A New Dihydrochromone Dimer and Other Secondary Metabolites from Cultures of the Marine Sponge-Associated Fungi Neosartorya fennelliae KUFA 0811 and Neosartorya tsunodae KUFC 9213

 , , , , , , , , and
, , , , , , , , and 
Abstract
:
1. Introduction
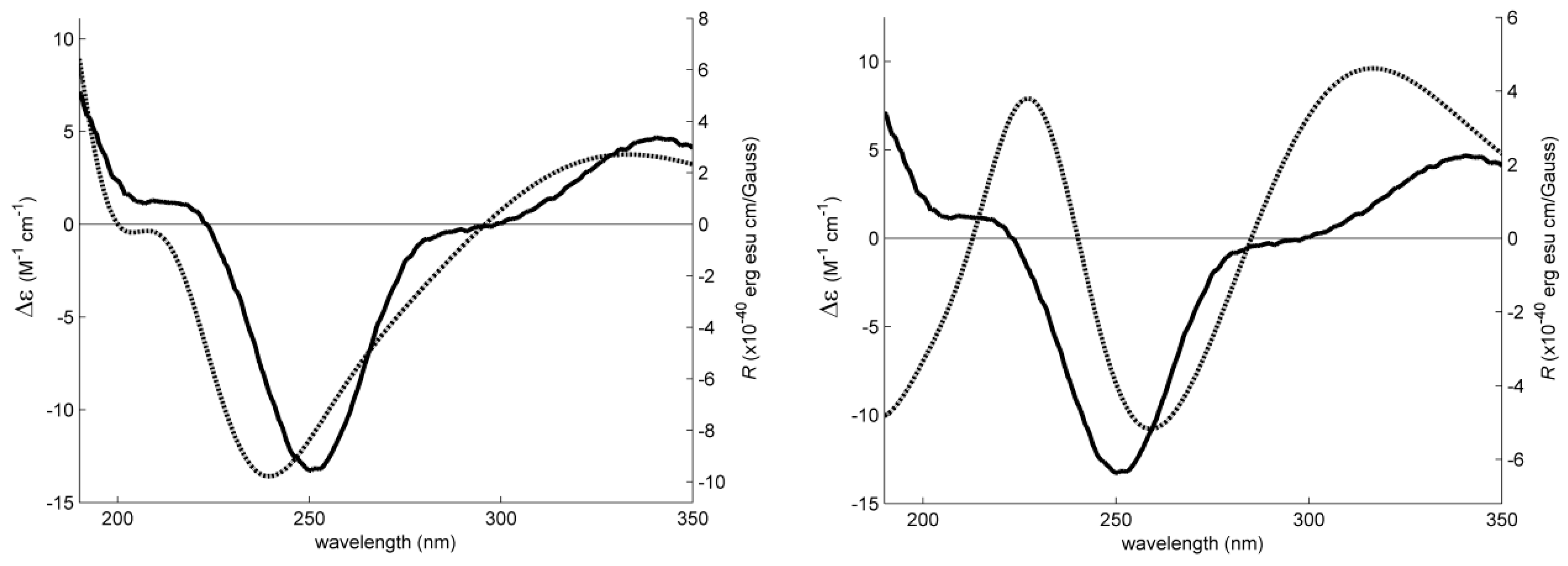
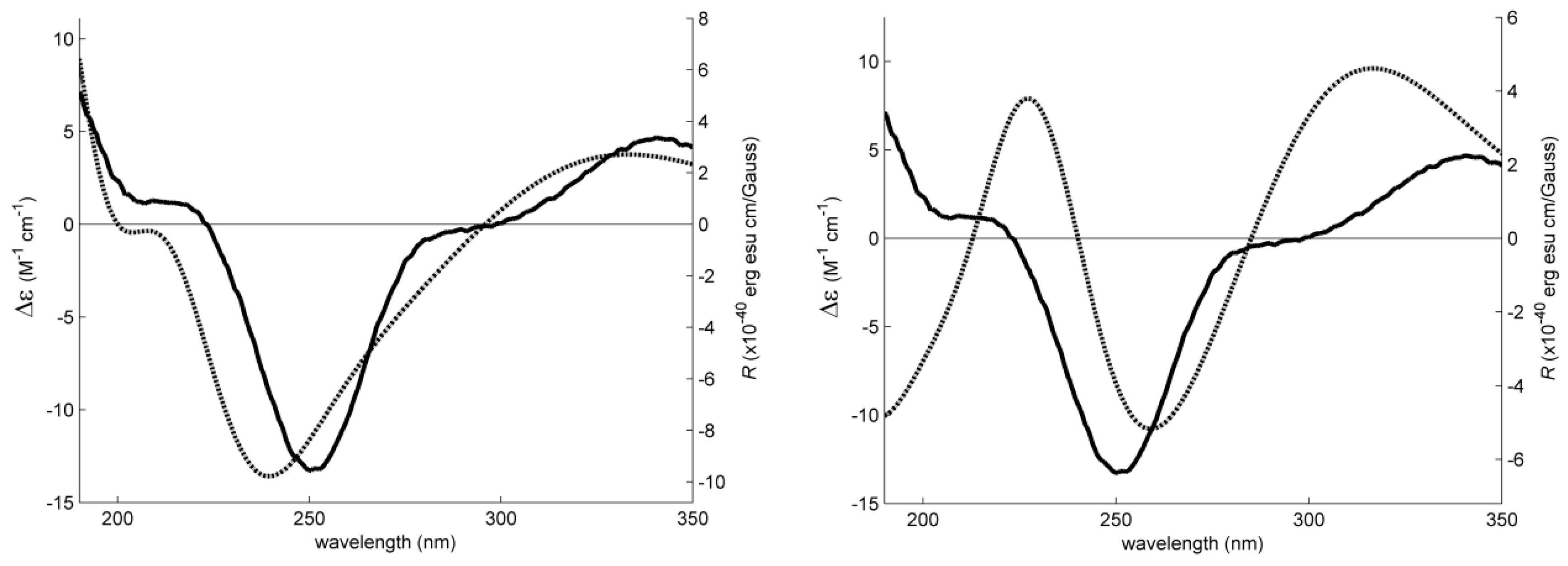
2. Results and Discussion
3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Extraction and Isolation
3.3.1. Paecilin E (1)
3.3.2. Dankasterone (2)
3.3.3. (1R, 8S, 9R)-1,9-Dihydroxy-8-(2-hydroxypropan-2-yl)-4-methoxy-5-methyl-1,7,8,9-tetrahydro-3H-furo[3,4-f]chromen-3-one (3)
3.3.4. (3β,5α,22E)-3,5-Dihydroxyergosta-7,22-dien-6-one (4)
3.4. Electronic Circular Dichroism (ECD)
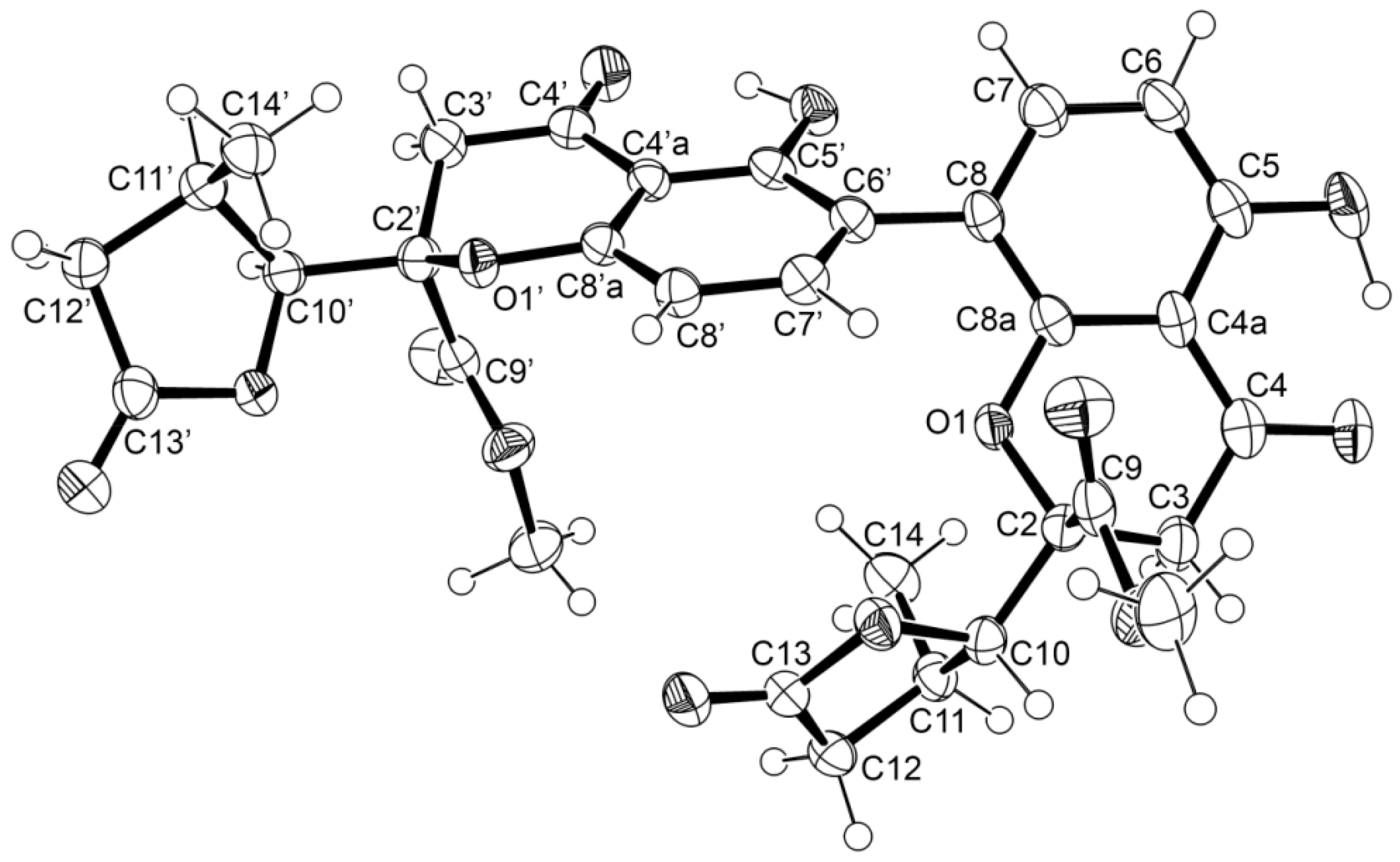
3.5. X-ray Crystal Structure of 1 and 3
3.6. Antibacterial Activity Bioassays
3.6.1. Bacterial Strains and Growth Conditions
3.6.2. Antimicrobial Susceptibility Testing
3.6.3. Biofilm Formation Inhibition Assay
3.6.4. Antibiotic Synergy Testing
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Prompanya, C.; Fernandes, C.; Cravo, S.; Pinto, M.M.M.; Dethoup, T.; Silva, A.M.S.; Kijjoa, A. A new cyclic hexapeptide and a new isocoumarin derivative from the marine sponge-associated fungus Aspergillus similanensis KUFA 0013. Mar. Drugs 2015, 13, 1432–1450. [Google Scholar] [CrossRef] [PubMed]
- Zin, W.W.; Prompanya, C.; Buttachon, S.; Kijjoa, A. Bioactive secondary metabolites from a Thai collection of soil and marine-derived fungi of the genera Neosartorya and Aspergillus. Curr. Drug Deliv. 2016, 13, 378–388. [Google Scholar] [PubMed]
- Bugni, T.S.; Ireland, C.M. Marine-derived fungi: A chemically and biologically diverse group of microorganisms. Nat. Prod. Rep. 2004, 21, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.M.; Bessa, L.J.; Buttachon, B.; Costa, P.M.; Buaruang, J.; Dethoup, T.; Silva, A.M.S.; Kijjoa, A. Antibacterial and antibiofilm activity of tryptoquivalines and meroditerpenes from marine-derived fungi Neosartorya paulistensis, N. laciniosa, N. tsunodae, and the soil fungi N. fischeri and N. siamensis. Mar. Drugs 2014, 12, 822–839. [Google Scholar] [CrossRef] [PubMed]
- Eamvijarn, A.; Gomes, N.M.; Dethoup, T.; Buaruang, J.; Manoch, L.; Silva, A.; Pedro, M.; Marini, I.; Roussis, V.; Kijjoa, A. Bioactive meroditerpenes and indole alkaloids from the soil fungus Neosartorya fischeri (KUFC 6344), and the marine-derived fungi Neosartorya laciniosa (KUFC 7896) and Neosartorya tsunodae (KUFC 9213). Tetrahedron 2013, 69, 8583–8591. [Google Scholar] [CrossRef]
- Krings, U.; Zelena, K.; Wu, S.; Berger, R.G. Thin-layer high-vacuum distillation to isolate volatile flavour compounds of cocoa powder. Eur. Food Res. Technol. 2006, 223, 675–681. [Google Scholar] [CrossRef]
- Szwalbe, A.J.; Williams, K.; O’Flynn, D.E.; Bailey, A.M.; Mulholland, N.P.; Vincent, J.L.; Willis, C.L.; Cox, R.J.; Simpson, T.J. Novel nonadride, heptadride and maleic acid metabolites from the byssochlamic acid producer Byssochlamys fulva IMI 40021—An insight into the biosynthesis of maleidrides. Chem. Commun. 2015, 51, 17088–17091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prachayasittikul, S.; Suphapong, S.; Worachartcheewan, A.; Lawung, R.; Ruchirawat, S.; Prachayasittikul, V. Bioactive metabolites from Spilanthes acmella Murr. Molecules 2009, 14, 850–867. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Krishna, M.M.; Ishida, K.; Anjaneyulu, V. Marine sterols. XXII. Occurrence of 3-oxo-4,6,8(14)-triunsaturated steroids in the sponge Dysidea herbacea. Chem. Pharm. Bull. 1992, 40, 72–74. [Google Scholar] [CrossRef]
- Kawahara, K.; Sekita, S.; Satake, M. Steroids from Calvatia cyathiformis. Phytochemistry 1994, 37, 213–215. [Google Scholar] [CrossRef]
- Amagata, T.; Tanaka, M.; Yamada, T.; Doi, M.; Minoura, K.; Ohishi, H.; Yamori, T.; Numata, A. Variation in cytostatic constituents of a sponge-derived Gymnascella dankaliensis by manipulating the carbon source. J. Nat. Prod. 2007, 70, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Zin, W.W.; Buttachon, S.; Buaruang, J.; Gales, L.; Pereira, J.A.; Pinto, M.M.M.; Silva, A.M.S.; Kijjoa, A. A new meroditerpene and a new tryptoquivaline from the algicolous fungus Neosartorya takakii KUFC 7898. Mar. Drugs 2015, 13, 3776–3790. [Google Scholar] [CrossRef] [PubMed]
- Zin, W.W.; Buttachon, S.; Dethoup, T.; Fernandes, C.; Cravo, S.; Pinto, M.M.M.; Gales, L.; Pereira, J.A.; Silva, A.M.S.; Sekeroglu, N.; et al. New cyclotetrapeptides and a new diketopiperzine derivative from the marine sponge-associated fungus Neosartorya glabra KUFA 0702. Mar. Drugs 2016, 14, 136. [Google Scholar] [CrossRef] [PubMed]
- Noinart, J.; Buttachon, S.; Dethoup, T.; Gales, L.; Pereira, J.A.; Urbatzka, R.; Freitas, S.; Lee, M.; Silva, A.M.S.; Pinto, M.M.M.; et al. A new ergosterol analog, a new bis-anthraquinone and anti-obesity activity of anthraquinones from the marine sponge-associated fungus Talaromyces stipitatus KUFA 0207. Mar. Drugs 2017, 15, 139. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, R.; Matsunaga, S. Saturated hopane and gammacerane triterpene-duils from the stem bark of Abies veitchii. Phytochemisty 1992, 31, 3535–3539. [Google Scholar] [CrossRef]
- Prata-Sena, M.; Ramos, A.A.; Buttachon, S.; Castro-Carvalho, B.; Marques, P.; Dethoup, T.; Kijjoa, A.; Rocha, E. Cytotoxic activity of secondary metabolites from marine-derived fungus Neosartorya siamensis in human cancer cells. Phytother. Res. 2016, 30, 1862–1971. [Google Scholar] [CrossRef] [PubMed]
- Achenbach, H.; Mülhlenfeld, A.; Weber, B.; Brillinger, G.U. Highly substituted chromanols from culture of Aspergillus duricaulis. Tetrahedron Lett. 1982, 45, 4659–4660. [Google Scholar] [CrossRef]
- Achenbach, H.; Mülhlenfeld, A.; Brillinger, G.U. Stiffwechselprodukte von mikroorganismen, XXX. Phthalide und chromanole aus Aspergillus duricaulis. Liebigs Ann. Chem. 1985, 1985, 1596–1628. [Google Scholar] [CrossRef]
- Fangkrathok, N.; Sripanidkulchai, B.; Umehara, K.; Noguchi, H. Bioactive ergostanoids and a new polyhydroxyoctane from Lentinus polychrous mycelia and their inhibitory effects on E2-enhanced cell proliferation of T47D cells. Nat. Prod. Res. 2013, 27, 1611–1619. [Google Scholar] [CrossRef] [PubMed]
- Kodani, S.; Imoto, A.; Mitsutani, A.; Murakami, M. Isolation and identification of the antialgal compound, harmane (1-methyl-β-carboline), produced by the algicidal bacterium, Pseudomonas sp. K44-1. J. Appl. Phycol. 2002, 14, 109–114. [Google Scholar] [CrossRef]
- Sasaki, M.; Takamatsu, H.; Oshita, K.; Yukio Kaneko, Y.; Yokotsuka, T. Isolation of lumichrome from the culture filtrate of Aspergillus oniki 1784. Nippon Nōgeikagaku Kaishi 1974, 48, 569–571. [Google Scholar] [CrossRef]
- Guo, Z.; She, Z.; Shao, C.; Wen, L.; Liu, F.; Zheng, Z.; Lin, Y. 1H and 13C NMR signal assignments of paecilin A and B, two new chromone derivatives from mangrove endophytic fungus Paeciliomyces sp. (tree 1–7). Magn. Reson. Chem. 2007, 45, 777–780. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Sun, Y.-L.; Zhang, X.-Y.; Han, Z.; Gao, H.-C.; He, F.; Qian, P.Y.; Qi, S.-H. Antifouling and antibacterial polyketides from marine gorgonian coral-associated fungus Penicillium sp. SCSDAF 0023. J. Antibiot. 2013, 66, 219–223. [Google Scholar] [CrossRef] [PubMed]
- El-Elimat, T.; Figueroa, M.; Adcock, A.F.; Kroll, D.J.; Swanson, S.M.; Wani, M.C.; Pearce, C.J.; Oberlies, N.H. Cytotoxic polyketides from an unidentified fungus (MSX 45109). Planta Med. 2013, 79-PL13. [Google Scholar] [CrossRef]
- El-Elimat, T.; Figueroa, M.; Raja, H.A.; Graf, T.N.; Swanson, S.M.; Falkinham, J.O., III; Wani, M.; Pearce, C.J.; Oberlies, N.H. Biosythetically distinct cytotoxic polyketides from Setophoma terrestris. Eur. J. Org. Chem. 2015, 2015, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Amagata, T.; Doi, M.; Tohgo, M.; Minoura, K.; Numanta, A. Dankasterone, a new class of cytotoxic steroid produced by Gymnascella species from a marine sponge. Chem. Commun. 1999, 30, 1321–1322. [Google Scholar] [CrossRef]
- Aiello, A.; Fattorusso, E.; Magno, S.; Menna, M. Isolation of five new 5α-hydroxy-6-keto-Δ7sterols from the marine sponge Oscarella lobularis. Steroids 1991, 56, 337–340. [Google Scholar] [CrossRef]
- Ishizuka, T.; Yaoita, Y.; Kikuchi, M. Sterol constituents from the fruit bodies of Grifola frondosa (Fr.) S.F. Gray. Chem. Pharm. Bull. 1997, 45, 1756–1760. [Google Scholar] [CrossRef]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: New York, NY, USA, 1990; pp. 315–322. [Google Scholar]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 72, 5463–5467. [Google Scholar] [CrossRef]
- Austin, A.; Petersson, G.A.; Frixch, M.J.; Dobek, F.J.; Scalmani, G.; Throssel, K. A density functional with spherical atom dispersion terms. J. Chem. Theory Comput. 2012, 8, 4989–5007. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Harada, N. ECD Cotton effect approximated by the Gaussian curve and other methods. Chirality 2010, 22, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short story of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Simões, R.R.; Aires-de-Sousa, M.; Conceicao, T.; Antunes, F.; da Costa, P.M.; de Lencastre, H. High prevalence of EMRSA-15 in Portuguese public buses: A worrisome finding. PLoS ONE 2011, 6, e17630. [Google Scholar] [CrossRef] [PubMed]
- Bessa, L.J.; Barbosa-Vasconcelos, A.; Mendes, A.; Vaz-Pires, P.; Martins da Costa, P. High prevalence of multidrug-resistant Escherichia coli and Enterococcus spp. in river water, upstream and downstream of a wastewater treatment plant. J. Water Health 2014, 12, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Zin, W.W.; Buttachon, S.; Dethoup, T.; Pereira, J.A.; Gales, L.; Inácio, A.; Costa, P.M.; Lee, M.; Sekeroglu, N.; Silva, A.M.S.; et al. Antibacterial and antibiofilm activities of the metabolites isolated from the culture of the mangrove-derived endophytic fungus Eurotium chevalieri KUFA 0006. Phytochemistry 2017, 141, 86–97. [Google Scholar] [CrossRef] [PubMed]
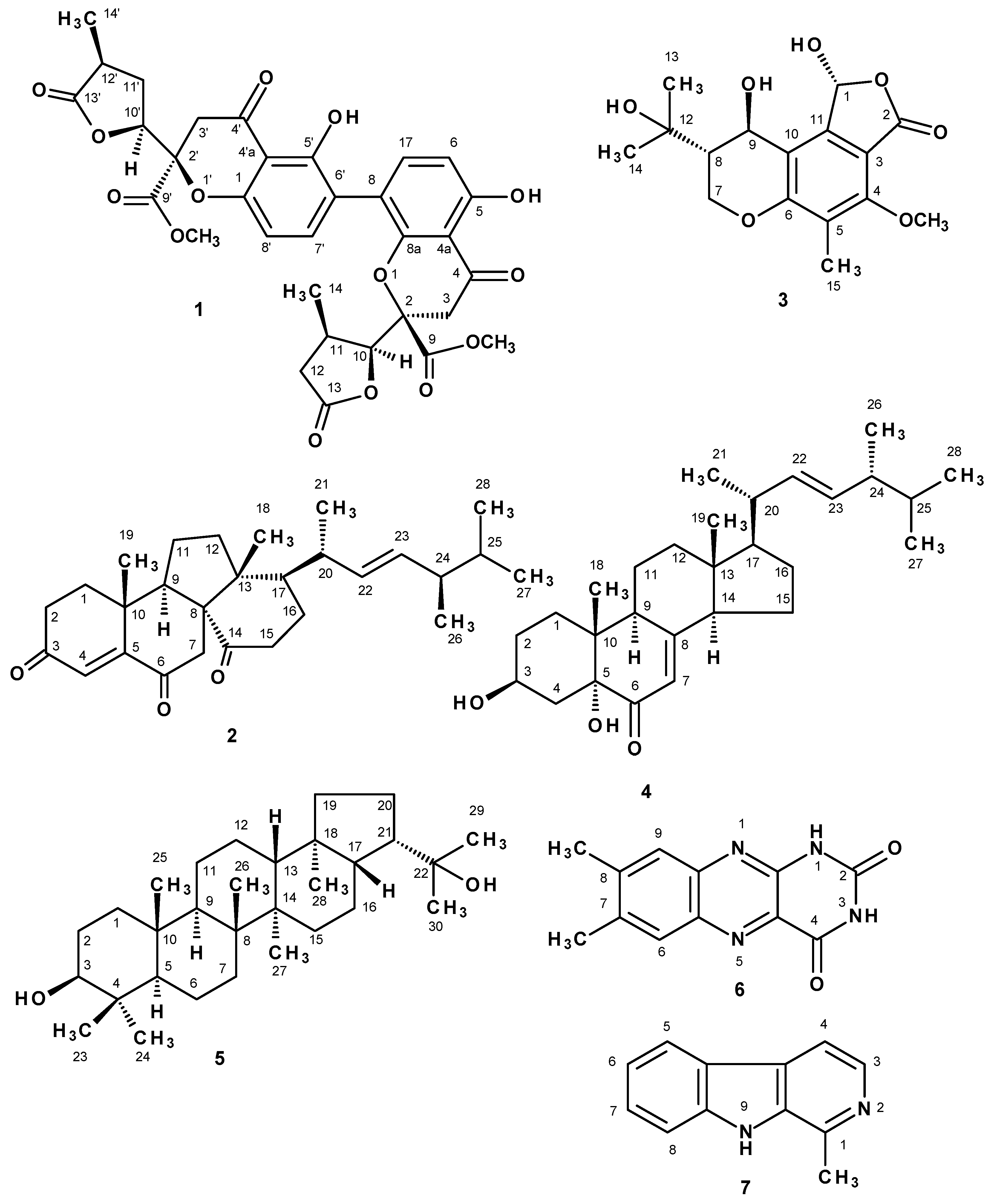
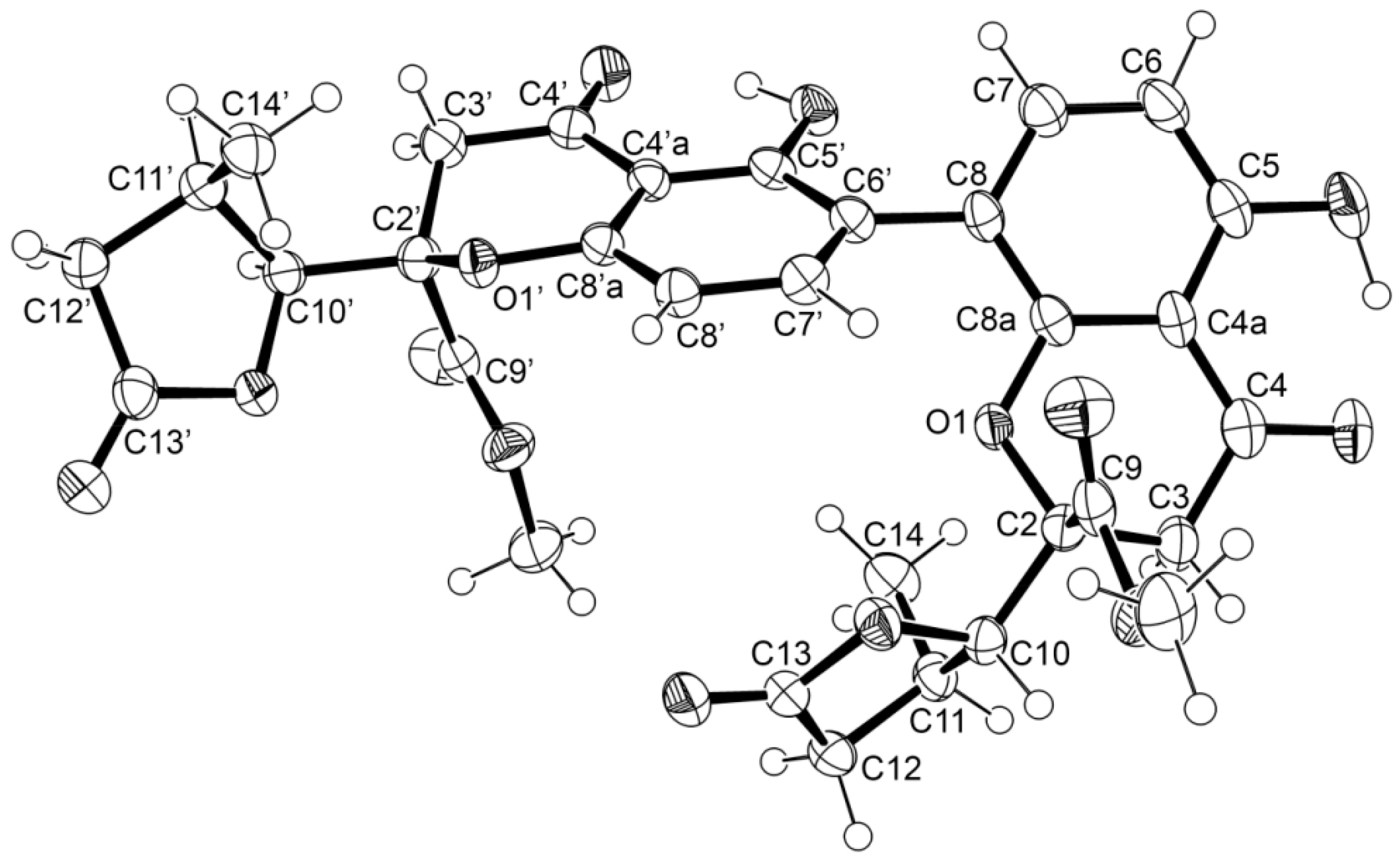
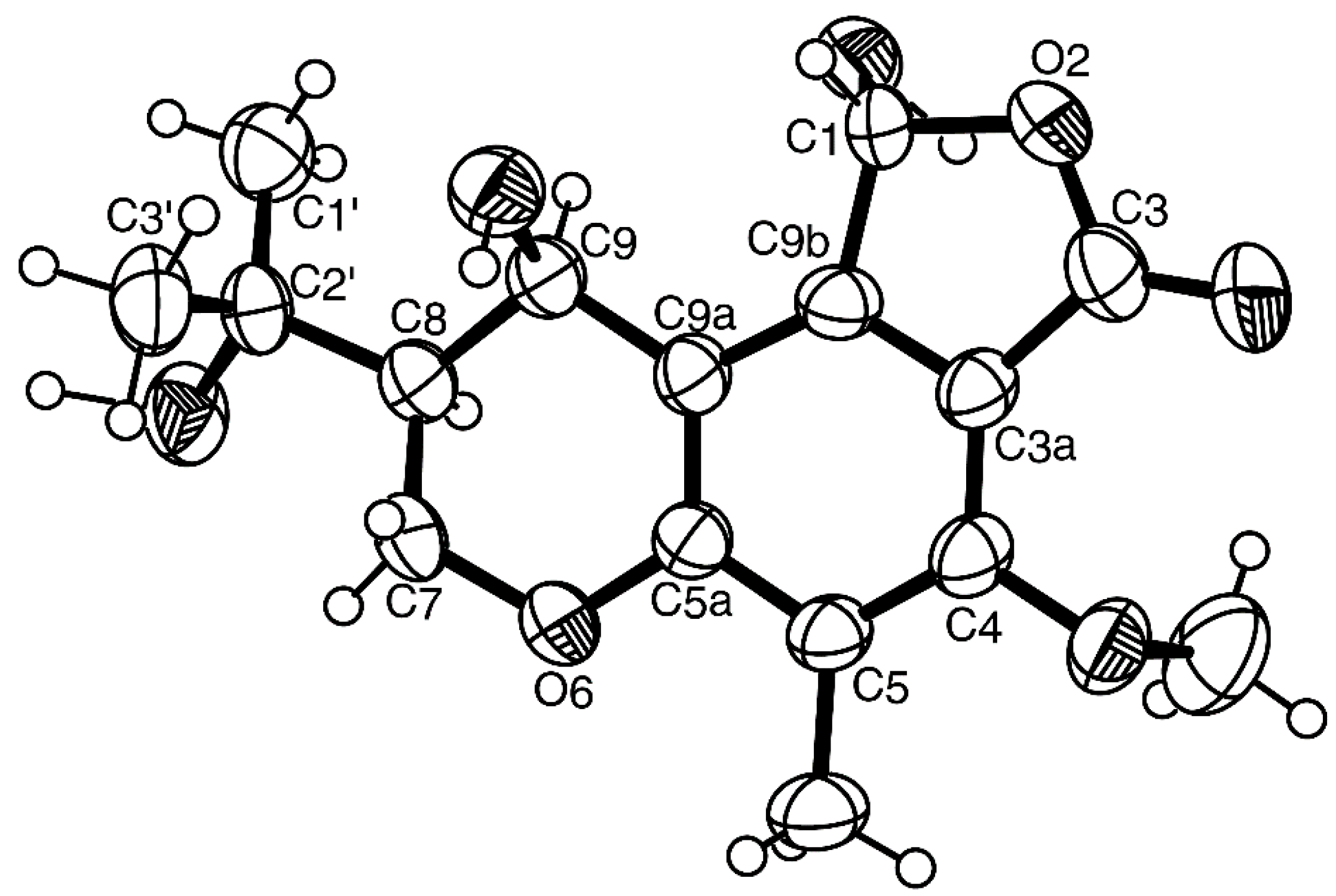
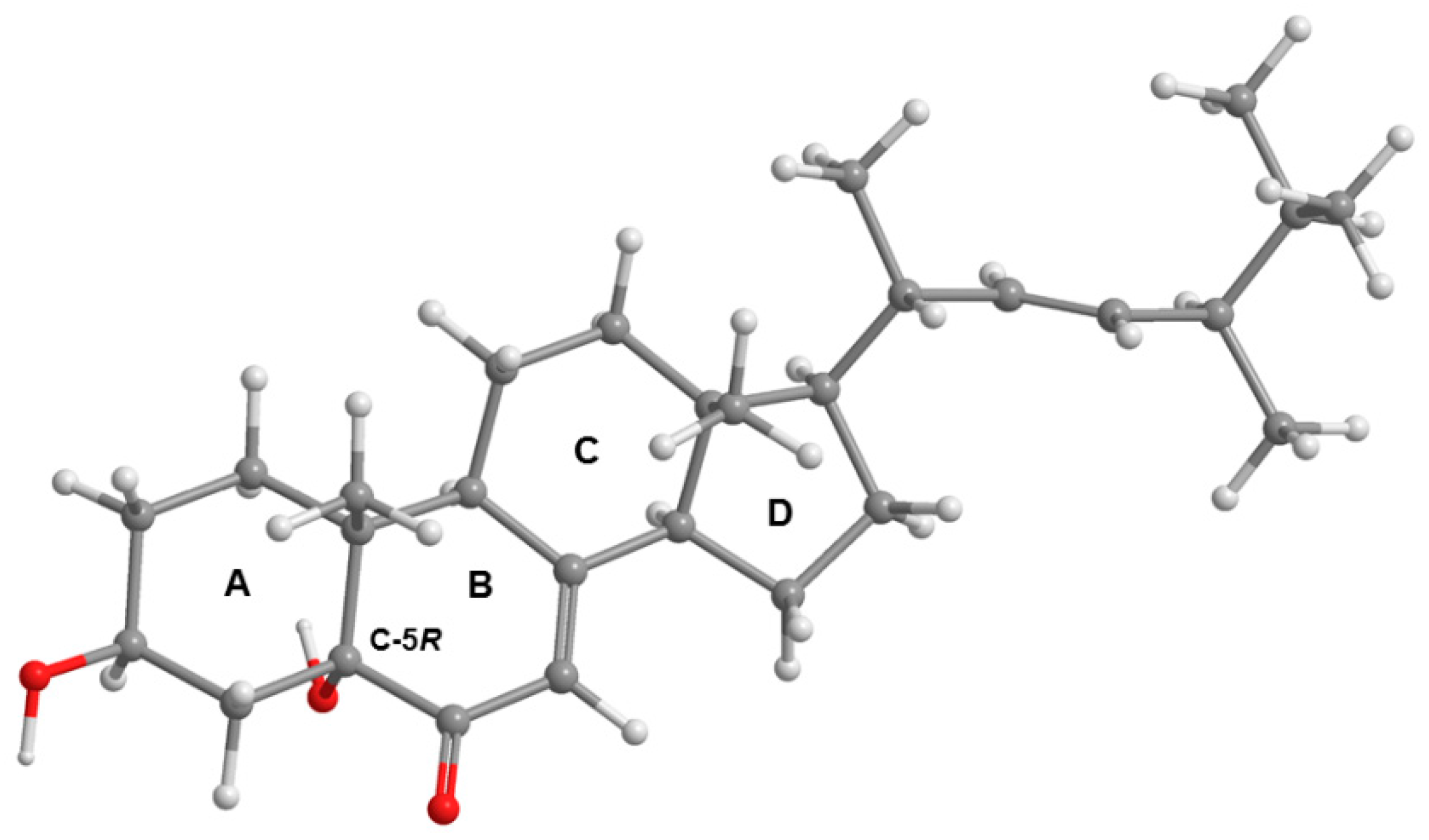

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δC, Type | δH, (J in Hz) | COSY | HMBC |
|---|---|---|---|---|
| 2 | 85.4, C | - | ||
| 3α | 32.9, CH2 | 3.58, d (17.4) | H-3β | C-2, 4, 9, 10 |
| β | 3.05, d (17.4) | H-3α | C-4, 4a | |
| 4 | 194.9, CO | - | ||
| 5 | 107.5, C | - | ||
| 6 | 109.2, CH | 6.61, d (8.6) | H-7 | C-4a, 8 |
| 7 | 140.9, CH | 7.50, d (8.6) | H-6 | C-5, 8a |
| 8 | 114.6, C | - | ||
| 9 | 168.9, CO (Ac) | - | ||
| OMe-9 | 53.3, CH3 | 3.70, s | C-9 | |
| 10 | 81.7, CH | 4.85, d (7.3) | H-11 | C-2, 3, 12, 13, 14 |
| 11 | 32.9, CH | 2.85, m | H-10, H2-12, Me-14 | C-2, 13, 14 |
| 12α | 35.3, CH2 | 1.75, dd (17.0, 9.9) | H-11, 12β | C-10, 13, 14 |
| β | 2.41, dd (17.0, 8.4) | H-11, 12 α | C-10, 13, 14 | |
| 13 | 174.9, CO | - | ||
| 14 | 14.1, CH3 | 1.06, d (7.1) | H-11 | C-10, 11, 12 |
| 2′ | 83.9, C | - | ||
| 3′α | 39.2, CH2 | 3.57, d (17.4) | H-3′β | C-2′, 4′, 9′, 10′ |
| β | 3.09, d (17.4) | H-3′α | C-4′, 4′a | |
| 4′ | 195.3, CO | - | ||
| 4′a | 107.0, C | - | ||
| 5′ | 158.1, C | - | ||
| 6′ | 116.6, C | - | ||
| 7′ | 140.6, CH | 7.61, d (8.6) | H-8′ | C-5′, 8′a, 8 |
| 8′ | 107.4, CH | 6.60, d (8.6) | H-7′ | C-6′, 8′a |
| 8′a | 158.2, C | - | ||
| 9′ | 169.3, CO (Ac) | - | ||
| OMe-9′ | 53.3, CH3 | 3.69, s | C-9′ | |
| 10′ | 81.9, CH | 4.97, d (6.7) | H-11′ | C-3′, 11′, 13′, 14′ |
| 11′ | 32.5, CH | 2.97, m | H-10′, 11′, 12′a, 12′β | C-2′, 13′, 14′ |
| 12′α | 36.9, CH2 | 2.33, dd (17.0, 5.4) | H-11′, 12′β | C-10′, 13′, 14′ |
| β | 2.86, dd (17.0, 8.1) | H-11′, 12′α | C-10′, 13′, 14′ | |
| 13′ | 175.5, CO | - | ||
| 14′ | 14.6, CH3 | 1.17, d (7.1) | H-11′ | C-10′, 11′, 12′ |
| OH-5 | - | 11.56, s | C-4a, 5, 6 | |
| OH-5′ | - | 11.83, s | C-4′a, 5′, 6′ |
| Position | δC, Type | δH, (J in Hz) | COSY | HMBC | NOESY |
|---|---|---|---|---|---|
| 1 | 95.6, CH | 6.64, s | - | C-3 | OH-1, H-9 |
| 3 | 166.1, CO | - | |||
| 3a | 109.4, C | - | |||
| 4 | 155.9, C | - | |||
| 5 | 120.0, C | - | |||
| 5a | 158.4, C | ||||
| 7α | 63.9, CH2 | 4.29, dd (12.0, 10.6) | H-7β, 8 | C-5a, 8, 9 | H-7β |
| β | 4.53, dd (11.6, 2.4) | H-7α | C-5a, 8, 9 | ||
| 8 | 46.6, CH | 1.79, dt (11.9, 2.8) | H-7α | C-2′, 7 | H-8, Me-1′, 3′ |
| 9 | 57.8, CH | 5.16, br | - | ||
| 9a | 146.8, C | - | |||
| 9b | 117.4, C | - | |||
| 10 | 8.6, CH3 | 2.05, s | - | C-3a, 4, 5, 5a, 9a | OMe-4 |
| 1′ | 28.4, CH3 | 1.27, s | - | C-2′, 3′, 8 | H-8, OH-2′, Me-3′ |
| 2′ | 69.9, C | - | |||
| 3′ | 27.7, CH3 | 1.24, s | C-1′, 2′, 8 | H-8, OH-2′, Me-1′ |
| Paecilin E (1) | Dankasterone A (2) | |||
|---|---|---|---|---|
| Bacterial strain | MIC | MBC | MIC | MBC |
| E.coli ATCC 25922 | >64 | >64 | >64 | >64 |
| E.coli SA/2 (ESBL) | >64 | >64 | >64 | >64 |
| P. aeruginosa ATCC 27853 | >64 | >64 | >64 | >64 |
| E. faecalis ATCC29212 | 16 | >64 | 32 | >64 |
| E. faecalis A5/102 (VRE) | 64 | >64 | 64 | >64 |
| E. faecalis B3/101 (VRE) | >64 | >64 | >64 | >64 |
| S. aureus ATCC 29213 | 32 | >64 | >64 | >64 |
| S. aureus 40/61/24 | >64 | >64 | >64 | >64 |
| S. aureus 66/1 (MRSA) | >64 | >64 | >64 | >64 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumla, D.; Shine Aung, T.; Buttachon, S.; Dethoup, T.; Gales, L.; Pereira, J.A.; Inácio, Â.; Costa, P.M.; Lee, M.; Sekeroglu, N.; et al. A New Dihydrochromone Dimer and Other Secondary Metabolites from Cultures of the Marine Sponge-Associated Fungi Neosartorya fennelliae KUFA 0811 and Neosartorya tsunodae KUFC 9213. Mar. Drugs 2017, 15, 375. https://doi.org/10.3390/md15120375
Kumla D, Shine Aung T, Buttachon S, Dethoup T, Gales L, Pereira JA, Inácio Â, Costa PM, Lee M, Sekeroglu N, et al. A New Dihydrochromone Dimer and Other Secondary Metabolites from Cultures of the Marine Sponge-Associated Fungi Neosartorya fennelliae KUFA 0811 and Neosartorya tsunodae KUFC 9213. Marine Drugs. 2017; 15(12):375. https://doi.org/10.3390/md15120375
Chicago/Turabian StyleKumla, Decha, Tin Shine Aung, Suradet Buttachon, Tida Dethoup, Luís Gales, José A. Pereira, Ângela Inácio, Paulo M. Costa, Michael Lee, Nazim Sekeroglu, and et al. 2017. "A New Dihydrochromone Dimer and Other Secondary Metabolites from Cultures of the Marine Sponge-Associated Fungi Neosartorya fennelliae KUFA 0811 and Neosartorya tsunodae KUFC 9213" Marine Drugs 15, no. 12: 375. https://doi.org/10.3390/md15120375