S.; Shin, S.M.; Kim, S.; Nam, Y.; Yoo, A.; Moon, M. Relationship between adult subventricular neurogenesis and Alzheimer’s disease: Pathologic roles and therapeutic implications. Front. Aging Neurosci. 2022, 14, 1002281. [Google Scholar] [CrossRef]
- Roy, E.; Cao, W. Glial interference: Impact of type I interferon in neurodegenerative diseases. Mol. Neurodegener. 2022, 17, 78. [Google Scholar] [CrossRef]
- Roy, E.R.; Wang, B.; Wan, Y.-w.; Chiu, G.; Cole, A.; Yin, Z.; Propson, N.E.; Xu, Y.; Jankowsky, J.L.; Liu, Z.; et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Investig. 2020, 130, 1912–1930. [Google Scholar] [CrossRef]
- Jana, A.; Wang, X.; Leasure, J.W.; Magana, L.; Wang, L.; Kim, Y.-M.; Dodiya, H.; Toth, P.T.; Sisodia, S.S.; Rehman, J. Increased Type I interferon signaling and brain endothelial barrier dysfunction in an experimental model of Alzheimer’s disease. Sci. Rep. 2022, 12, 16488. [Google Scholar] [CrossRef]
- Duarte, N.; Shafi, A.M.; Penha-Gonçalves, C.; Pais, T.F. Endothelial type I interferon response and brain diseases: Identifying STING as a therapeutic target. Front. Cell Dev. Biol. 2023, 11, 1249235. [Google Scholar] [CrossRef]
- Udeochu, J.C.; Amin, S.; Huang, Y.; Fan, L.; Torres, E.R.S.; Carling, G.K.; Liu, B.; McGurran, H.; Coronas-Samano, G.; Kauwe, G.; et al. Tau activation of microglial cGAS–IFN reduces MEF2C-mediated cognitive resilience. Nat. Neurosci. 2023, 26, 737–750. [Google Scholar] [CrossRef]
- Hur, J.Y.; Frost, G.R.; Wu, X.; Crump, C.; Pan, S.J.; Wong, E.; Barros, M.; Li, T.; Nie, P.; Zhai, Y.; et al. The innate immunity protein IFITM3 modulates γ-secretase in Alzheimer’s disease. Nature 2020, 586, 735–740. [Google Scholar] [CrossRef]
- Prelli Bozzo, C.; Nchioua, R.; Volcic, M.; Koepke, L.; Krüger, J.; Schütz, D.; Heller, S.; Stürzel, C.M.; Kmiec, D.; Conzelmann, C.; et al. IFITM proteins promote SARS-CoV-2 infection and are targets for virus inhibition in vitro. Nat. Commun. 2021, 12, 4584. [Google Scholar] [CrossRef]
- Shi, G.; Kenney, A.D.; Kudryashova, E.; Zani, A.; Zhang, L.; Lai, K.K.; Hall-Stoodley, L.; Robinson, R.T.; Kudryashov, D.S.; Compton, A.A.; et al. Opposing activities of IFITM proteins in SARS-CoV-2 infection. EMBO J. 2021, 40, e106501. [Google Scholar] [CrossRef]
- Vavougios, G.D.; Breza, M.; Mavridis, T.; Krogfelt, K.A. FYN, SARS-CoV-2, and IFITM3 in the neurobiology of Alzheimer’s disease. Brain Disord. 2021, 3, 100022. [Google Scholar] [CrossRef]
- Magusali, N.; Graham, A.C.; Piers, T.M.; Panichnantakul, P.; Yaman, U.; Shoai, M.; Reynolds, R.H.; Botia, J.A.; Brookes, K.J.; Guetta-Baranes, T.; et al. Genetic variability associated with OAS1 expression in myeloid cells increases the risk of Alzheimer’s disease and severe COVID-19 outcomes. bioRxiv 2021. [Google Scholar] [CrossRef]
- Yang, A.C.; Kern, F.; Losada, P.M.; Agam, M.R.; Maat, C.A.; Schmartz, G.P.; Fehlmann, T.; Stein, J.A.; Schaum, N.; Lee, D.P.; et al. Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature 2021, 595, 565–571. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, J.; Hou, Y.; Leverenz, J.B.; Kallianpur, A.; Mehra, R.; Liu, Y.; Yu, H.; Pieper, A.A.; Jehi, L.; et al. Network medicine links SARS-CoV-2/COVID-19 infection to brain microvascular injury and neuroinflammation in dementia-like cognitive impairment. Alzheimer’s Res. Ther. 2021, 13, 110. [Google Scholar] [CrossRef]
- Vavougios, G.D.; Nday, C.; Pelidou, S.H.; Zarogiannis, S.G.; Gourgoulianis, K.I.; Stamoulis, G.; Doskas, T. Double hit viral parasitism, polymicrobial CNS residency and perturbed proteostasis in Alzheimer’s disease: A data driven, in silico analysis of gene expression data. Mol. Immunol. 2020, 127, 124–135. [Google Scholar] [CrossRef]
- Daroische, R.; Hemminghyth, M.S.; Eilertsen, T.H.; Breitve, M.H.; Chwiszczuk, L.J. Cognitive Impairment After COVID-19—A Review on Objective Test Data. Front. Neurol. 2021, 12, 1238. [Google Scholar] [CrossRef]
- Pirker-Kees, A.; Platho-Elwischger, K.; Hafner, S.; Redlich, K.; Baumgartner, C. Hyposmia Is Associated with Reduced Cognitive Function in COVID-19: First Preliminary Results. Dement. Geriatr. Cogn. Disord. 2021, 50, 68–73. [Google Scholar] [CrossRef]
- Baek, M.S.; Cho, H.; Lee, H.S.; Lee, J.H.; Ryu, Y.H.; Lyoo, C.H. Effect of A/T/N imaging biomarkers on impaired odor identification in Alzheimer’s disease. Sci. Rep. 2020, 10, 11556. [Google Scholar] [CrossRef]
- Lafaille-Magnan, M.-E.; Poirier, J.; Etienne, P.; Tremblay-Mercier, J.; Frenette, J.; Rosa-Neto, P.; Breitner, J.C.S.; For the PREVENT-AD Research Group. Odor identification as a biomarker of preclinical AD in older adults at risk. Neurology 2017, 89, 327–335. [Google Scholar] [CrossRef]
- de Erausquin, G.A.; Snyder, H.; Brugha, T.S.; Seshadri, S.; Carrillo, M.; Sagar, R.; Huang, Y.; Newton, C.; Tartaglia, C.; Teunissen, C.; et al. Chronic neuropsychiatric sequelae of SARS-CoV-2: Protocol and methods from the Alzheimer’s Association Global Consortium. Alzheimer’s Dement. 2022, 8, e12348. [Google Scholar] [CrossRef]
- Douaud, G.; Lee, S.; Alfaro-Almagro, F.; Arthofer, C.; Wang, C.; McCarthy, P.; Lange, F.; Andersson, J.L.R.; Griffanti, L.; Duff, E.; et al. SARS-CoV-2 is associated with changes in brain structure in UK Biobank. Nature 2022, 604, 697–707. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Miao, V.N.; Owings, A.H.; Navia, A.W.; Tang, Y.; Bromley, J.D.; Lotfy, P.; Sloan, M.; Laird, H.; Williams, H.B.; et al. Impaired local intrinsic immunity to SARS-CoV-2 infection in severe COVID-19. Cell 2021, 184, 4713–4733.e4722. [Google Scholar] [CrossRef]
- Hatton, C.F.; Botting, R.A.; Dueñas, M.E.; Haq, I.J.; Verdon, B.; Thompson, B.J.; Spegarova, J.S.; Gothe, F.; Stephenson, E.; Gardner, A.I.; et al. Delayed induction of type I and III interferons mediates nasal epithelial cell permissiveness to SARS-CoV-2. Nat. Commun. 2021, 12, 7092. [Google Scholar] [CrossRef]
- Mavrikaki, M.; Lee, J.D.; Solomon, I.H.; Slack, F.J. Severe COVID-19 is associated with molecular signatures of aging in the human brain. Nature Aging 2022, 2, 1130–1137. [Google Scholar] [CrossRef]
- Meinhardt, J.; Radke, J.; Dittmayer, C.; Franz, J.; Thomas, C.; Mothes, R.; Laue, M.; Schneider, J.; Brünink, S.; Greuel, S.; et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat. Neurosci. 2021, 24, 168–175. [Google Scholar] [CrossRef]
- Schreiber, G. The Role of Type I Interferons in the Pathogenesis and Treatment of COVID-19. Front. Immunol. 2020, 11, 595739. [Google Scholar] [CrossRef]
- Ahn, J.H.; Kim, J.; Hong, S.P.; Choi, S.Y.; Yang, M.J.; Ju, Y.S.; Kim, Y.T.; Kim, H.M.; Rahman, M.D.T.; Chung, M.K.; et al. Nasal ciliated cells are primary targets for SARS-CoV-2 replication in the early stage of COVID-19. J. Clin. Investig. 2021, 131, e148517. [Google Scholar] [CrossRef]
- Das, S.; Li, Z.; Wachter, A.; Alla, S.; Noori, A.; Abdourahman, A.; Tamm, J.A.; Woodbury, M.E.; Talanian, R.V.; Biber, K.; et al. Distinct transcriptomic responses to Aβ plaques, neurofibrillary tangles, and APOE in Alzheimer’s disease. Alzheimer’s Dement. 2024, 20, 74–90. [Google Scholar] [CrossRef]
- Serrano, G.E.; Walker, J.E.; Tremblay, C.; Piras, I.S.; Huentelman, M.J.; Belden, C.M.; Goldfarb, D.; Shprecher, D.; Atri, A.; Adler, C.H.; et al. SARS-CoV-2 Brain Regional Detection, Histopathology, Gene Expression, and Immunomodulatory Changes in Decedents with COVID-19. J. Neuropathol. Exp. Neurol. 2022, 81, 666–695. [Google Scholar] [CrossRef]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.H. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017, 21, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Crook, H.; Ramirez, A.; Hosseini, A.; Vavougyios, G.; Lehmann, C.; Bruchfeld, J.; Schneider, A.; D’Avossa, G.; Lo Re, V.; Salmoiraghi, A.; et al. European Working Group on SARS-CoV-2: Current Understanding, Unknowns, and Recommendations on the Neurological Complications of COVID-19. Brain Connect. 2023, 13, 178–210. [Google Scholar] [CrossRef]
- Bayat, A.H.; Azimi, H.; Hassani Moghaddam, M.; Ebrahimi, V.; Fathi, M.; Vakili, K.; Mahmoudiasl, G.R.; Forouzesh, M.; Boroujeni, M.E.; Nariman, Z.; et al. COVID-19 causes neuronal degeneration and reduces neurogenesis in human hippocampus. Apoptosis 2022, 27, 852–868. [Google Scholar] [CrossRef]
- Muccioli, L.; Sighinolfi, G.; Mitolo, M.; Ferri, L.; Jane Rochat, M.; Pensato, U.; Taruffi, L.; Testa, C.; Masullo, M.; Cortelli, P.; et al. Cognitive and functional connectivity impairment in post-COVID-19 olfactory dysfunction. Neuroimage Clin. 2023, 38, 103410. [Google Scholar] [CrossRef]
- Deer, R.R.; Rock, M.A.; Vasilevsky, N.; Carmody, L.; Rando, H.; Anzalone, A.J.; Basson, M.D.; Bennett, T.D.; Bergquist, T.; Boudreau, E.A.; et al. Characterizing Long COVID: Deep Phenotype of a Complex Condition. EBioMedicine 2021, 74, 103722. [Google Scholar] [CrossRef]
- Baruch, K.; Deczkowska, A.; David, E.; Castellano, J.M.; Miller, O.; Kertser, A.; Berkutzki, T.; Barnett-Itzhaki, Z.; Bezalel, D.; Wyss-Coray, T.; et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 2014, 346, 89–93. [Google Scholar] [CrossRef]
- Vavougios, G.D. Potentially irreversible olfactory and gustatory impairments in COVID-19: Indolent vs. fulminant SARS-CoV-2 neuroinfection. Brain Behav. Immun. 2020, 87, 107–108. [Google Scholar] [CrossRef]
- Lee, J.S.; Shin, E.-C. The type I interferon response in COVID-19: Implications for treatment. Nat. Rev. Immunol. 2020, 20, 585–586. [Google Scholar] [CrossRef]
- Wu, B.; Ramaiah, A.; Garcia, G., Jr.; Hasiakos, S.; Arumugaswami, V.; Srikanth, S. ORAI1 Limits SARS-CoV-2 Infection by Regulating Tonic Type I IFN Signaling. J. Immunol. 2022, 208, 74–84. [Google Scholar] [CrossRef]
- Yang, R.-C.; Huang, K.; Zhang, H.-P.; Li, L.; Zhang, Y.-F.; Tan, C.; Chen, H.-C.; Jin, M.-L.; Wang, X.-R. SARS-CoV-2 productively infects human brain microvascular endothelial cells. J. Neuroinflammation 2022, 19, 149. [Google Scholar] [CrossRef]
- Wenzel, J.; Lampe, J.; Müller-Fielitz, H.; Schuster, R.; Zille, M.; Müller, K.; Krohn, M.; Körbelin, J.; Zhang, L.; Özorhan, Ü.; et al. The SARS-CoV-2 main protease Mpro causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nat. Neurosci. 2021, 24, 1522–1533. [Google Scholar] [CrossRef]
- Vavougios, G.D.; Zarogiannis, S.G.; Hadjigeorgiou, G.; Krogfelt, K.A.; Gourgoulianis, K.I. SARS-CoV-2 and type I interferon signaling in brain endothelial cells: Blurring the lines between friend or foe. Stem Cell Rep. 2022, 17, 1012–1013. [Google Scholar] [CrossRef]
- Kong, W.; Montano, M.; Corley, M.J.; Helmy, E.; Kobayashi, H.; Kinisu, M.; Suryawanshi, R.; Luo, X.; Royer, L.A.; Roan, N.R.; et al. Neuropilin-1 Mediates SARS-CoV-2 Infection of Astrocytes in Brain Organoids, Inducing Inflammation Leading to Dysfunction and Death of Neurons. mBio 2022, 13, e0230822. [Google Scholar] [CrossRef]
- Kong, D.; Park, K.H.; Kim, D.-H.; Kim, N.G.; Lee, S.-E.; Shin, N.; Kook, M.G.; Kim, Y.B.; Kang, K.-S. Cortical-blood vessel assembloids exhibit Alzheimer’s disease phenotypes by activating glia after SARS-CoV-2 infection. Cell Death Discov. 2023, 9, 32. [Google Scholar] [CrossRef]
- Wang, Z.X.; Wan, Q.; Xing, A. HLA in Alzheimer’s Disease: Genetic Association and Possible Pathogenic Roles. Neuromolecular Med. 2020, 22, 464–473. [Google Scholar] [CrossRef]
- Haure-Mirande, J.-V.; Audrain, M.; Ehrlich, M.E.; Gandy, S. Microglial TYROBP/DAP12 in Alzheimer’s disease: Transduction of physiological and pathological signals across TREM2. Mol. Neurodegener. 2022, 17, 55. [Google Scholar] [CrossRef]
- Vavougios, G.D.; Mavridis, T.; Artemiadis, A.; Krogfelt, K.A.; Hadjigeorgiou, G. Trained immunity in viral infections, Alzheimer’s disease and multiple sclerosis: A convergence in type I interferon signalling and IFNβ-1a. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166430. [Google Scholar] [CrossRef]
- Magusali, N.; Graham, A.C.; Piers, T.M.; Panichnantakul, P.; Yaman, U.; Shoai, M.; Reynolds, R.H.; Botia, J.A.; Brookes, K.J.; Guetta-Baranes, T.; et al. A genetic link between risk for Alzheimer’s disease and severe COVID-19 outcomes via the OAS1 gene. Brain 2021, 144, 3727–3741. [Google Scholar] [CrossRef]
- Michalovicz, L.T.; Lally, B.; Konat, G.W. Peripheral challenge with a viral mimic upregulates expression of the complement genes in the hippocampus. J. Neuroimmunol. 2015, 285, 137–142. [Google Scholar] [CrossRef]
- Petrisko, T.J.; Bloemer, J.; Pinky, P.D.; Srinivas, S.; Heslin, R.T.; Du, Y.; Setti, S.E.; Hong, H.; Suppiramaniam, V.; Konat, G.W.; et al. Neuronal CXCL10/CXCR3 Axis Mediates the Induction of Cerebral Hyperexcitability by Peripheral Viral Challenge. Front. Neurosci. 2020, 14, 220. [Google Scholar] [CrossRef]
- Grabrucker, S.; Marizzoni, M.; Silajdžić, E.; Lopizzo, N.; Mombelli, E.; Nicolas, S.; Dohm-Hansen, S.; Scassellati, C.; Moretti, D.V.; Rosa, M.; et al. Microbiota from Alzheimer’s patients induce deficits in cognition and hippocampal neurogenesis. Brain 2023, 146, 4916–4934. [Google Scholar] [CrossRef]
- Viengkhou, B.; Hofer, M.J. Breaking down the cellular responses to type I interferon neurotoxicity in the brain. Front. Immunol. 2023, 14, 1110593. [Google Scholar] [CrossRef]
- Jin, M.; Shiwaku, H.; Tanaka, H.; Obita, T.; Ohuchi, S.; Yoshioka, Y.; Jin, X.; Kondo, K.; Fujita, K.; Homma, H.; et al. Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat. Commun. 2021, 12, 6565. [Google Scholar] [CrossRef]
- Roy, E.R.; Chiu, G.; Li, S.; Propson, N.E.; Kanchi, R.; Wang, B.; Coarfa, C.; Zheng, H.; Cao, W. Concerted type I interferon signaling in microglia and neural cells promotes memory impairment associated with amyloid β plaques. Immunity 2022, 55, 879–894.e6. [Google Scholar] [CrossRef]
- Yu, L.; Liu, P. Cytosolic DNA sensing by cGAS: Regulation, function, and human diseases. Signal Transduct. Target. Ther. 2021, 6, 170. [Google Scholar] [CrossRef]
- Choi, U.Y.; Kang, J.-S.; Hwang, Y.S.; Kim, Y.-J. Oligoadenylate synthase-like (OASL) proteins: Dual functions and associations with diseases. Exp. Mol. Med. 2015, 47, e144. [Google Scholar] [CrossRef]
- Kuang, M.; Zhao, Y.; Yu, H.; Li, S.; Liu, T.; Chen, L.; Chen, J.; Luo, Y.; Guo, X.; Wei, X.; et al. XAF1 promotes anti-RNA virus immune responses by regulating chromatin accessibility. Sci. Adv. 2023, 9, eadg5211. [Google Scholar] [CrossRef]
- Sanford, S.A.I.; Miller, L.V.C.; Vaysburd, M.; Keeling, S.; Tuck, B.J.; Clark, J.; Neumann, M.; Syanda, V.; James, L.C.; McEwan, W.A. The type-I interferon response potentiates seeded tau aggregation and exacerbates tau pathology. Alzheimer’s Dement. 2024, 10, 1013–1025. [Google Scholar] [CrossRef]
- Di Primio, C.; Quaranta, P.; Mignanelli, M.; Siano, G.; Bimbati, M.; Scarlatti, A.; Piazza, C.R.; Spezia, P.G.; Perrera, P.; Basolo, F.; et al. Severe acute respiratory syndrome coronavirus 2 infection leads to Tau pathological signature in neurons. PNAS Nexus 2023, 2, pgad282. [Google Scholar] [CrossRef]
- Ramani, A.; Muller, L.; Ostermann, P.N.; Gabriel, E.; Abida-Islam, P.; Muller-Schiffmann, A.; Mariappan, A.; Goureau, O.; Gruell, H.; Walker, A.; et al. SARS-CoV-2 targets neurons of 3D human brain organoids. EMBO J. 2020, 39, e106230. [Google Scholar] [CrossRef]
- Käufer, C.; Schreiber, C.S.; Hartke, A.S.; Denden, I.; Stanelle-Bertram, S.; Beck, S.; Kouassi, N.M.; Beythien, G.; Becker, K.; Schreiner, T.; et al. Microgliosis and neuronal proteinopathy in brain persist beyond viral clearance in SARS-CoV-2 hamster model. EBioMedicine 2022, 79, 103999. [Google Scholar] [CrossRef]
- Hou, Y.; Li, C.; Yoon, C.; Leung, O.W.; You, S.; Cui, X.; Chan, J.F.; Pei, D.; Cheung, H.H.; Chu, H. Enhanced replication of SARS-CoV-2 Omicron BA.2 in human forebrain and midbrain organoids. Signal Transduct. Target Ther. 2022, 7, 381. [Google Scholar] [CrossRef]
- Levine, K.S.; Leonard, H.L.; Blauwendraat, C.; Iwaki, H.; Johnson, N.; Bandres-Ciga, S.; Ferrucci, L.; Faghri, F.; Singleton, A.B.; Nalls, M.A. Virus exposure and neurodegenerative disease risk across national biobanks. Neuron 2023, 111, 1086–1093.e2. [Google Scholar] [CrossRef]
- Green, R.; Mayilsamy, K.; McGill, A.R.; Martinez, T.E.; Chandran, B.; Blair, L.J.; Bickford, P.C.; Mohapatra, S.S.; Mohapatra, S. SARS-CoV-2 infection increases the gene expression profile for Alzheimer’s disease risk. Mol. Ther.-Methods Clin. Dev. 2022, 27, 217–229. [Google Scholar] [CrossRef]
- Domizio, J.D.; Gulen, M.F.; Saidoune, F.; Thacker, V.V.; Yatim, A.; Sharma, K.; Nass, T.; Guenova, E.; Schaller, M.; Conrad, C.; et al. The cGAS–STING pathway drives type I IFN immunopathology in COVID-19. Nature 2022, 603, 145–151. [Google Scholar] [CrossRef]
- Wu, Z.; Tang, W.; Ibrahim, F.; Chen, X.; Yan, H.; Tao, C.; Wang, Z.; Guo, Y.; Fu, Y.; Wang, Q.; et al. Aβ Induces Neuroinflammation and Microglial M1 Polarization via cGAS-STING-IFITM3 Signaling Pathway in BV-2 Cells. Neurochem. Res. 2023, 48, 2881–2894. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Ramshorn, K.; Pinci, F.; Zuber, S.; O’Duill, F.; Schmid-Burgk, J.L.; Hoss, F.; Buhmann, R.; et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 2017, 171, 1110–1124.e18. [Google Scholar] [CrossRef]
- Wang, W.; Hu, D.; Wu, C.; Feng, Y.; Li, A.; Liu, W.; Wang, Y.; Chen, K.; Tian, M.; Xiao, F.; et al. STING promotes NLRP3 localization in ER and facilitates NLRP3 deubiquitination to activate the inflammasome upon HSV-1 infection. PLoS Pathog. 2020, 16, e1008335. [Google Scholar] [CrossRef]
- Stancu, I.-C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brône, B.; Huaux, F.; Octave, J.-N.; et al. Aggregated Tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef]
- Reiken, S.; Sittenfeld, L.; Dridi, H.; Liu, Y.; Liu, X.; Marks, A.R. Alzheimer’s-like signaling in brains of COVID-19 patients. Alzheimers’s Dement. 2022, 18, 955–965. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, L.; Bao, L.; Liu, J.; Zhu, H.; Lv, Q.; Liu, R.; Chen, W.; Tong, W.; Wei, Q.; et al. SARS-CoV-2 crosses the blood–brain barrier accompanied with basement membrane disruption without tight junctions alteration. Signal Transduct. Target. Ther. 2021, 6, 337. [Google Scholar] [CrossRef]
- Krasemann, S.; Haferkamp, U.; Pfefferle, S.; Woo, M.S.; Heinrich, F.; Schweizer, M.; Appelt-Menzel, A.; Cubukova, A.; Barenberg, J.; Leu, J.; et al. The blood-brain barrier is dysregulated in COVID-19 and serves as a CNS entry route for SARS-CoV-2. Stem Cell Rep. 2022, 17, 307–320. [Google Scholar] [CrossRef]
- Tang, K.; Wu, L.; Luo, Y.; Gong, B. Quantitative assessment of SARS-CoV-2 RNAemia and outcome in patients with coronavirus disease 2019. J. Med. Virol. 2021, 93, 3165–3175. [Google Scholar] [CrossRef]
- Scott, A.M.; Jager, A.C.; Gwin, M.; Voth, S.; Balczon, R.; Stevens, T.; Lin, M.T. Pneumonia-induced endothelial amyloids reduce dendritic spine density in brain neurons. Sci. Rep. 2020, 10, 9327. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Balczon, R.; Pittet, J.F.; Wagener, B.M.; Moser, S.A.; Morrow, K.A.; Voth, S.; Francis, C.M.; Leavesley, S.; Bell, J.; et al. Nosocomial Pneumonia Elicits an Endothelial Proteinopathy: Evidence for a Source of Neurotoxic Amyloids in Critically Ill Patients. Am. J. Respir. Crit. Care Med. 2018, 198, 1575–1578. [Google Scholar] [CrossRef] [PubMed]
- White, M.R.; Kandel, R.; Tripathi, S.; Condon, D.; Qi, L.; Taubenberger, J.; Hartshorn, K.L. Alzheimer’s associated β-amyloid protein inhibits influenza A virus and modulates viral interactions with phagocytes. PLoS ONE 2014, 9, e101364. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Raimondi, F.; Rajendran, L.; Temussi, P.A. Why does the Aβ peptide of Alzheimer share structural similarity with antimicrobial peptides? Commun. Biol. 2020, 3, 135. [Google Scholar] [CrossRef] [PubMed]
- Shafran, N.; Shafran, I.; Ben-Zvi, H.; Sofer, S.; Sheena, L.; Krause, I.; Shlomai, A.; Goldberg, E.; Sklan, E.H. Secondary bacterial infection in COVID-19 patients is a stronger predictor for death compared to influenza patients. Sci. Rep. 2021, 11, 12703. [Google Scholar] [CrossRef] [PubMed]
- Rhoades, N.S.; Pinski, A.N.; Monsibais, A.N.; Jankeel, A.; Doratt, B.M.; Cinco, I.R.; Ibraim, I.; Messaoudi, I. Acute SARS-CoV-2 infection is associated with an increased abundance of bacterial pathogens, including Pseudomonas aeruginosa in the nose. Cell Rep. 2021, 36, 109637. [Google Scholar] [CrossRef]
- Voth, S.; Gwin, M.; Francis, C.M.; Balczon, R.; Frank, D.W.; Pittet, J.-F.; Wagener, B.M.; Moser, S.A.; Alexeyev, M.; Housley, N.; et al. Virulent Pseudomonas aeruginosa infection converts antimicrobial amyloids into cytotoxic prions. FASEB J. 2020, 34, 9156–9179. [Google Scholar] [CrossRef]
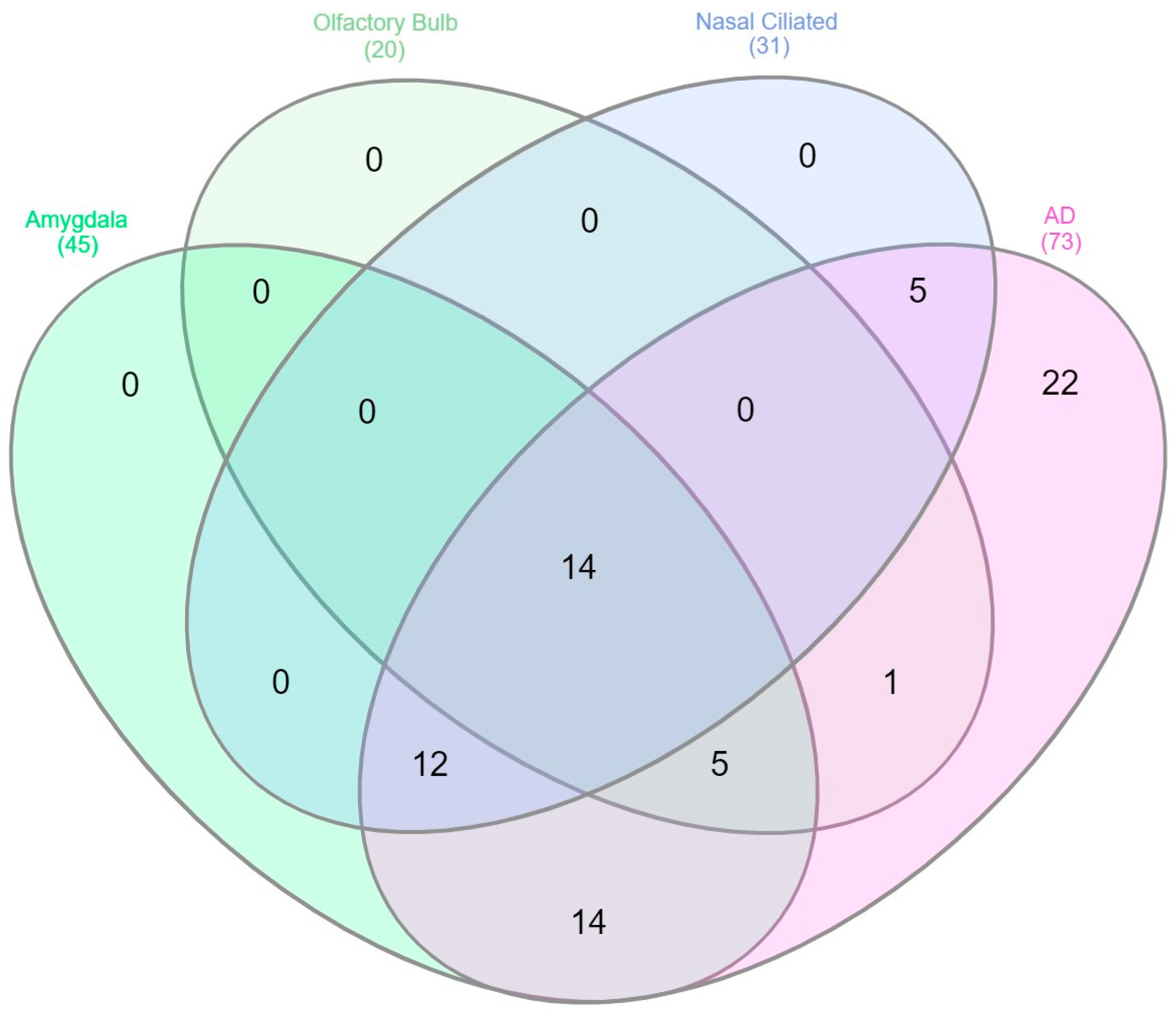
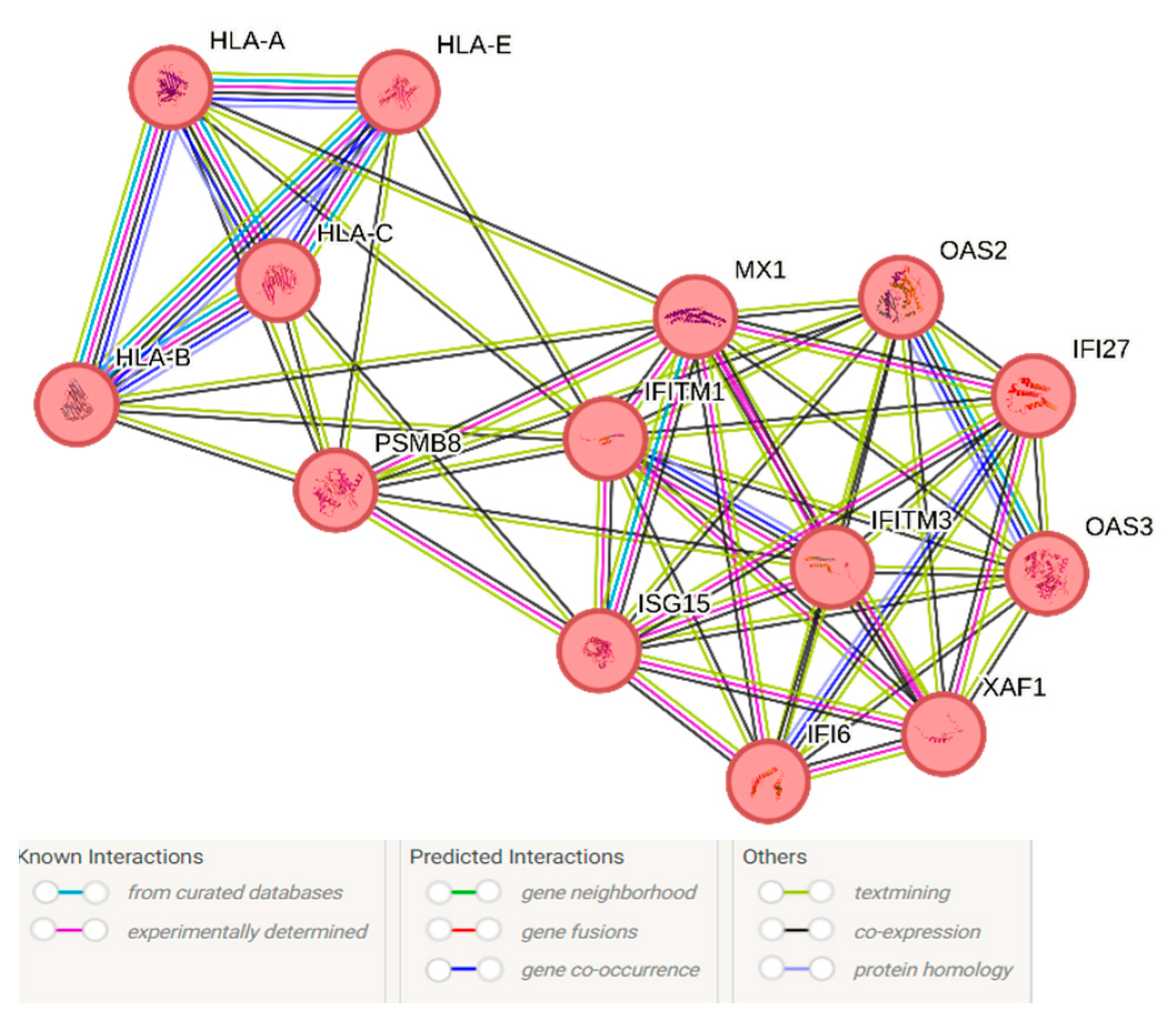

{kind=link}
{kind=link}
{kind=link}
| Reactome Term ID | Term Description | False Discovery Rate |
|---|---|---|
| R-HSA-909733 | Interferon alpha/beta signaling | 5.68 × 10−31 |
| R-HSA-877300 | Interferon-gamma signaling | 1.51 × 10−8 |
| R-HSA-1236977 | Endosomal/Vacuolar pathway | 8.23 × 10−8 |
| R-HSA-1236974 | ER-Phagosome pathway | 1.40 × 10−6 |
| R-HSA-983170 | Antigen Presentation: Folding, assembly, and peptide loading of class I MHC | 1.84 × 10−6 |
| R-HSA-9705671 | SARS-CoV-2 activates/modulates innate and adaptive immune responses | 4.65 × 10−6 |
| R-HSA-198933 | Immunoregulatory interactions between a Lymphoid and a non-Lymphoid cell | 5.53 × 10−6 |
| R-HSA-1169410 | Antiviral mechanism by IFN-stimulated genes | 5.74 × 10−5 |
| R-HSA-2172127 | DAP12 interactions | 0.00049 |
| R-HSA-1280218 | Adaptive Immune System | 0.00097 |
| R-HSA-5663205 | Infectious disease | 0.0027 |
| R-HSA-8983711 | OAS antiviral response | 0.0029 |
| R-HSA-168249 | Innate Immune System | 0.005 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vavougios, G.D.; Mavridis, T.; Doskas, T.; Papaggeli, O.; Foka, P.; Hadjigeorgiou, G. SARS-CoV-2-Induced Type I Interferon Signaling Dysregulation in Olfactory Networks Implications for Alzheimer’s Disease. Curr. Issues Mol. Biol. 2024, 46, 4565-4579. https://doi.org/10.3390/cimb46050277
Vavougios GD, Mavridis T, Doskas T, Papaggeli O, Foka P, Hadjigeorgiou G. SARS-CoV-2-Induced Type I Interferon Signaling Dysregulation in Olfactory Networks Implications for Alzheimer’s Disease. Current Issues in Molecular Biology. 2024; 46(5):4565-4579. https://doi.org/10.3390/cimb46050277
Chicago/Turabian StyleVavougios, George D., Theodoros Mavridis, Triantafyllos Doskas, Olga Papaggeli, Pelagia Foka, and Georgios Hadjigeorgiou. 2024. "SARS-CoV-2-Induced Type I Interferon Signaling Dysregulation in Olfactory Networks Implications for Alzheimer’s Disease" Current Issues in Molecular Biology 46, no. 5: 4565-4579. https://doi.org/10.3390/cimb46050277
APA StyleVavougios, G. D., Mavridis, T., Doskas, T., Papaggeli, O., Foka, P., & Hadjigeorgiou, G. (2024). SARS-CoV-2-Induced Type I Interferon Signaling Dysregulation in Olfactory Networks Implications for Alzheimer’s Disease. Current Issues in Molecular Biology, 46(5), 4565-4579. https://doi.org/10.3390/cimb46050277