Boron Monochalcogenides; Stable and Strong Two-Dimensional Wide Band-Gap Semiconductors



Abstract
1. Introduction
2. Computational Method
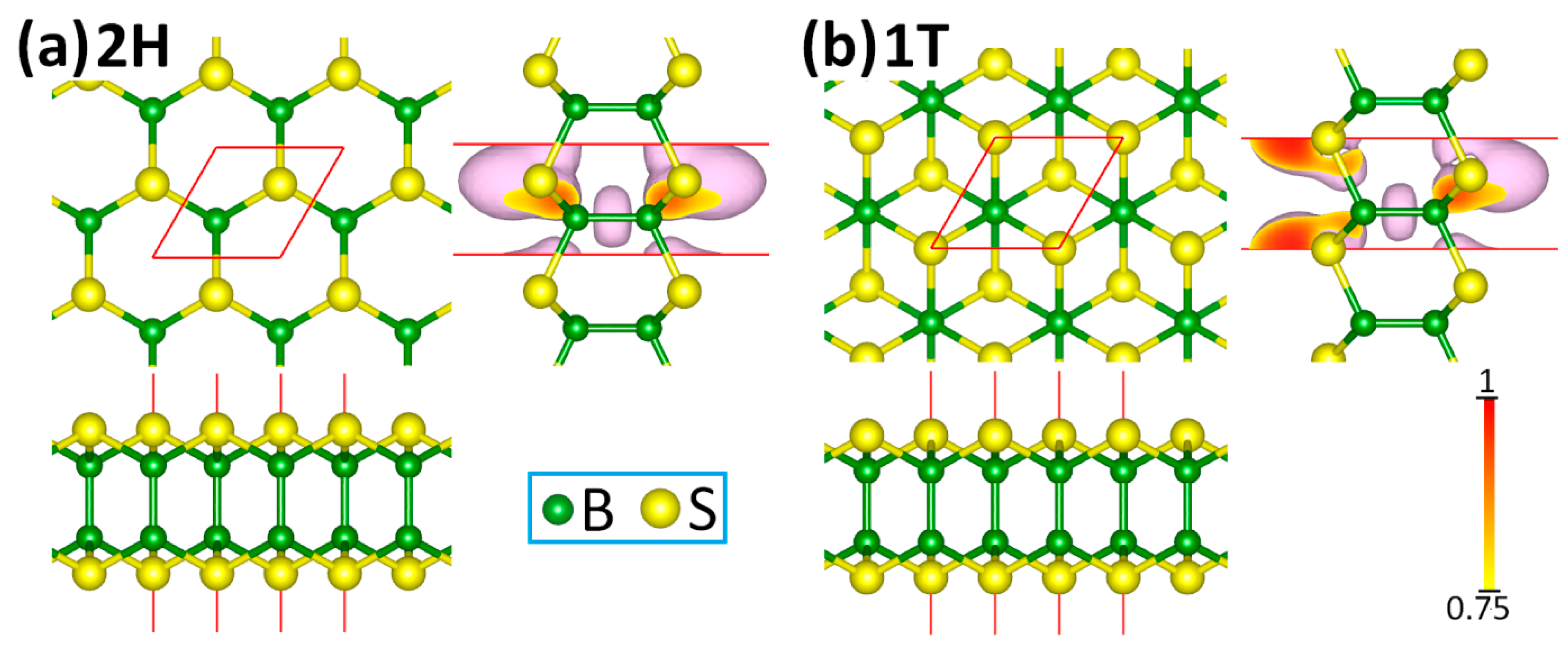
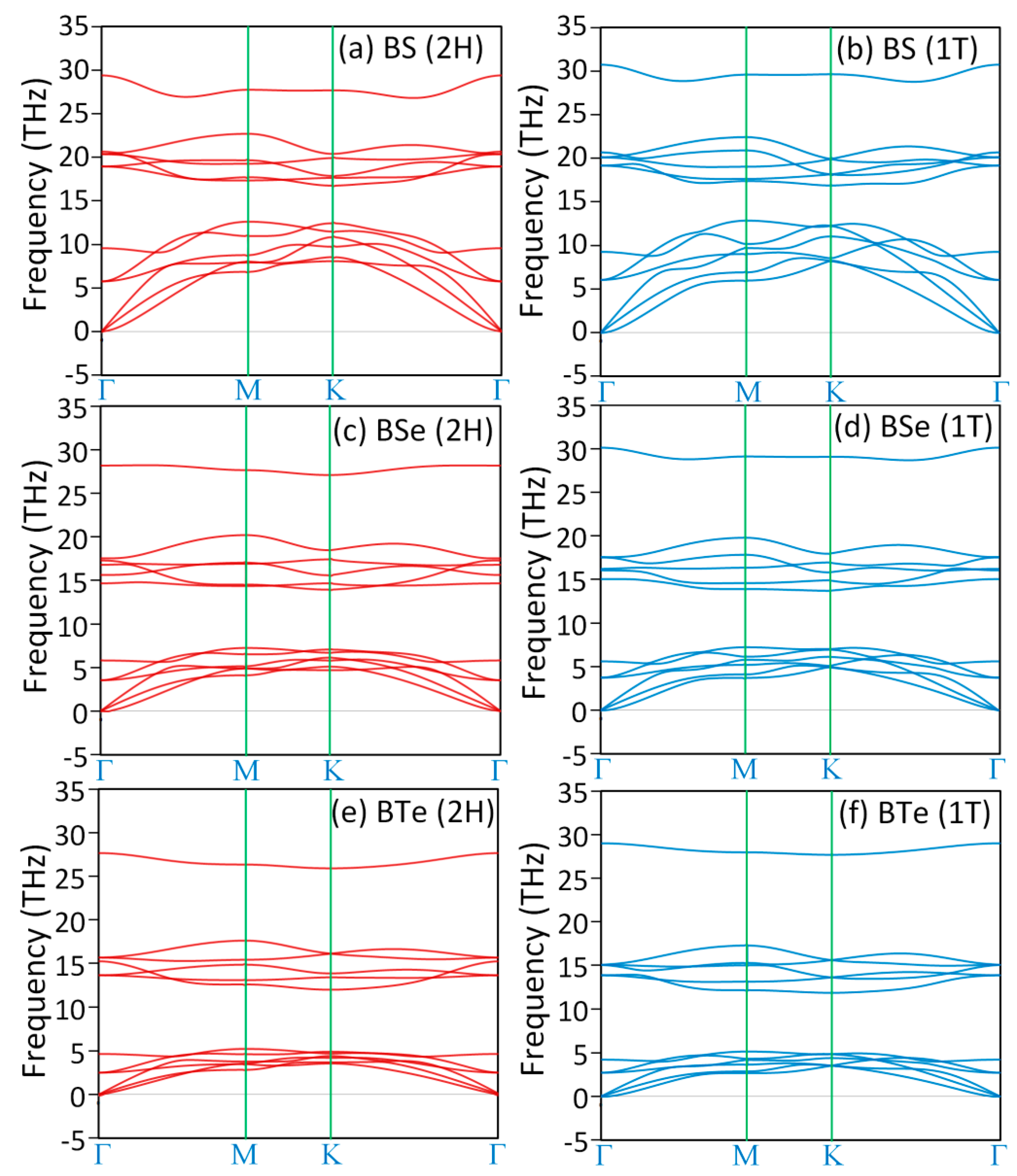
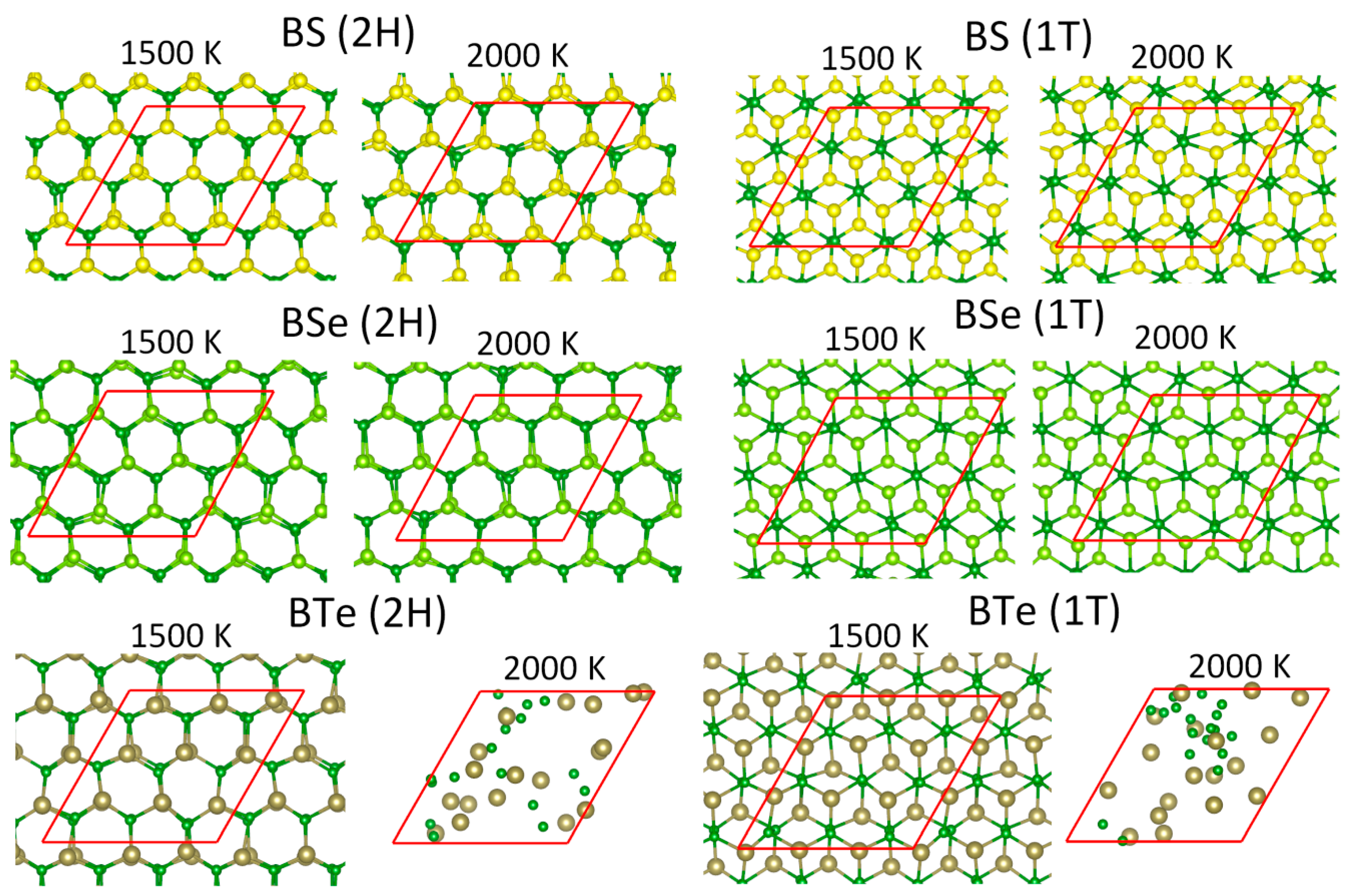
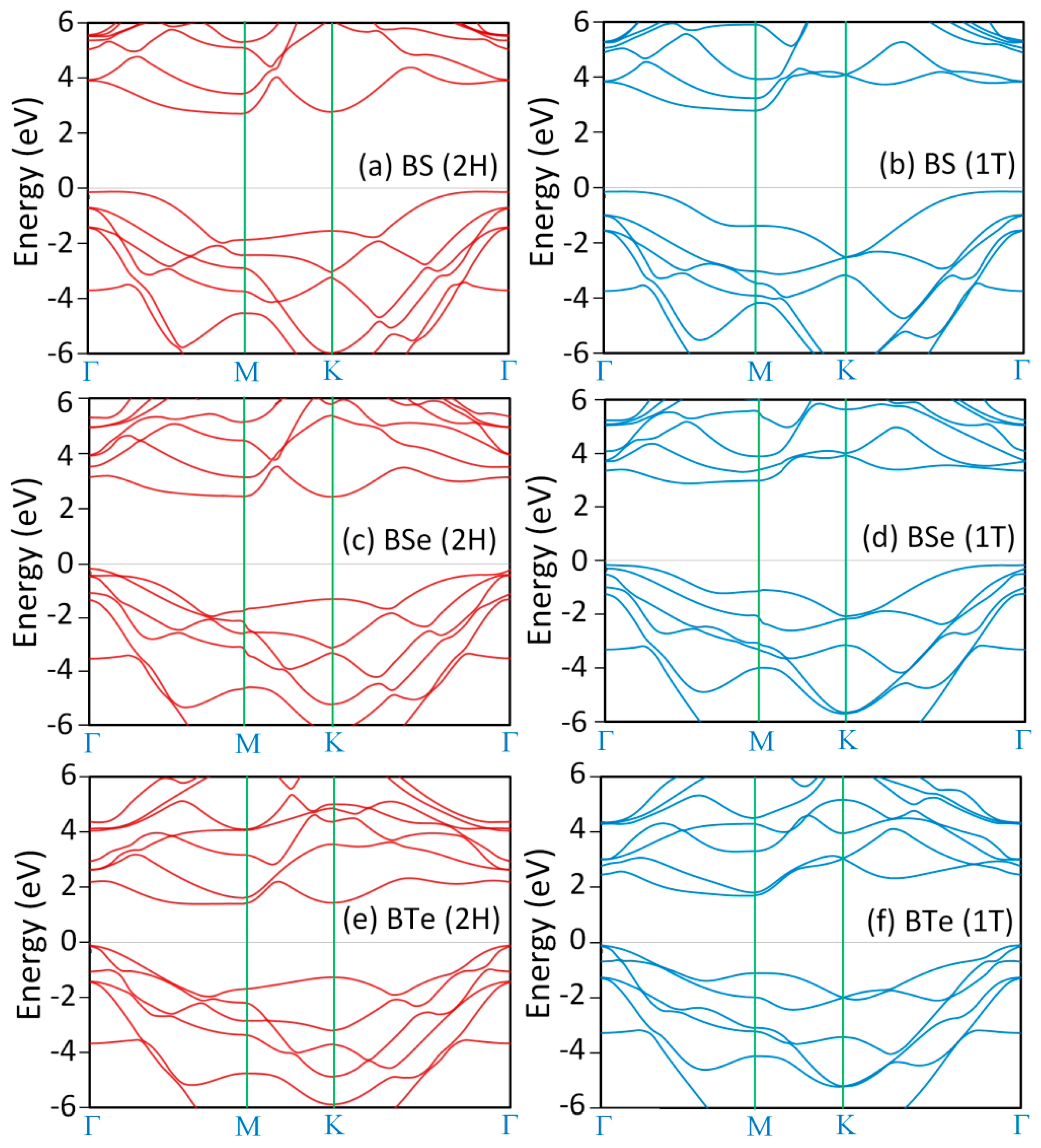
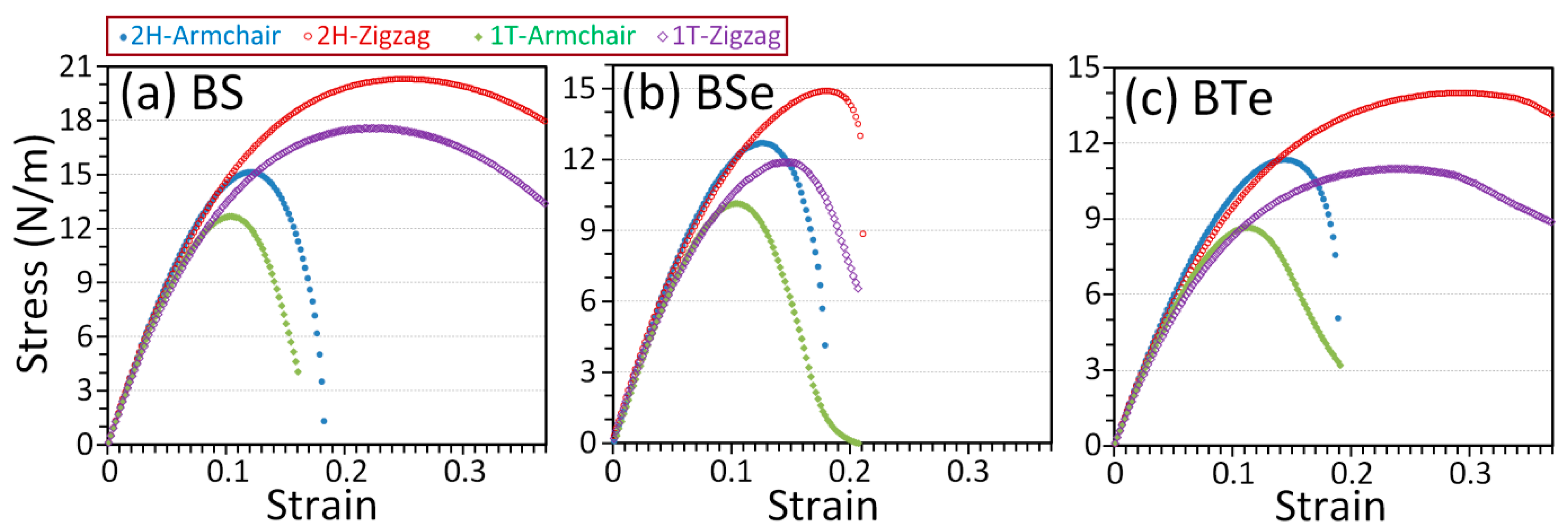
3. Results and Discussions
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.A.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Novoselov, K.S.; Mishchenko, A.; Carvalho, A.; Neto, A.H.C. 2D materials and van der Waals heterostructures. Science 2016, 353, aac9439. [Google Scholar] [CrossRef] [PubMed]
- Mounet, N.; Gibertini, M.; Schwaller, P.; Campi, D.; Merkys, A.; Marrazzo, A.; Sohier, T.; Castelli, I.E.; Cepellotti, A.; Pizzi, G.; et al. Two-dimensional materials from high-throughput computational exfoliation of experimentally known compounds. Nat. Nanotechnol. 2018, 13, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Oganov, A.R.; Glass, C.W. Crystal structure prediction using ab initio evolutionary techniques: Principles and applications. J. Chem. Phys. 2006, 124, 244704. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.W.; Oganov, A.R.; Hansen, N. USPEX-Evolutionary crystal structure prediction. Comput. Phys. Commun. 2006, 175, 713–720. [Google Scholar] [CrossRef]
- Oganov, A.R.; Lyakhov, A.O.; Valle, M. How evolutionary crystal structure prediction works-and why. Acc. Chem. Res. 2011, 44, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Mannix, A.J.; Zhou, X.F.; Kiraly, B.; Wood, J.D.; Alducin, D.; Myers, B.D.; Liu, X.; Fisher, B.L.; Santiago, U.; Guest, J.R.; et al. Synthesis of borophenes: Anisotropic, two-dimensional boron polymorphs. Science 2015, 350, 1513–1516. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Zhang, J.; Zhong, Q.; Li, W.; Li, S.; Li, H.; Cheng, P.; Meng, S.; Chen, L.; Wu, K. Experimental Realization of Two-Dimensional Boron Sheets. Nat. Chem. 2016, 8, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.F.; Dong, X.; Oganov, A.R.; Zhu, Q.; Tian, Y.; Wang, H.T. Semimetallic two-dimensional boron allotrope with massless Dirac fermions. Phys. Rev. Lett. 2014, 112, 085502. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, Y.; Gao, G.; Yakobson, B.I. Two-Dimensional Boron Monolayers Mediated by Metal Substrates. Angew. Chem. Int. Ed. 2015, 127, 13214–13218. [Google Scholar] [CrossRef]
- Kubota, Y.; Watanabe, K.; Tsuda, O.; Taniguchi, T. Deep ultraviolet light-emitting hexagonal boron nitride synthesized at atmospheric pressure. Science 2007, 317, 932–934. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Ci, L.; Lu, H.; Sorokin, P.B.; Jin, C.; Ni, J.; Kvashnin, A.G.; Kvashnin, D.G.; Lou, J.; Yakobson, B.I.; et al. Large scale growth and characterization of atomic hexagonal boron nitride layers. Nano Lett. 2010, 10, 3209–3215. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, J.; Lee, E.K.; Jung, M.; Shin, D.; Choi, H.J.; Seo, J.M.; Jung, S.M.; Kim, D.; Li, F.; Lah, M.S.; et al. Two-dimensional polyaniline (C3N) from carbonized organic single crystals in solid state. Proc. Natl. Acad. Sci. USA 2016, 113, 7414–7419. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, J.; Lee, E.K.; Jung, M.; Shin, D.; Jeon, I.Y.; Jung, S.M.; Choi, H.J.; Seo, J.M.; Bae, S.Y.; Sohn, S.D.; et al. Nitrogenated holey two-dimensional structures. Nat. Commun. 2015, 6, 6486. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Fischer, A.; Goettmann, F.; Antonietti, M.; Müller, J.O.; Schlögl, R.; Carlsson, J.M. Graphitic carbon nitride materials: Variation of structure and morphology and their use as metal-free catalysts. J. Mater. Chem. 2008, 18, 4893–4908. [Google Scholar] [CrossRef]
- Algara-Siller, G.; Severin, N.; Chong, S.Y.; Björkman, T.; Palgrave, R.G.; Laybourn, A.; Antonietti, M.; Khimyak, Y.Z.; Krasheninnikov, A.V.; Rabe, J.P.; et al. Triazine-based graphitic carbon nitride: A two-dimensional semiconductor. Angew. Chem. 2014, 53, 7450–7455. [Google Scholar] [CrossRef] [PubMed]
- Aufray, B.; Kara, A.; Vizzini, S.; Oughaddou, H.; Leandri, C.; Ealet, B.; Le Lay, G. Graphene-like silicon nanoribbons on Ag(110): A possible formation of silicene. Appl. Phys. Lett. 2010, 96, 183102. [Google Scholar] [CrossRef]
- Vogt, P.; De Padova, P.; Quaresima, C.; Avila, J.; Frantzeskakis, E.; Asensio, M.C.; Resta, A.; Ealet, B.; Le Lay, G. Silicene: Compelling experimental evidence for graphenelike two-dimensional silicon. Phys. Rev. Lett. 2012, 108, 155501. [Google Scholar] [CrossRef] [PubMed]
- Bianco, E.; Butler, S.; Jiang, S.; Restrepo, O.D.; Windl, W.; Goldberger, E.J. Stability and exfoliation of germanane: A germanium graphane analogue. ACS Nano 2013, 7, 4414–4421. [Google Scholar] [CrossRef] [PubMed]
- Geim, A.K.; Grigorieva, I.V. Van der Waals heterostructures. Nature 2013, 499, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.H.; Kalantar-Zadeh, K.; Kis, A.; Coleman, J.N.; Strano, M.S. Electronics and optoelectronics of two-dimensional transition metal dichalcogenides. Nat. Nanotechnol. 2012, 7, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Single-layer MoS2 transistors. Nat. Nanotechnol. 2011, 6, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Demarteau, M.; Roelofs, A. Ambipolar phosphorene field effect transistor. ACS Nano 2014, 8, 11730–11738. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yu, Y.; Ye, G.J.; Ge, Q.; Ou, X.; Wu, H.; Feng, D.; Chen, X.H.; Zhang, Y. Black phosphorus field-effect transistors. Nat. Nanotechnol. 2014, 9, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Bandurin, D.A.; Tyurnina, A.V.; Geliang, L.Y.; Mishchenko, A.; Zólyomi, V.; Morozov, S.V.; Kumar, R.K.; Gorbachev, R.V.; Kudrynskyi, Z.R.; Pezzini, S.; et al. High electron mobility, quantum Hall effect and anomalous optical response in atomically thin InSe. Nat. Nano 2017, 12, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Cherednichenko, K.A.; Kruglov, I.A.; Oganov, A.R.; le Godec, Y.; Mezouar, M.; Solozhenko, V.L. Boron monosulfide: Equation of state and pressure-induced phase transition. J. Appl. Phys. 2018, 123, 135903. [Google Scholar] [CrossRef]
- Zólyomi, V.; Drummond, N.D.; Fal’Ko, V.I. Electrons and phonons in single layers of hexagonal indium chalcogenides from ab initio calculations. Phys. Rev. B Condens. Matter Mater. Phys. 2014, 89, 205416. [Google Scholar] [CrossRef]
- Fan, D.; Yang, C.; Lu, S.; Hu, X. Two-Dimensional Boron Monosulfides: Semiconducting and Metallic Polymorphs. arXiv, 2018; arXiv:1803.03459. [Google Scholar]
- Kresse, G. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223–16233. [Google Scholar] [CrossRef]
- Monkhorst, H.; Pack, J. Special points for Brillouin zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef] [PubMed]
- Togo, A.; Oba, F.; Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B Condens. Matter Mater. Phys. 2008, 78, 134106. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Dumcenco, D.O.; Huang, Y.-S.; Suenaga, K. Atomic mechanism of the semiconducting-to-metallic phase transition in single-layered MoS2. Nat. Nanotechnol. 2014, 9, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Mattheiss, L.F. Band structures of transition-metal-dichalcogenide layer compounds. Phys. Rev. B 1973, 8, 3719–3740. [Google Scholar] [CrossRef]
- Wypych, F.; Schöllhorn, R.; Schollhorn, R.; Schöllhorn, R. 1T-MoS2, a new metallic modification of molybdenum disulfide. J. Chem. Soc. Chem. Commun. 1992, 19, 1386–1388. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of Chemical-Bonds Based on Topological Analysis of Electron Localization Functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Sadeghzadeh, S. Borophene sheets with in-plane chain-like boundaries; a reactive molecular dynamics study. Comput. Mater. Sci. 2018, 143, 1–14. [Google Scholar] [CrossRef]
- Sadeghzadeh, S. The creation of racks and nanopores creation in various allotropes of boron due to the mechanical loads. Superlattices Microstruct. 2017, 111, 1145–1161. [Google Scholar] [CrossRef]
- Le, M.-Q.; Batra, R.C. Mode-I stress intensity factor in single layer graphene sheets. Comput. Mater. Sci. 2016, 118, 251–258. [Google Scholar] [CrossRef]
- Ganji, A.R.P.; Armat, M.R.; Tabatabaeichehr, M.; Mortazavi, H. The Effect of Self-Management Educational Program on Pain Intensity in Elderly Patients with Knee Osteoarthritis: A Randomized Clinical Trial. Open Access Maced. J. Med. Sci. 2018. [Google Scholar] [CrossRef]
- Mortazavi, H. Designing a multidimensional pain assessment tool for critically Ill elderly patients: An agenda for future research. Indian J. Crit. Care Med. 2018, 22, 390–391. [Google Scholar] [CrossRef]
- Le, M.-Q. Reactive molecular dynamics simulations of the mechanical properties of various phosphorene allotropes. Nanotechnology 2018, 29, 195701. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.; Le, M.Q.; Nguyen, V.T.; Bui, T.L. Effects of various defects on the mechanical properties of black phosphorene. Superlattices Microstruct. 2017, 112, 186–199. [Google Scholar] [CrossRef]
- Shirazi, A.H.N.; Abadi, R.; Izadifar, M.; Alajlan, N.; Rabczuk, T. Mechanical responses of pristine and defective C3N nanosheets studied by molecular dynamics simulations. Comput. Mater. Sci. 2018, 147, 316–321. [Google Scholar] [CrossRef]
- Abadi, R.; Shirazi, A.H.N.; Izadifar, M.; Sepahi, M.; Rabczuk, T. Fabrication of nanopores in polycrystalline boron-nitride nanosheet by using Si, SiC and diamond clusters bombardment. Comput. Mater. Sci. 2018, 145, 280–290. [Google Scholar] [CrossRef]
- Shi, L.B.; Zhang, Y.Y.; Xiu, X.M.; Dong, H.K. Structural Characteristics and Strain Behaviors of Two-Dimensional C3N: First Principles Calculations. Carbon 2018, 134, 103–111. [Google Scholar] [CrossRef]
- Mortazavi, H.; Tabatabaeichehr, M.; Taherpour, M.; Masoumi, M. Relationship Between Home Safety and Prevalence of Falls and Fear of Falling Among Elderly People: A Cross-Sectional Study. Mater. Soc. Med. 2018, 30, 103–107. [Google Scholar] [CrossRef]
- Liu, F.; Ming, P.; Li, J. Ab initio calculation of ideal strength and phonon instability of graphene under tension. Phys. Rev. B Condens. Matter Mater. Phys. 2007, 76, 064120. [Google Scholar] [CrossRef]
- Peng, Q.; Ji, W.; De, S. Mechanical properties of the hexagonal boron nitride monolayer: Ab initio study. Comput. Mater. Sci. 2012, 56, 11–17. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Lattice Constant (Å) | LB-B (Å) | LB-X (Å) | Eunit-cell (eV) | ΔQ | |
|---|---|---|---|---|---|---|
| BS | 2H | 3.041 | 1.728 | 1.950 | −5.813 | 1.88 |
| 1T | 3.056 | 1.704 | 1.954 | −5.818 | 1.86 | |
| BSe | 2H | 3.259 | 1.712 | 2.087 | −5.241 | 0.68 |
| 1T | 3.274 | 1.686 | 2.087 | −5.256 | 0.58 | |
| BTe | 2H | 3.565 | 1.713 | 2.314 | −4.720 | −0.1 |
| 1T | 3.589 | 1.681 | 2.316 | −4.746 | −0.11 | |
| Structure | Yarmchair | Yzigzag | Parmchair | Pzigzag | UTSarmchair | UTSzigzag | SUTSarmchair | SUTSzigzag | |
|---|---|---|---|---|---|---|---|---|---|
| BS | 2H | 203 | 201 | 0.13 | 0.12 | 15.17 | 20.31 | 0.12 | 0.26 |
| 1T | 195 | 193 | 0.13 | 0.12 | 12.69 | 17.60 | 0.11 | 0.23 | |
| BSe | 2H | 162 | 159 | 0.17 | 0.14 | 12.72 | 14.92 | 0.13 | 0.18 |
| 1T | 155 | 155 | 0.17 | 0.14 | 10.15 | 11.89 | 0.11 | 0.15 | |
| BTe | 2H | 131 | 130 | 0.17 | 0.14 | 11.36 | 14.01 | 0.15 | 0.3 |
| 1T | 122 | 121 | 0.17 | 0.14 | 11.00 | 8.66 | 0.12 | 0.27 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mortazavi, B.; Rabczuk, T. Boron Monochalcogenides; Stable and Strong Two-Dimensional Wide Band-Gap Semiconductors. Energies 2018, 11, 1573. https://doi.org/10.3390/en11061573
Mortazavi B, Rabczuk T. Boron Monochalcogenides; Stable and Strong Two-Dimensional Wide Band-Gap Semiconductors. Energies. 2018; 11(6):1573. https://doi.org/10.3390/en11061573
Chicago/Turabian StyleMortazavi, Bohayra, and Timon Rabczuk. 2018. "Boron Monochalcogenides; Stable and Strong Two-Dimensional Wide Band-Gap Semiconductors" Energies 11, no. 6: 1573. https://doi.org/10.3390/en11061573
APA StyleMortazavi, B., & Rabczuk, T. (2018). Boron Monochalcogenides; Stable and Strong Two-Dimensional Wide Band-Gap Semiconductors. Energies, 11(6), 1573. https://doi.org/10.3390/en11061573