4. Materials and Methods
4.1. General Procedures
IR spectra were recorded on a Perkin Elmer BX FT-IR spectrometer (Perkin Elmer, Waltham, MA, USA). NMR spectra were recorded on a Bruker Avance 300 spectrometer or a Bruker Avance 500 spectrometer (Bruker, Billerica, MA, USA). The chemical shifts were reported in ppm relative to the residual signal solvent (MeOH-d4: δH 3.31, δC 49.15; CDCl3: δH 7.26, δC 77.16). High-resolution mass spectra were obtained on a LCT Premier XE spectrometer (Waters Corporation, Milford, NY, USA) in electrospray ionization mode by direct infusion of the purified compounds. Medium-Pressure Liquid Chromatography (MPLC) was performed on a Puriflash apparatus (Interchim, Montluçon, France) using a Silica gel Interchim cartridge PF-50SIHP (Interchim, Montluçon, France). The synthesis reactions were monitored by thin-layer chromatography (TLC). The TLC was performed on aluminum precoated silica gel 60 F254 plates (Merck, Darmstadt, Germany) and revealed with a solution of sulfuric vanillin. Preparative HPLC purifications were performed on an autoprep system: Waters 600 controller and Waters 600 pump with a Waters 996 photodiode array detector (Waters Corporation, Milford, NY, USA), equipped with a Waters Sunfire C18 (19 × 150 mm, 5 μm) column. Organic solvents were purchased from Carlo Erba (Val de Reuil, France). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) or Fluorochem (Hadfield, UK).
4.2. Animal Material
Sponges were collected by hand using SCUBA in Wallis and Futuna during the sampling cruise Wallis 2018 aboard the R/V Alis off the coast of Wallis Island (18 July 2018–31, 13°16.367′ S; 176°12.283′ W) between 6 and 50 m deep [
19]. Voucher samples are deposited at the Queensland Museum (Brisbane, Australia) and were identified by Dr. Merrick Ekins, accession numbers available in the
Supplementary Materials (Table S1). Sponges were deep-frozen on board until workup. They were then freeze-dried and grounded before extraction.
The sponge Pericharax heteroraphis Poléjaeff 1883 was collected in the lagoon of Wallis Island (13°16.177’ S, 176°08.120’ W) at 15 m deep in July 2018. The corresponding voucher specimen is available under accessing number G339027.
4.3. HPLC-QTOF-ESI-MS/MS Analysis
LC-MS/MS analyses were performed using the methods described in the work of Schilling et al. [
38].
4.4. Network Metabolomic Analyses
The molecular network of the
Pericharax samples was constructed according to the procedure described by Schilling et al. [
38].
4.5. Extraction and Isolation of Compounds 3, 5, 6 and 9
The 47 sponge extracts preliminary tested were obtained by a pressurized liquid extraction process with a mixture of CH
2Cl
2/MeOH (1:1), as previously described [
39].
The freeze-dried sponge sample of Pericharax heteroraphis (93 g) was extracted by ASE at 40 °C under pressure (100 bar) three times with a mixture of CH2Cl2/MeOH (1:1). The extracts were combined and dried under reduced pressure to afford a brown residue (6 g), and 5.5 g of this residue was partitioned between n-BuOH and H2O. The butanolic extract (1.4 g) was fractioned by C18 reverse-phase MPLC eluted with successive solvents (H2O, H2O/CH3CN gradient, CH3CN, CH2Cl2 and THF) to afford fractions F1 to F4.
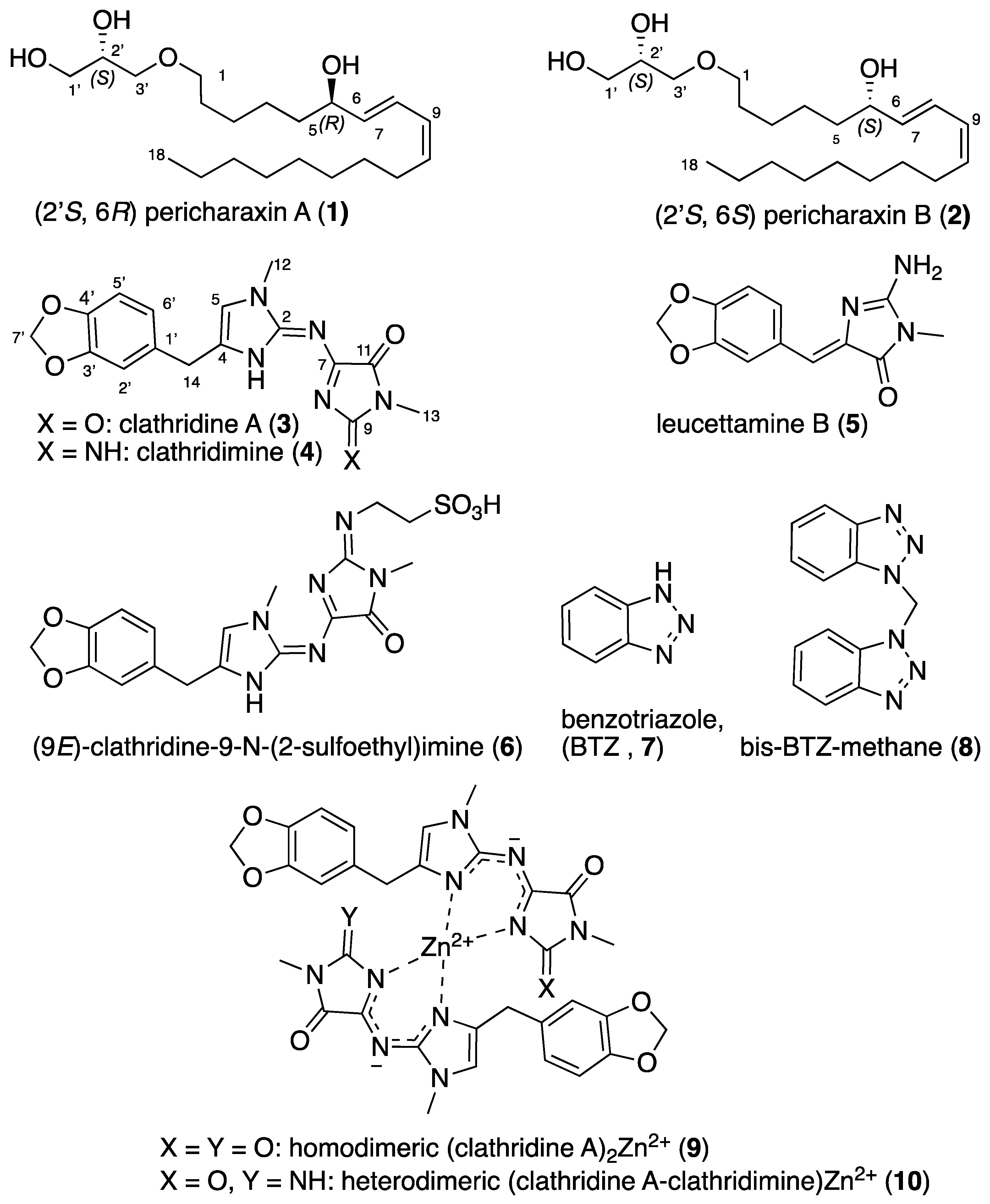
Fraction F2 (H2O/CH3CN gradient) was purified by preparative reverse-phase HPLC (column: Waters Sunfire C18, 19 mm × 150 mm, 5 μm, H2O + 0.1% formic acid/CH3CN + 0.1% formic acid) to obtained four 2-aminoimidazoles: clathridine A (3) (1.9 mg), leucettamine B (5) (1.2 mg), (9E)-clathridine-9-N-(2-sulfoethyl)imine (6) (1.2 mg) and homodimeric (clathridine A)2 Zn2+ (9) (2.4 mg).
Clathridine A (3): yellow solid (1.9 mg); UV (MeOH) λmax (log ε) 203 (0.18), 215 (0.18), 283 (0.01), 388 (0.03) nm; 1H NMR (500 MHz, CDCl3): δ (ppm) 6.75 (1H, d, J = 7.8 Hz, H-5′), 6.73 (1H, s, H-2′), 6.71 (1H, d, J = 7.8 Hz, H-6′), 6.53 (1H, s, H-5), 5.94 (2H, s, H-7′), 3.81 (2H, s, H-14), 3.72 (3H, s, H-12), 3.19 (3H, s, H-13); 13C NMR (125 MHz, CDCl3): δ (ppm) 161.8 (C-9), 154.6 (C-11), 147.6 (C-5′), 146.5 (C-2), 146.3 (C-3′), 139.5 (C-4), 132.6 (C-1′), 121.6 (C-6′), 117.5 (C-5), 109.2 (C-2′), 108.2 (C-5′), 100.8 (C-7′), 34.4 (C-14), 32.0 (C-12), 24.6 (C-13) (C-7 non detected); HRESIMS m/z 342.1172 [M + H]+ (calcd for C16H16N5O4, 342.1202).
Leucettamine B (5): clear oil (1.2 mg); UV (MeOH) λmax (log ε) 234 (0.38), 367 (0.72) nm; 1H NMR (500 MHz, CDCl3): δ (ppm) 7.41 (1H, s, H-1 ar), 7.21 (1H, d, J = 8.0 Hz, H-2 ar), 6.65 (1H, d, J = 8.0 Hz, H-3 ar), 6.83 (1H, s, CH), 6.02 (2H, s, CH2 benzodioxol), 3.30 (3H, s, CH3); 13C NMR (125 MHz, CDCl3): δ (ppm) 165.2 (CO), 157.4 (CNH2), 149.6 (C-ar), 149.2 (C-ar), 126.6 (C-2), 119.1 (CH) 109.8 (C-1), 109.0 (C-3), 101.8 (CH2), 26.0 (CH3); HRESIMS m/z 246.0846 [M + H]+ (calcd for C12H12N3O3, 246.0879).
(9E)-Clathridine-9-N-(2-sulfoethyl)imine B (6): clear oil (1.2 mg); UV (MeOH) λmax (log ε) 217 (0.38), 261 (0.25), 385 (0.38) nm; 1H NMR (500 MHz, CDCl3): δ (ppm) 6.84 (1H, s, H-2′), 6.77 (2H, s, H-5′ and H-6′), 6.35 (1H, s, H-5), 5.95 (2H, s, H-7′), 4.25 (2H, m, H-16), 4.09 (2H, s, H-14), 3.71 (3H, S, H-12), 3.35 (3H, S, H-13), 3.33 (2H, m, H-17); 13C NMR (125 MHz, CDCl3): δ (ppm) 165.2 (CO), 157.4 (CNH2), 149.6 (C-ar), 149.2 (C-ar), 126.6 (C-2), 119.1 (CH) 109.8 (C-1), 109.0 (C-3), 101.8 (CH2), 26.0 (CH3); HRESIMS m/z 449.1257 [M + H]+ (calcd for C18H21N6O6S, 449.1243).
Homodimeric (clathridine A)2 Zn2+ (9): yellow solid (2.4 mg); UV (MeOH) λmax (log ε) 220 (0.33), 286 (0.03), 368 (0.09) nm; 1H NMR (500 MHz, CDCl3): δ (ppm) 6.63 (2H, s, H-5), 6.50 (2H, d, J = 7.9 Hz, H-5′), 6.26 (2H, t, J = 7.9 Hz, H-6′), 6.25 (2H, s, H-2′), 5.88 (4H, dd, J = 13.6, 1.1 Hz, H-7′), 3.80 (6H, s, H-12), 3.51 (2H, d, J = 16.3 Hz, H-14a), 3.37 (2H, d, J = 16.3 Hz, H-14b), 3.04 (2H, s, H-13); 13C NMR (125 MHz, CDCl3): δ (ppm) 164.9 (C-9), 161.4 (C-11), 149.0 (C-2), 147.7 (C-3′), 146.5 (C-4′), 136.3 (C-4), 131.1 (C-1′), 121.7 (C-2′), 117.7 (C-5), 108.6 (C-6′), 107.9 (C-5′), 101.2 (C-7′), 33.3 (C-14), 32.7 (C-12), 24.7 (C-13); HRESIMS m/z 745.1479 [M + H]+ (calcd for C32H29N10O8Zn, 745.1461).
4.6. Total Synthesis of Clathridine A (3) and Clathridimine (4)
Piperonal (12): To a suspension of pyridinium dichromate (18.55 g, 49 mmol, 1.5 eq.) in dry DCM (230 mL), trichloromethylsilane (14.6 mL, 115 mmol, 3.5 eq.) was added dropwise at 0 °C. After 15 min, piperonyl alcohol (5 g, 33 mmol, 1 eq.) was added, and the reaction mixture was stirred for 1 h at room temperature. Moist silica gel was then added, and the suspension was filtered through a pad of silica gel. The solvent was evaporated under reduced pressure, and the crude product was purified by flash column chromatography on silica gel (gradient 100% Heptane to70:30 Heptane/EtOAc) to give 12 as an oil (3.76 g, 75% yield). 1H NMR (300 MHz, CDCl3): δ (ppm) 9.78 (1H, s, H-14), 7.38 (1H, dd, J = 7.8, 1.6 Hz, H-6′), 7.26 (1H, d, J = 1.6 Hz, H-2′), 6.86 (1H, d, J = 7.8 Hz, H-5′), 6.00 (2H, s, H-7′); 13C NMR (75 MHz, CDCl3): δ (ppm) 190.2 (C-14), 153.1 (C-4′), 148.7 (C-3′), 132.0 (C-1′), 128.6 (C-6′), 108.3 (C-5′), 106.8 (C-2′), 102.1 (C-7′); ESIMS m/z 151.03 [M+H]+.
Preclathridine A (
13): Prepared in four steps according to the procedure described by Koswatta et al. [
28,
40]. 4-iodo-1-methyl-1
H-imidazole
11 (5.5 g, 27 mmol, 1 eq.) in anhydrous THF (96 mL), EtMgBr (3.0 M solution in ether, 9.4 mL, 28 mmol, 1.05 eq.) and piperonal
12 (4.2 g, 28 mmol, 1.05 eq.) gave intermediate
4-(Benzo[1,3]dioxol-5-yl)hydroxymethyl-1-methyl-1H-imidazole (4.31 g, 70% yield) as an orange solid. (
1H NMR (300 MHz, CDCl3):
δ (ppm) 7.41 (1H, br s, H-2), 6.94 (1H, br s, H-2′), 6.91 (1H, dd,
J = 7.9, 1.6 Hz, H-6′), 6.76 (1H, d,
J = 7.9 Hz, H-5′), 5.93 (2H, s, H-7′), 5.70 (1H, s, H-14), 3.60 (3H, s, H-12);
13C NMR (75 MHz, CDCl3):
δ (ppm) 147.6 (C-4′), 146.0 (C-4), 145.9 (C-3′), 137.5 (C-2), 137.1 (C-1′), 120.1 (C-6′), 116.9 (C-5), 107.9 (C-5′), 107.3 (C-2′), 100.9 (C-7′), 70.4 (C-14), 33.4 (C-12); HRESIMS
m/
z 233.0926 [M+H]
+ (calcd for C
12H
13N
2O
3, 233.0926)). Then, this coupling product (4.3 g, 18.5 mmol, 1 eq.) in anhydrous DCM (140 mL), TFA (5.7 mL, 74 mmol, 4 eq.) and Et
3SiH (13.6 mL, 85 mmol, 4.5 eq.) gave
4-(Benzo[1,3]dioxol-5-yl)methyl-1-methyl-1H-imidazole (2.3 g, 60% yield) as a yellow solid. (
1H NMR (300 MHz, CDCl3):
δ (ppm) 7.34 (1H, br s, H-2), 6.76 (1H, br s, H-2′), 6.72 (2H, s, H-5′, H-6′), 6.51 (1H, s, H-5), 5.89 (2H, s, H-7′), 3.81 (2H, s, H-14), 3.58 (3H, s, H-12);
13C NMR (75 MHz, CDCl3):
δ (ppm) 147.5 (C-4′), 145.7 (C-3′), 142.6 (C-4), 137.3 (C-2), 134.2 (C-1′), 121.5 (C-6′), 116.9 (C-5), 109.3 (C-5′), 108.0 (C-2′), 100.7 (C-7′), 34.6 (C-14), 33.1 (C-12); HRESIMS
m/
z 217.0931 [M+H]
+ (calcd for C
12H
13N
2O
2, 217.0977)). Then, the previous product (2.2 g, 10.2 mmol, 1 eq.) in anhydrous THF (40 mL),
n-BuLi (1.6 M in hexane, 7.0 mL, 11.2 mmol, 1.1 eq.) and tosyl azide (30%
w/w in toluene, 9.0 mL, 12.2 mmol, 1.2 eq.) gave the azide intermediate (1.9 g, 71% yield) as a brownish oil. (
1H NMR (300 MHz, CDCl3):
δ (ppm) 6.76 (1H, br s, H-2′), 6.72 (2H, s, H-5′, H-6′), 6.25 (1H, s, H-5), 5.90 (2H, s, H-7′), 3.75 (2H, s, H-14), 3.32 (3H, s, H-12);
13C NMR (75 MHz, CDCl3):
δ (ppm) 147.6 (C-4′), 145.9 (C-3′), 140.1 (C-2), 139.9 (C-4), 133.5 (C-1′), 121.7 (C-6′), 116.0 (C-5), 109.4 (C-5′), 108.1 (C-2′), 100.8 (C-7′), 34.7 (C-14), 31.4 (C-12); HRESIMS
m/
z 258.0991 [M+H]
+ (calcd for C
12H
12N
5O
2, 258.0991)). This azide intermediate (1.85 g, 7.2 mmol, 1 eq.) was dissolved in EtOH (58 mL) in the presence of 10% Pd/C on charcoal (0.38 g, 0.36 mmol, 5 mol%) to give
13 (1.57 g, 95% yield) as a yellow solid.
1H NMR (300 MHz, CDCl3):
δ (ppm) 6.72 (1H, br s, H-1), 6.70 (2H, s, H-5′, H-6′), 6.05 (1H, s, H-5), 5.89 (2H, s, H-7′), 3.63 (2H, s, H-14), 3.34 (3H, s, H-12);
13C NMR (75 MHz, CDCl3):
δ (ppm) 147.8 (C-2), 147.7 (C-4′), 146.1 (C-3′), 134.5 (C-4), 133.3 (C-1′), 121.8 (C-6′), 112.9 (C-5), 109.5 (C-5′), 108.3 (C-2′), 101.0 (C-7′), 33.7 (C-14), 31.8 (C-12); HRESIMS
m/
z 232.1100 [M+H]
+ (calcd for C
12H
14N
3O
2, 232.1086).
Clathridine A (
3): Prepared using the method described by Ohta [
41]. To a solution of 1-methylparabanic acid (
14) (0.3 g, 2.4 mmol, 1 eq.) and DMAP (cat.) in anhydrous CHCl
3 (2.4 mL), triethylamine (0.7 mL, 5 mmol, 2.1 eq.) and then TMS-Cl (0.63 mL, 5 mmol, 2.1 eq.) were added, and the resulting mixture was stirred at room temperature for 2 h under Ar. Then, a solution of preclathridine A (
13) (0.55 g, 2.4 mmol, 1 eq.) in anhydrous CHCl
3 (2 mL) was added, and the reaction mixture was stirred at reflux for 24 h in a sealed tube. After returning to room temperature, the reaction was quenched with water (2 mL), and the layers separated. The aqueous layer was extracted with CHCl
3 (3 × 10 mL), and the combined organic phases were dried over MgSO
4, filtered and concentrated. The crude product was purified by flash column chromatography on silica gel (gradient 100% DCM to 70:30 DCM/EtOAc) to give
3 (0.250 g, 30% yield) as a yellow solid.
1H NMR (300 MHz, CDCl3):
δ (ppm) 6.74 (1H, br s, H-2′), 6.72 (2H, s, H-5′, H-6′), 6.52 (1H, s, H-5), 5.93 (2H, s, H-7′), 3.82 (2H, s, H-14), 3.71 (3H, s, H-12), 3.18 (3H, s, H-13);
13C NMR (75 MHz, CDCl3):
δ (ppm) 161.8 (C-11), 155.1 (C-9), 152.3 (C-7), 147.6 (C-3′), 146.5 (C-2), 146.3 (C-4′), 138.9 (C-4), 132.3 (C-1′), 121.7 (C-6′), 117.5 (C-5), 109.2 (C-5′), 108.3 (C-2′), 100.9 (C-7′), 34.0 (C-14), 32.2 (C-12), 24.8 (C-13); HRESIMS
m/
z 342.1167 [M+H]
+ (calcd for C
16H
16N
5O
4, 342.1202).
tert-butyl-(1-methyl-4-oxo-4,5-dihydro-1H-imidazol-2-yl)carbamate (16): To a solution of creatinine (15) (2.0 g, 17.7 mmol, 1 eq.) in anhydrous DMF (7 mL) was added Boc2O (4.2 g, 19.4 mmol, 1.1 eq.). The resulting mixture was then stirred at 60 °C overnight. After returning to room temperature, the reaction was quenched with water (10 mL), and the layers separated. The aqueous layer was extracted with EtOAc (3 × 15 mL), and the combined organic layers were dried over MgSO4, filtered and concentrated to give 16 (3.5 g, 93% yield) as a pale yellow oil. The crude product was used for the next step without any further purification. 1H NMR (300 MHz, CDCl3): δ (ppm) 10.44 (1H, br s, NH-Boc), 3.90 (2H, s, H-11), 3.10 (3H, s, N-CH3), 1.51 (9H, s, CH3 t-Bu); 13C NMR (75 MHz, CDCl3): δ (ppm) 169.5 (C-7), 162.4 (C=O Boc), 159.0 (C-9), 80.1 (Cq t-Bu), 51.4 (C-11), 30.7 (C-13), 28.1 (CH3 t-Bu); ESIMS m/z 228.15 [M+H]+ (calcd for C9H14N3O3, 228.10).
tert-butyl-(1-methyl-4,5-dioxo-4,5-dihydro-1H-imidazol-2-yl)carbamate (17): To a solution of 16 (1.15 g, 5.4 mmol, 1 eq.) in EtOAc (30 mL), a solution of sodium periodate NaIO4 (3.5 g, 16.3 mmol, 3 eq.) in water (30 mL) was added, followed by the introduction of ruthenium(III) chloride (1.17 g, 0.80 mmol, 0.15 eq.). The reaction mixture was stirred vigorously at room temperature for 2 h. The layers were then separated, and the aqueous layer was extracted with EtOAc (2 × 50 mL). The combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated. The crude product was purified by flash column chromatography on silica gel (gradient 99:1 Heptane/EtOAc to 60:40 Heptane/EtOAc) to give 17 (0.480 g, 40% yield) as a pale yellow solid. 1H NMR (300 MHz, CDCl3): δ (ppm) 10.38 (1H, br s, NH-Boc), 3.26 (3H, s, N-CH3), 1.55 (9H, s, CH3 t-Bu); 13C NMR (75 MHz, CDCl3): δ (ppm) 160.5 (C=O Boc), 156.0 (C-9), 155.1 (C-7), 154.4 (C-11), 83.3 (Cq t-Bu), 26.3 (CH3 t-Bu), 28.1 (C-13); HRESIMS m/z 226.0846 [M-H]- (calcd for C9H12N3O4, 226.0828).
Clathridimine (4): (a) To a solution of compound 17 (0.295 g, 1.3 mmol, 1 eq.) and DMAP (cat.) in anhydrous CHCl3 (1.3 mL), triethylamine (0.380 mL, 2.7 mmol, 2.1 eq.) and then TMS-Cl (0.346 mL, 2.7 mmol, 2.1 eq.) were added, and the resulting mixture was stirred at room temperature for 2 h under Ar. Then, a solution of preclathridine A (13) (0.300 g, 1.3 mmol, 1 eq.) in anhydrous CHCl3 (1.3 mL) was added, and the reaction mixture was stirred at reflux for 24 h in a sealed tube. After returning to room temperature, the reaction was quenched with water (2 mL), and the layers separated. The aqueous layer was extracted with CHCl3 (3 × 10 mL), and the combined organic phases were dried over MgSO4, filtered and concentrated to give 0.550 g of the crude product 18 as a pale yellow solid, which was used in the next step without any further purification. 1H NMR (300 MHz, CDCl3): δ (ppm) 6.89 (1H, d, J = 1.6 Hz, H-2′), 6.85 (1H, dd, J = 7.9, 1.6 Hz, H-6′), 6.73 (1H, d, J = 7.9 Hz, H-5′), 6.60 (1H, s, H-5), 5.90 (2H, s, H-7′), 3.82 (2H, s, H-14), 3.70 (3H, s, H-12), 3.25 (3H, s, H-13), 1.59 (9H, s, CH3 t-Bu); 13C NMR (75 MHz, CDCl3): δ (ppm) 161.6 (C=O Boc), 160.3 (C-11), 155.0 (C-9), 147.7 (C-3′), 146.9 (C-2), 146.1 (C-4′), 141.1 (C-7), 140.8 (C-4), 133.3 (C-1′), 121.8 (C-6′), 117.6 (C-5), 109.5 (C-2′), 108.2 (C-5′), 100.8 (C-7′), 81.5 (Cq t-Bu Boc), 34.8 (C-14), 32.1 (C-12), 28.8 (CH3 t-Bu BOC), 25.8 (C-13); HRESIMS m/z 441.1930 [M+H]+ (calcd for C21H25N6O5, 441.1886).
(b) The crude product 18 was dissolved in DCM (4.7 mL), trifluoroacetic acid (4.7 mL) was added and the resulting mixture was stirred at room temperature for 2 h. Then, the solvent was evaporated under reduced pressure, and the resulting product was dissolved in EtOAc and washed with a 10% aqueous solution of NaHCO3. The aqueous layer was extracted with EtOAc (3 × 10 mL), and the combined organic phases were washed with water and brine and then dried over MgSO4, filtered and concentrated. The crude product was purified by flash column chromatography on silica gel (gradient 100% DCM to 100% EtOAc) to give 4 (0.280 g, 64% yield) as a pale orange solid. 1H NMR (300 MHz, CDCl3): δ (ppm) 6.77 (1H, d, J = 7.9 Hz, H-5′), 6.74 (1H, d, J = 1.7 Hz, H-2′), 6.71 (1H, dd, J = 7.9, 1.7 Hz, H-6′), 6.41 (1H, s, H-5), 5.95 (2H, s, H-7′), 3.87 (2H, s, H-14), 3.69 (3H, s, H-12), 3.26 (3H, s, H-13); 13C NMR (75 MHz, CDCl3): δ (ppm) 163.2 (C-11), 157.2 (C-9), 147.7 (C-3′), 147.7 (C-2), 146.5 (C-4′), 139.7 (C-7), 134.7 (C-4), 131.6 (C-1′), 121.8 (C-6′), 115.9 (C-5), 109.2 (C-2′), 108.5 (C-5′), 101.0 (C-7′), 33.1 (C-14), 32.3 (C-12), 25.5 (C-13); HRESIMS m/z 341.1363 [M+H]+ (calcd for C16H17N6O3, 341.1362).
4.7. Complexation of Clathridine A (3) and Clathridimine (4)
Homodimeric (clathridine A)
2 Zn
2+ (
9): Prepared according to the literature [
22]. To a stirred solution of clathridine A (
3) (5.3 mg, 0.015 mmol) in CH
2Cl
2 (10 mL) was added a 0.1 M aqueous ZnSO
4 solution (10 mL). The reaction mixture was kept under stirring at room temperature for 1 h. The organic layer was separated, dried over anhydrous MgSO
4 and concentrated to obtain
5 (5.6 mg) as a yellow solid, which was identical to natural
9 by comparison of their spectral and chromatographic data.
Homodimeric (clathridimine)2 Zn2+ (27): To a stirred solution of clathridimine (4) (5.1 mg, 0.015 mmol) in CH2Cl2 (10 mL) was added a 0.1 M aqueous ZnSO4 solution (10 mL). The reaction mixture was kept under stirring at room temperature for 1 h. The organic layer was separated, dried over anhydrous MgSO4 and concentrated to give 27 (5.6 mg) as a yellow solid. 1H NMR (500 MHz, CDCl3): δ (ppm) 6.63 (2H, s, H-5), 6.49 (2H, d, J = 8.3 Hz, H-5′), 6.23 (2H, d, J = 3.9 Hz, H-6′), 6.22 (2H, s, H-2′), 5.90 (4H, dd, J = 15.4, 0.9 Hz, H-7′), 3.81 (6H, s, H-12), 3.51 (2H, d, J = 16.2 Hz, H-14a), 3.34 (2H, d, J = 16.2 Hz, H-14b), 3.16 (2H, s, H-13); 13C NMR (125 MHz, CDCl3): δ (ppm) 164.0 (C-9), 158.7 (C-11), 149.5 (C-2), 147.8 (C-3′), 146.3 (C-4′), 135.4 (C-4), 131.1 (C-1′), 121.2 (C-6′), 117.2 (C-5), 108.4 (C-6′), 107.9 (C-5′), 101.3 (C-7′), 33.3 (C-14), 32.7 (C-12), 25.6 (C-13); HRESIMS m/z 743.1780 [M + H]+ (calcd for C32H31N12O6Zn, 743.1781).
Heterodimeric (clathridine A-clathridimine) Zn2+ (
10): To a stirred solution of clathridine A (
3) (5.3 mg, 0.015 mmol) and clathridimine (
4) (5.1 mg, 0.015 mmol) in CH
2Cl
2 (20 mL) was added a 0.1 M aqueous ZnSO
4 solution (20 mL). The reaction mixture was kept under stirring at room temperature for 1 h. The organic layer was separated, dried over anhydrous MgSO
4 and concentrated to give a mixture of
9,
10 and
27 (7.0 mg) as a yellow solid. NMR data: see
Table 1; HRESIMS
m/
z 744.1639 [M+H]
+ (calcd for C
32H
30N
11O
7Zn, 744.1621).
4.8. Total Synthesis of Leucettamine B (5)
L-DOPA methyl ester (20): To a solution of L-DOPA (5.0 g, 25 mmol, 1 eq.) in MeOH (30 mL) at 0 °C, SOCl2 (2.0 mL, 28 mmol, 1.1 eq.) was added dropwise. The reaction mixture was allowed to warm to room temperature and was then refluxed for 2 h. After completion of the reaction, the mixture was co-evaporated with toluene three times to give 20 as a white solid (6.8 g, 99%). 1H NMR (300 MHz, MeOD): δ (ppm) 6.77 (1H, d, J = 8.0 Hz, H-8), 6.69 (1H, d, J = 2.0 Hz, H-5), 6.57 (1H, dd, J = 8.0, 2.0 Hz, H-9), 4.22 (1H, t, J = 13.0, 6.8 Hz, H-2), 3.82 (3H, s, O-CH3), 3.08 (2H, ddd, J = 23.0, 13.0, 7.0, 5.9 Hz, H-3); 13C NMR (75 MHz, MeOD): δ (ppm) 170.6 (C-1), 146.9 (C-6), 146.3 (C-7), 126.4 (C-4), 122.0 (C-9), 117.5 (C-5), 117.0 (C-8), 55.5 (C-2), 53.7 (O-CH3), 36.9 (C-3); HRESIMS m/z 212.0910 [M+H]+ (calcd for C10H14NO4, 212.0923).
N-Boc-L-DOPA methyl ester (21): To a solution of L-DOPA methyl ester (6.3 g, 25.5 mmol, 1 eq.) in MeOH (130 mL), triethylamine (7.0 mL, 51 mmol, 2 eq.) was added, followed by the introduction of BOC2O (6.12 g, 28 mmol, 1.1 eq). The reaction mixture was stirred at room temperature for 3 h. Then, the solvent was evaporated under reduced pressure, and the resulting residue was neutralized with a solution of 1 M HCl (5 mL). The aqueous solution was extracted with EtOAc (3 × 10 mL), and the combined organic layers were dried over MgSO4, filtered and concentrated to give 21 as a white solid (7.4 g, 94%), which was used in the next step without further purification. 1H NMR (300 MHz, MeOD): δ (ppm) 6.68 (1H, d, J = 8.0 Hz, H-8), 6.63 (1H, d, J = 2.0 Hz, H-5), 6.50 (1H, dd, J = 8.0, 2.0 Hz, H-9), 4.28 (1H, dd, J = 13.6, 8.1, 6.2 Hz, H-2), 3.67 (3H, s, O-CH3), 2.92 (1H, dd, J = 19.8, 13.6, 6.2 Hz, H-3a), 2.77 (1H, dd, J = 21.7, 13.6, 8.1 Hz, H-3b), 1.39 (9H, s, CH3 BOC); 13C NMR (75 MHz, MeOD): δ (ppm) 174.5 (C-1), 157.8 (C=O BOC), 146.3 (C-6), 145.3 (C-7), 129.6 (C-4), 121.7 (C-9), 117.4 (C-5), 116.4 (C-8), 80.8 (Cq BOC), 56.8 (C-2), 52.7 (O-CH3), 38.3 (C-3), 28.7 (CH3 BOC); HRESIMS m/z 312.1462 [M+H]+ (calcd for C15H22NO6, 312.1447).
Methyl-(S)-3-(benzo[d][1,3]dioxol-5-yl)-2-((tert-butoxycarbonyl)amino)propanoate (22): To a solution of 21 (3.5 g, 11.5 mmol, 1 eq.) in anhydrous DMF (7 mL), cesium fluoride (8.7 g, 57 mmol, 5 eq.) was added, and the reaction mixture was stirred at room temperature for 1 h. Then, dichloromethane (3.5 mL, 52 mmol, 4.5 eq.) was added, and the mixture was stirred at 110 °C for 2 h. After returning to room temperature, the reaction mixture was diluted with Et2O and washed with a cold saturated solution of NaHCO3 (3 × 50 mL). The organic layers were dried over MgSO4, filtered and concentrated. The crude product was purified by flash column chromatography on silica gel (gradient 100% Heptane to 50% EtOAc) to give 22 (1.1 g, 30% yield) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ (ppm) 6.72 (1H, d, J = 8.0 Hz, H-8), 6.59 (1H, d, J = 1.5 Hz, H-5), 6.55 (1H, dd, J = 8.0, 1.5 Hz, H-9), 5.92 (2H, s, H-10), 5.00 (1H, d, J = 7.0 Hz, NH-BOC), 4.51 (1H, dd, J = 13.5, 8.1, 6.0 Hz, H-2), 3.71 (3H, s, O-CH3), 2.98 (2H, m, H-3), 1.42 (9H, s, CH3 BOC); 13C NMR (75 MHz, CDCl3): δ (ppm) 174.5 (C-1), 157.8 (C=O BOC), 146.3 (C-6), 145.3 (C-7), 129.8 (C-4), 122.5 (C-9), 105.9 (C-5), 108.4 (C-8), 101.5 (C-10), 88.9 (Cq BOC), 53.6 (C-2, O-CH3), 38.1 (C-3), 28.3 (CH3 BOC); HRESIMS m/z 346.1255 [M+Na]+ (calcd for C16H21NO6Na, 346.1267).
Methyl-3-(benzo[d][1,3]dioxol-5-yl)-2-((2,2,2-trifluoroacetyl)-λ4-azaneyl)propanoate (23): To a solution of 22 (1.0 g, 3.3 mmol, 1 eq.) in dichloromethane (5.5 mL), trifluoroacetic acid (1.4 mL, C = 2.5 M) was added, and the reaction mixture was stirred at room temperature for 2 h. Then, the solvent was evaporated under reduced pressure, and the resulting residue was dissolved in EtOAc. After the addition of a few drops of Heptane, a precipitate appeared and was filtered. The obtained solid was dried to give 23 (0.97 g, 93% yield) as a white solid. 1H NMR (300 MHz, MeOD): δ (ppm) 6.80 (1H, d, J = 8.0 Hz, H-8), 6.75 (1H, d, J = 1.8 Hz, H-5), 6.70 (1H, dd, J = 8.0, 1.8 Hz, H-9), 5.95 (2H, s, H-10), 4.26 (1H, dd, J = 7.5, 5.8 Hz, H-2), 3.82 (3H, s, O-CH3), 3.19 (1H, dd, J = 14.5, 5.8 Hz, H-3a), 3.06 (1H, dd, J = 14.5, 7.5 Hz, H-3b); 13C NMR (75 MHz, MeOD): δ (ppm) 170.5 (C-1), 149.7 (C-6), 149.0 (C-7), 128.8 (C-4), 123.9 (C-9), 110.5 (C-5), 109.8 (C-8), 102.8 (C-10), 55.4 (C-2), 53.7 (O-CH3), 37.2 (C-3); HRESIMS m/z 224.0907 [M+H]+ (calcd for C11H14NO4, 224.0923).
Pyr-DOPA-OMe (24): To a solution of 23 (0.96 g, 3.0 mmol, 1 eq.) and trichloroacetyl pyrrole (0.7 g, 3.3 mmol, 1.1 eq.) in anhydrous acetonitrile (9 mL), triethylamine acid (1.0 mL, 6.9 mmol, 2.3 eq.) was added. The reaction mixture was stirred at room temperature for 60 h and then filtered and washed with acetonitrile. The solvent was evaporated under reduced pressure, and the resulting residue was dissolved in EtOAc (30 mL) and washed with water (30 mL). The aqueous layer was saturated with NaCl and extracted with EtOAc (3 × 30 mL). The combined organic layers were dried over MgSO4, filtered and concentrated. The crude product was purified by flash column chromatography on silica gel (gradient 100% Heptane to 100% EtOAc) to give 24 (0.95 g, 74% yield) as a yellow amorphous solid. 1H NMR (300 MHz, CDCl3): δ (ppm) 9.80 (1H, br s, NH pyrrole), 6.92 (1H, ddd, J = 4.0, 2.6, 1.4 Hz, H-2′), 6.72 (1H, d, J = 8.0 Hz, H-8), 6.62 (1H, d, J = 1.7 Hz, H-5), 6.58 (2H, m, H-9, H-4′), 6.41 (1H, d, J = 8.0 Hz, NH amide), 6.21 (1H, dt, J = 5.2, 2.6 Hz, H-3′), 5.92 (2H, s, H-10), 4.99 (1H, dt, J = 11.3, 5.6 Hz, H-2), 3.75 (3H, s, O-CH3), 3.12 (2H, ddd, J = 14.0, 5.6 Hz, H-3); 13C NMR (75 MHz, CDCl3): δ (ppm) 172.3 (C-1), 160.7 (C-6′), 148.1 (C-6), 147.0 (C-7), 129.6 (C-4), 125.5 (C-5′), 122.6 (C-9), 122.2 (C-2′), 110.1 (C-3′), 110.0 (C-4′), 109.7 (C-5), 108.5 (C-8), 101.2 (C-10), 53.3 (C-2), 52.6 (O-CH3), 38.1 (C-3); HRESIMS m/z 317.1132 [M+H]+ (calcd for C16H17N2O5, 317.1137).
Diketopiperazine Pyr-DOPA (25): Product 24 (0.300 g, 0.95 mmol, 1 eq.) was dissolved in degassed anhydrous THF (20 mL, C = 0.05 M), and the solution was cooled to 0 °C. NaH (95%, 0.034 g, 1.3 mmol, 1.4 eq.) was added, and the reaction mixture was stirred at this temperature for 5 min. Then, it was allowed to warm to room temperature and stirred for 1 h. After completion of the reaction, the reaction mixture was poured slowly into an acetate buffer (pH = 3.8, 30 mL), and the aqueous layer was gently washed with EtOAc (3 × 40 mL). The combined organic layers were dried over MgSO4, filtered and concentrated to give 25 (0.270 g, 98% yield) as a yellow amorphous solid, which was used in the next step without further purification. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.52 (1H, dd, J = 1.52 Hz, H-2′), 7.06 (1H, dd, J = 1.52 Hz, H-4′), 6.74 (1H, d, J = 7.8 Hz, H-8), 6.69 (1H, d, J = 1.5 Hz, H-5), 6.66 (1H, dd, J = 7.8, 1.5 Hz, H-9), 6.50 (1H, t, J = 3.35 Hz, H-3′), 6.30 (1H, br s, NH amide), 5.92 (2H, dd, J = 2.6, 1.3 Hz, H-10), 4.56 (1H, ddd, J = 8.8, 3.8, 1.9 Hz, H-2), 3.40 (1H, dd, J = 13.8, 3.8 Hz, H-3a), 3.00 (1H, dd, J = 13.8, 8.8 Hz, H-3b); 13C NMR (75 MHz, CDCl3): δ (ppm) ND (C-1), ND (C-6′), 148.4 (C-6), 147.5 (C-7), 127.9 (C-4), 125.2 (C-5′), 122.6 (C-9), 119.9 (C-2′), 119.1 (C-4′), 115.8 (C-3′), 109.7 (C-5), 109.0 (C-8), 101.4 (C-10), 58.9 (C-2), 41.0 (C-3); HRESIMS m/z 285.0851 [M+H]+ (calcd for C15H13N2O4, 285.0875).
Leucettamine B (5) and its regioisomer 26: To a solution of diketopiperazine 25 (0.206 g, 0.73 mmol, 1 eq.) in anhydrous DMF (10 mL, C = 0.08 M), diethyl sulfide (0.31 mL, 2.9 mmol, 4 eq.) was added, and the reaction mixture was stirred under O2 atmosphere at room temperature for 48 h. After the formation of diketopiperazine-OH, methylguanidine hydrochloride (0.090 g, 0.8 mmol, 1.1 eq.) and potassium tert-butanolate (0.100 g, 0.87 mmol, 1.2 eq.) were added, and the reaction mixture was stirred at room temperature for 30 min before heating to 60 °C for 24 h. After returning to room temperature, the mixture was co-evaporated with toluene three times. The crude product was purified by flash column chromatography on silica gel (gradient 100% DCM to 8:20 DCM/MeOH) to give 4 fractions, which were further purified by reverse-phase HPLC (column: Waters Sunfire C18, 10 mm × 150 mm, 5 μm; H2O + 0.1% formic acid/CH3CN + 0.1% formic acid, 90:10 to 60:40) to give leucettamine B (28 mg, 12% yield) and its regioisomer 26 (11 mg, 5%).
Leucettamine B (5): 1H NMR (300 MHz, CDCl3): δ (ppm) 8.04 (d, J = 1.5 Hz, 1H, H-9), 7.33 (dd, J = 8.1, 1.5 Hz, 1H, H-13), 6.82 (d, J = 8.1 Hz, 1H, H-12), 6.43 (s, 1H, H-7), 6.01 (s, 2H, H-14), 3.15 (s, 3H, 3- N-CH3); 13C NMR (75 MHz, CDCl3): δ (ppm) 170.6 (C-4), 160.4 (C-2), 148.9 (C-10), 147.9 (C-11), 132.0 (C-6), 126.4 (C-13), 126.1 (C-5), 114.8 (C-7), 110.9 (C-9), 108.9 (C-12), 102.2 (C-14), 25.8 (3-N-CH3); HRESIMS m/z 246.0870 [M+H]+ (calcd for C12H12N3O3, 246.0879).
Regioisomer 26: 1H NMR (300 MHz, CDCl3): δ (ppm) 8.04 (d, J = 1.5 Hz, 1H, H-9), 7.33 (dd, J = 8.1, 1.5 Hz, 1H, H-13), 6.82 (d, J = 8.1 Hz, 1H, H-12), 6.34 (s, 1H, H-7), 6.01 (s, 2H, H-14), 3.05 (s, 3H, 3- N-CH3); HRESIMS m/z 246.0858 [M+H]+ (calcd for C12H2N3O3, 246.0879).
4.9. Biological Screening
hCol X–promoter activity in a murine ATDC5 pre-chondrocyte line cultured under endochondral differentiation conditions was assessed by the measurement of luminometric luciferase signals. The human Col X promoter–luciferase reporter plasmid (Col X–Luc) was constructed by cloning the full promoter of the human type X collagen gene into pMet-Luc2. A stably transfected monoclonal cell line resistant to neomycin (G418) was created by transfecting Col X–Luc into the mouse chondrogenic cell line (ATDC5).
The expression of type X collagen is confined within hypertrophic chondrocytes and precedes the embark of endochondral bone formation [
17]. Type X collagen facilitates endochondral ossification by regulating matrix mineralization and compartmentalizing matrix components.
The stimulation of Col X transcription means the stimulation of endochondral ossification, involved in bone repair. Conversely, the reduction and the inhibition of Col X transcription can be of interest to stop the final steps of endochondral ossification during the chondrogenic differentiation of mesenchymal stem cells [
42].
4.10. ATDC5 Micromass Model
Pre-chondrogenic ATDC5 cells derived from mouse tetracarcinoma (AT805) were cultured in growth medium (1:1 Dulbecco’s modified Eagle’s medium (DMEM):Ham’s F-12 mix (Gibco)) containing 1% (vol/vol) antibiotic–antimycotic (Gibco), 5% fetal bovine serum (FBS) (Gibco), 10 μg/mL human transferrin (Sigma-Aldrich products, France) and 30 nM sodium selenite (Sigma). Cells were maintained in a humidified atmosphere of 5% CO2 at 37 °C.
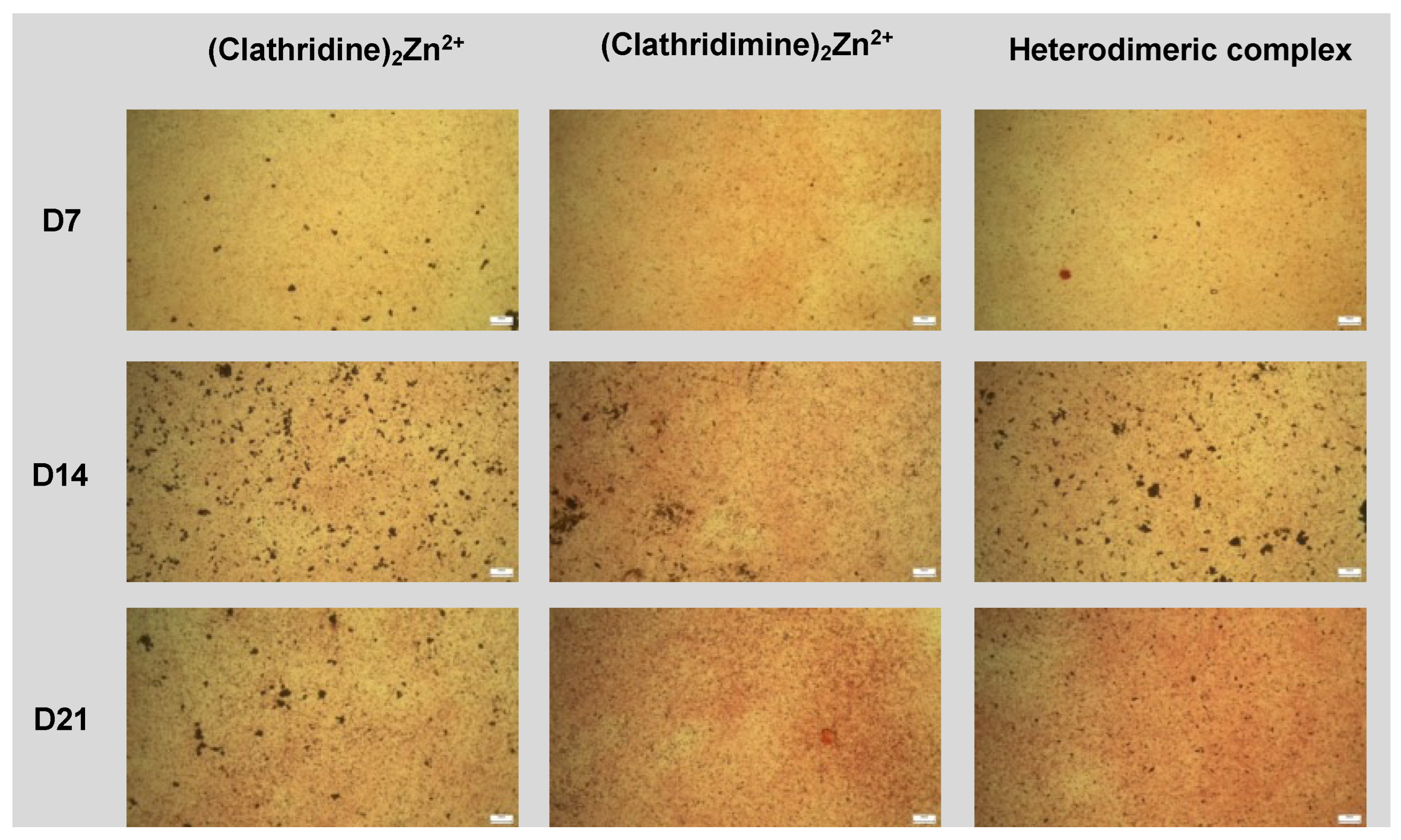
High-density micromass cultures of ATDC5 cells were grown to study endochondral differentiation. Cells were trypsinised, washed and resuspended at 2 × 107 cells/mL in a chondrogenic medium made of DMEM-F12 enriched by 1% (vol/vol) antibiotic–antimycotic, 5% FBS and 1× of ITS (insuline, transferin and selenite) premix (resulting in 10 μg/mL insulin, 5 μg/mL human transferrin and 30 nM sodium selenite) (Life Technologies). Three droplets of 106 cells (10 μL each) were carefully placed in the center of each well of a 6-well plate. Cells were allowed to adhere for 2 h at 37 °C, followed by the addition of 500 μL chondrogenic medium. Hypertrophic differentiation and mineralization were induced by a mineralization medium made of α-minimum essential medium Eagle (Gibco) containing 1% (vol/vol) antibiotic–antimycotic, 5% FBS, 1× of ITS premix, 50 μg/mL ascorbic acid-2-phosphate (Sigma) and 10 mM β-glycerophosphate (Sigma). Micromasses were collected at time points 7, 14 and 21 days. Each time point was processed with three technical replicates.
Matrix mineralization was assessed by Alizarin red staining. Micromasses were treated with 100 μg/mL of compound and cultured in osteogenic differentiation medium for 7, 14 and 21 days. Cells were then fixed with 4% paraformaldehyde for 30 min at 4 °C and incubated with 1% (w/v) Alizarin red solution (Sigma-Aldrich products, France, A5533) for 5 min. Staining was observed under a light microscope (ZEISS axiovert).
For Alizarin red quantification, 10% acetic acid was added to the cells and incubated for 30 min at room temperature. Cells were gently scraped. Lysates were heated at 85 °C for 10 min and centrifuged for 15 min at 4 °C. The supernatant was recovered and treated with 10% ammonium hydroxide, and the absorbance representative of a mineralized nodule formation was measured at 425 nm.
4.11. DFT Calculations
The geometries of all intermediate structures involved in the hydrolysis mechanism of clathridimine were optimized in the gas phase using the Gaussian 09 package [
43] with Becke’s three-parameter hybrid exchange functional (B3LYP) [
44,
45] and the 6–31G(d,p) basis set. Subsequent vibrational frequency calculations confirmed that these conformations were local minima. The complete characterization of these structures can be found in the
Supplementary Materials (Figures S49 and S50).

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}