3.1. Elastic Constants
The calculated lattice constants and equilibrium volumes in the ground state are listed in
Table 1, where the experimental results of wurtzite and zinc-blende GaN crystals obtained from previous studies are also given for comparison. Obviously, the calculation results show good consistency with the experimental data, thus demonstrating the effectiveness of the calculations in this study. Moreover, the lattice parameters of wurtzite and zinc-blende structures are commonly overestimated by GGA while underestimated by LDA. The calculation results of the lattice constants obtained by LDA are closer to the experimental results as the maximum deviation of the lattice constant (herein,
a,
b and
c) is about 1%, which is slightly less than that obtained by the GGA method.
The elastic constant is an essential property that can provide a link between the mechanical and dynamic behavior of crystals and give important information concerning the nature of the chemical bonding operating in solids [
30]. In the stiffness matrix
Cij of hexagonal crystals, there are five independent elastic constants,
C11,
C12,
C13,
C33 and
C44, and the elastic constants for the hexagonal wurtzite GaN crystal should satisfy the following stability criteria [
42]:
In the stiffness of cubic zinc-blende GaN, there are three independent elastic constants,
C11,
C44 and
C12; restrictions on the elastic constants are [
42]:
The calculated elastic constants
Cij and
Sij for the two GaN crystals are listed in
Table 2 and
Table 3, respectively, where the previous experimental data of
Cij are consistent with the results obtained in this work, thus proving the correctness of the simulation carried out in this paper. Clearly, according to the result shown in
Table 2, the elastic constants of the two GaN crystals obey the above stability criteria.
3.2. Elastic Properties
The bulk modulus (
B) is a parameter to measure how incompressible/resistant to compressibility the materials are.
B is defined as the ratio of the increase in infinite small pressure to the resulting relative volume change and can be predicted from the calculated elastic constants within the Voigt–Reuss scheme [
42]:
where
BV,
BR and
GV,
GR stand for the upper and lower bounds of the bulk moduli and shear moduli of the polycrystalline aggregate, respectively. Moreover, the effective bulk modulus (
B) and effective shear modulus (
G) can be calculated using the Voigt-Reuss-Hill approximation [
46], which are considered arithmetic means:
The effective elastic modulus (
E) and Poisson’s ratio (
ν) of the GaN polycrystalline aggregate can be expressed as follows [
47]:
The calculated
B,
G,
E,
ν and
B/
G for the two GaN polycrystals are listed in
Table 4. Generally, the two GaN polycrystals have many common mechanical properties. The buck moduli of the two crystals are similar, while the other modulus values (i.e.,
G and
E) for wurtzite GaN are a little larger, indicating that, compared to zinc-blende GaN, the stiffness of wurtzite GaN is slightly higher. According to the Pugh criterion, if
B/
G > 1.75, then ductile behavior may occur, otherwise the material behaves in a brittle mode [
48]. Obviously, the ratio of
B to
G for hexagonal wurtzite GaN is 1.31 (GGA) or 1.61 (LDA), which is much less than 1.75, as given in
Table 4, implying that wurtzite GaN may act like a brittle material. For zinc-blende GaN, the ratio is 1.59 (GGA) or 1.76 (LDA), i.e., it is close to 1.75. This implies that zinc-blende GaN might also be a brittle material while its brittleness is less than that of wurtzite GaN. Moreover, Poisson’s ratio (
ν) is widely applied to evaluate the stability of the crystals under shear deformation, and a larger
ν indicates that the materials would present a better plasticity. Zinc-blende GaN exhibits a better plastic behavior because of the larger
ν (0.24 for GGA and 0.26 for LDA), while wurtzite GaN is more stable against shear stress for the smaller
ν (0.20 for GGA and 0.24 for LDA). Furthermore, the value of
ν implies the degree of directionality of the covalent bond. Its value is small (about 0.1) for covalent materials, while, for ionic materials, the typical value is 0.25 [
49]. The calculated value of
ν ranges from 0.20 to 0.27 for the two structures, indicating they are both ionic materials.
3.3. Elastic Anisotropy
The elastic anisotropy of materials has a significant effect on their physical properties, such as anisotropic deformation, crack propagation and elastic instability. In this section, the elastic anisotropy factor (
A) is applied to explore the anisotropy degree of the two GaN monocrystals [
56], as given in
Table 5. If the value of anisotropy factor
A is close to 1, then the mechanical behavior of the plane is prone to isotropy. The calculated results show that, in wurtzite GaN,
A of planes containing the [001] axis (i.e., [0001] axis or orientation) is far from 1, indicating wurtzite GaN displays obvious anisotropic characteristics in these planes. In addition, zinc-blende GaN presents strong anisotropy in its crystal planes {100} and {110}.
To further investigate the anisotropic characteristics of monocrystal GaN crystals, three-dimensional surfaces revealing the elastic anisotropy were analyzed. The direction dependence of the elastic modulus of general monocrystals is given as follows [
42]:
where
l1,
l2 and
l3 are the direction cosines concerning the
a,
b and
c directions of the crystal lattices, respectively, and
Sij (
i,
j = 1, 2, 3) are the compliance coefficients, which are listed in
Table 3. According to Equation (11) and the crystal features of hexagonal and cubic monocrystals, the direction dependences of the elastic moduli of the wurtzite and zinc-blende can be expressed as in Equations (12) and (13), respectively.
The simulation results of direction dependences of the elastic moduli calculated by GGA and LDA are similar, and LDA method was applied for simulating the direction dependences of the elastic moduli for the higher accuracy in calculating structural parameters and elastic constants.
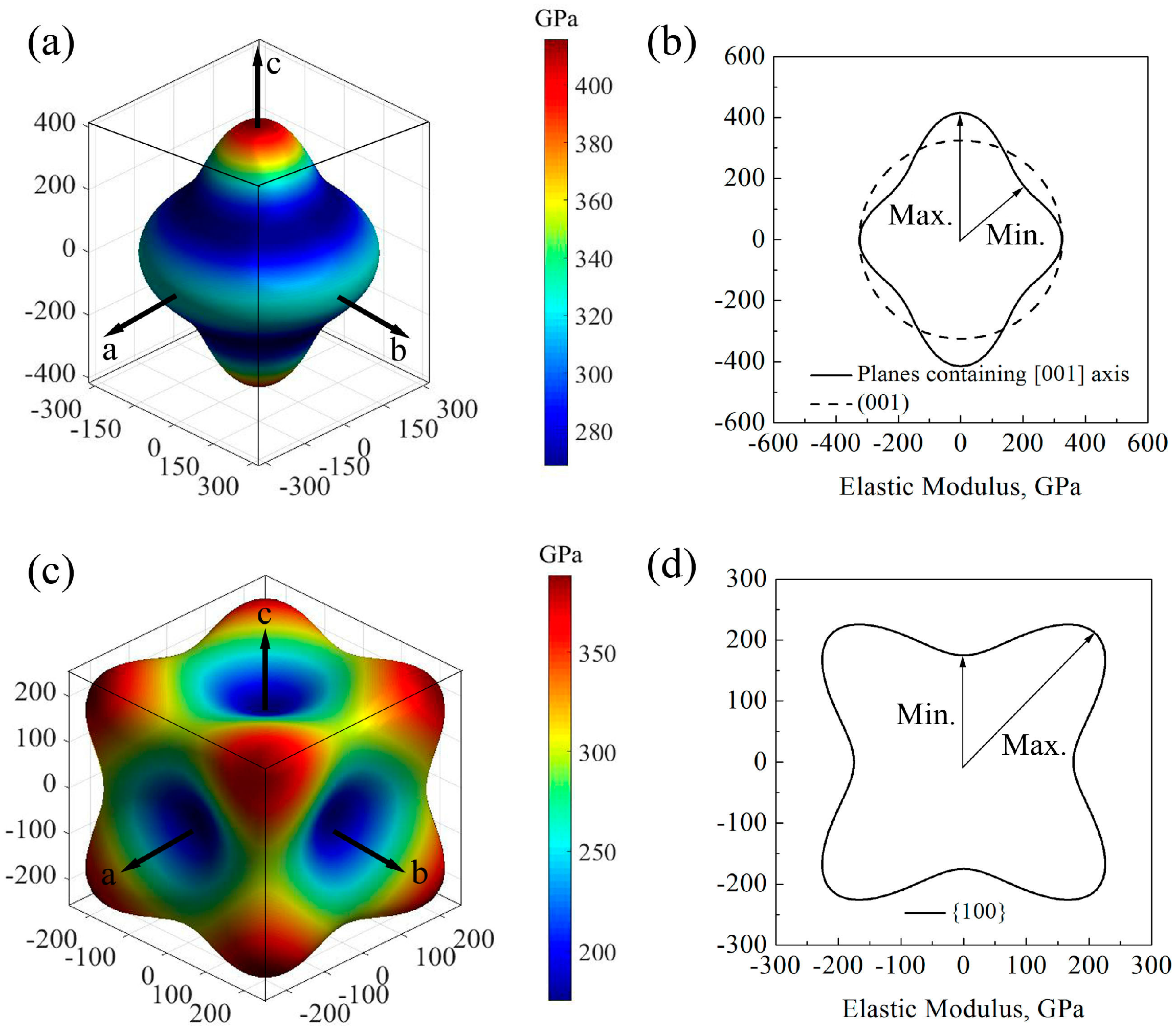
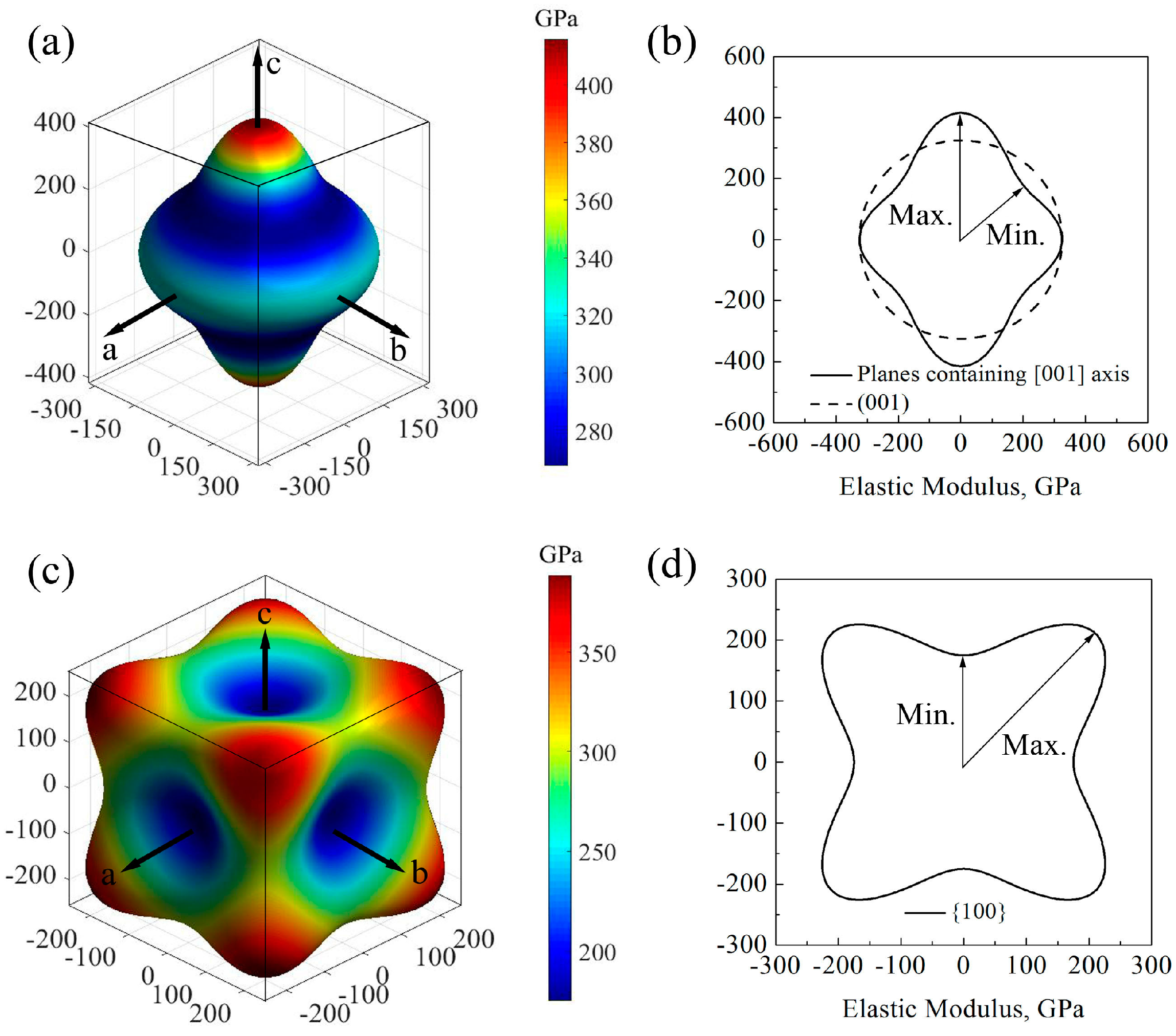
Figure 2 shows the direction dependences of the elastic moduli and their projections on the main planes of GaN crystals. For wurtzite GaN, the shape of elastic modulus at plane (001) is a circle and the value is 324.8 GPa, while it is far from a circle at planes containing [001] axis (e.g., planes (
) and (
)) (
Figure 2a,b); therefore, the elastic modulus of wurtzite GaN may exhibit an isotropic characteristic at the plane (001) while it shows significant anisotropy at the planes containing [001] axis, where the maximum and minimum values are 415.8 GPa and 267.8 GPa, respectively. For zinc-blende GaN, the elastic moduli along the [100] axis (i.e.,
Ex), [010] axis (i.e.,
Ey) and the [001] axis (i.e.,
Ez) are all the minimum value 175.0 GPa, and the maximum elastic moduli are located at the orientations <111> and the value is 388.5 GPa, as shown in
Figure 2c. In addition, in the zinc-blende GaN, there are obvious anisotropic characteristics for all small miller indices crystallographic planes, and
Figure 2d shows that the maximum and minimum elastic moduli in the projection of planes {100} are 297.7 and 175.0 GPa, respectively.
Moreover, shear moduli of hexagonal wurtzite GaN and cubic zinc-blende GaN can be expressed as Equation (14) [
57] and Equations (15)–(17), respectively,
where
θ and
φ are the Euler angles in the measurement systems.
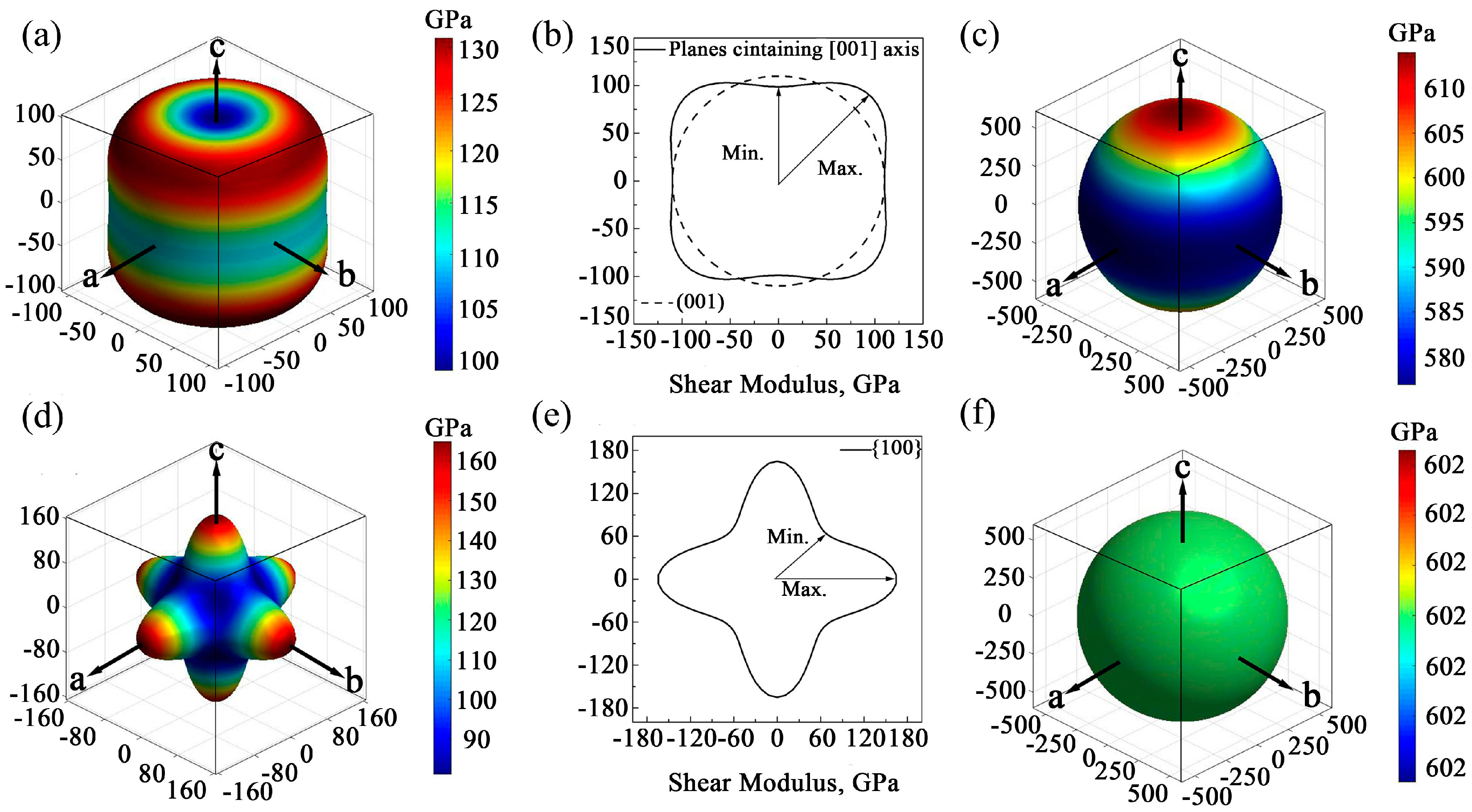
Figure 3a,b,d shows the direction dependences of the shear moduli and their projections on the main planes of GaN crystals. For wurtzite GaN, it is found that the shape of directional shear modulus at the (001) plane is a circle, while it is far from the circle at the planes containing the [001] axis. Therefore, the shear modulus of wurtzite GaN may exhibit an isotropic characteristic at the plane (001) and the isotropic value is 110.0 GPa, while it shows significant anisotropy at planes containing [001] axis, and the maximum and minimum values are 130.8 GPa and 98.9 GPa, respectively (see
Figure 3a,b). For zinc-blende GaN, the shear moduli along the [100] axis (i.e.,
Ex), [010] axis (i.e.,
Ey) and the [001] axis (i.e.,
Ez) are all the maximum value 165.0 GPa, and the minimum shear moduli are located at the orientations <111> and the value is 81.0 GPa (see
Figure 3d). In addition, in the zinc-blende GaN, there are also obvious anisotropic characteristics for all small miller indices crystallographic planes, and
Figure 3e shows that the maximum and minimum shear moduli in the projection of planes {100} are 165.0 and 92.8 GPa, respectively. Obviously, to some extent, the shear modulus of GaN monocrystals has an inverse direction dependence compared to the elastic modulus. In summary, the elastic moduli and shear moduli of GaN monocrystals exhibit obvious anisotropy behavior, while the following calculation shows that the bulk moduli of the two monocrystals are nearly isotropic. Directional dependences of bulk moduli of hexagonal and cubic GaN monocrystal can be calculated using Equations (18) and (19), respectively,
For wurtzite GaN, the maximum bulk modulus is located at crystal orientations which are vertical to [001] ([0001]) axis, e.g., [100] (
), [010] (
), etc., and its value is 614.0 GPa. Besides, the minimum bulk modulus is in the direction of [100] axis and the value is 576.8 GPa, as shown in
Figure 3c. Obviously, there is very slight anisotropy in the wurtzite GaN. For zinc-blende GaN, there is no anisotropy of bulk modulus, and the value is 602.4 GPa in all orientations (see
Figure 3f).
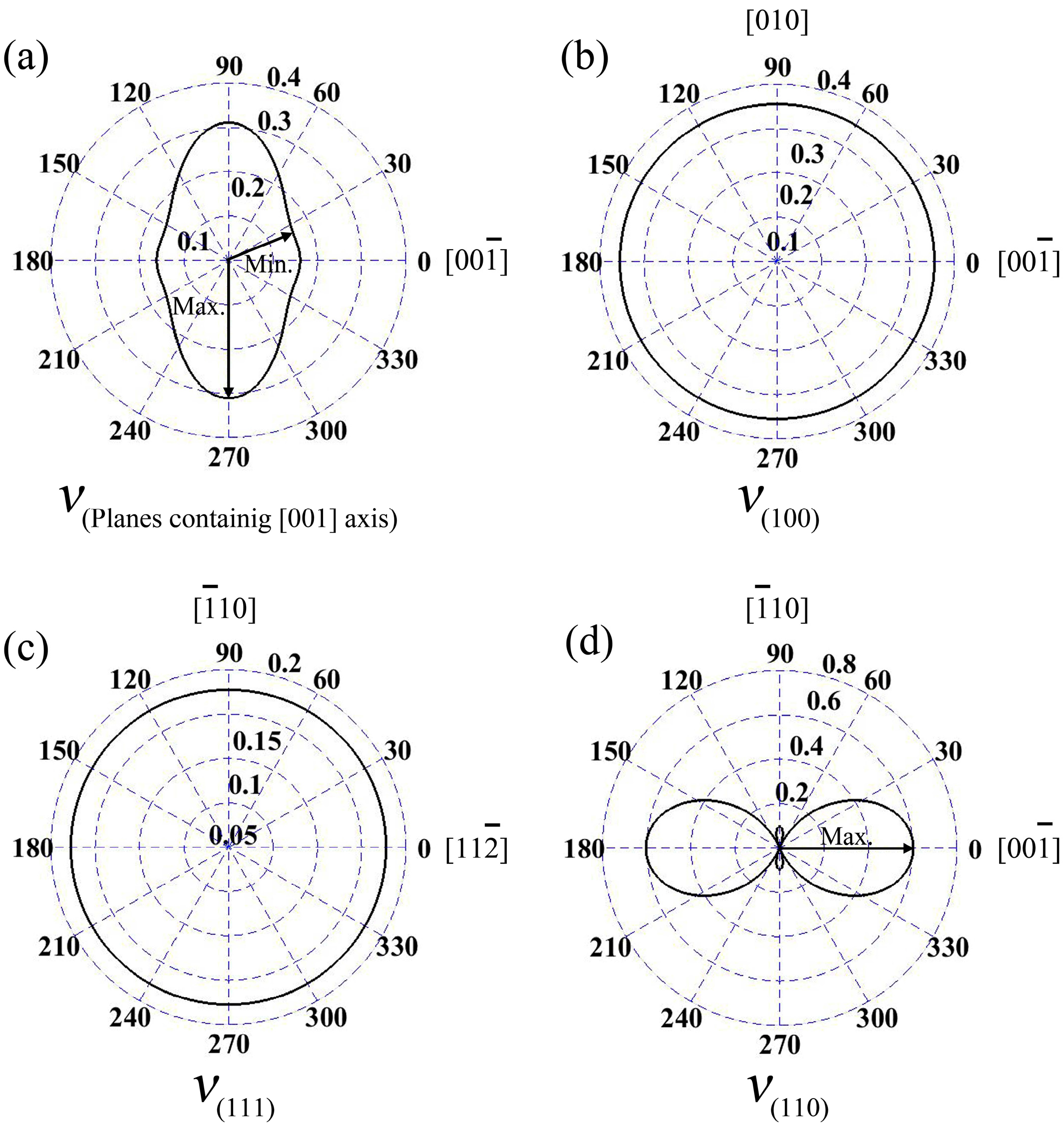
Further, the curves of Poisson’s ratios at the planes containing [001] axis
v(
θ) in the wurtzite GaN can be expressed as Equation (20) [
58], and Poisson’s ratios at the plane (
hkl) of zinc-blende GaN can be described as Equation (21) [
59],
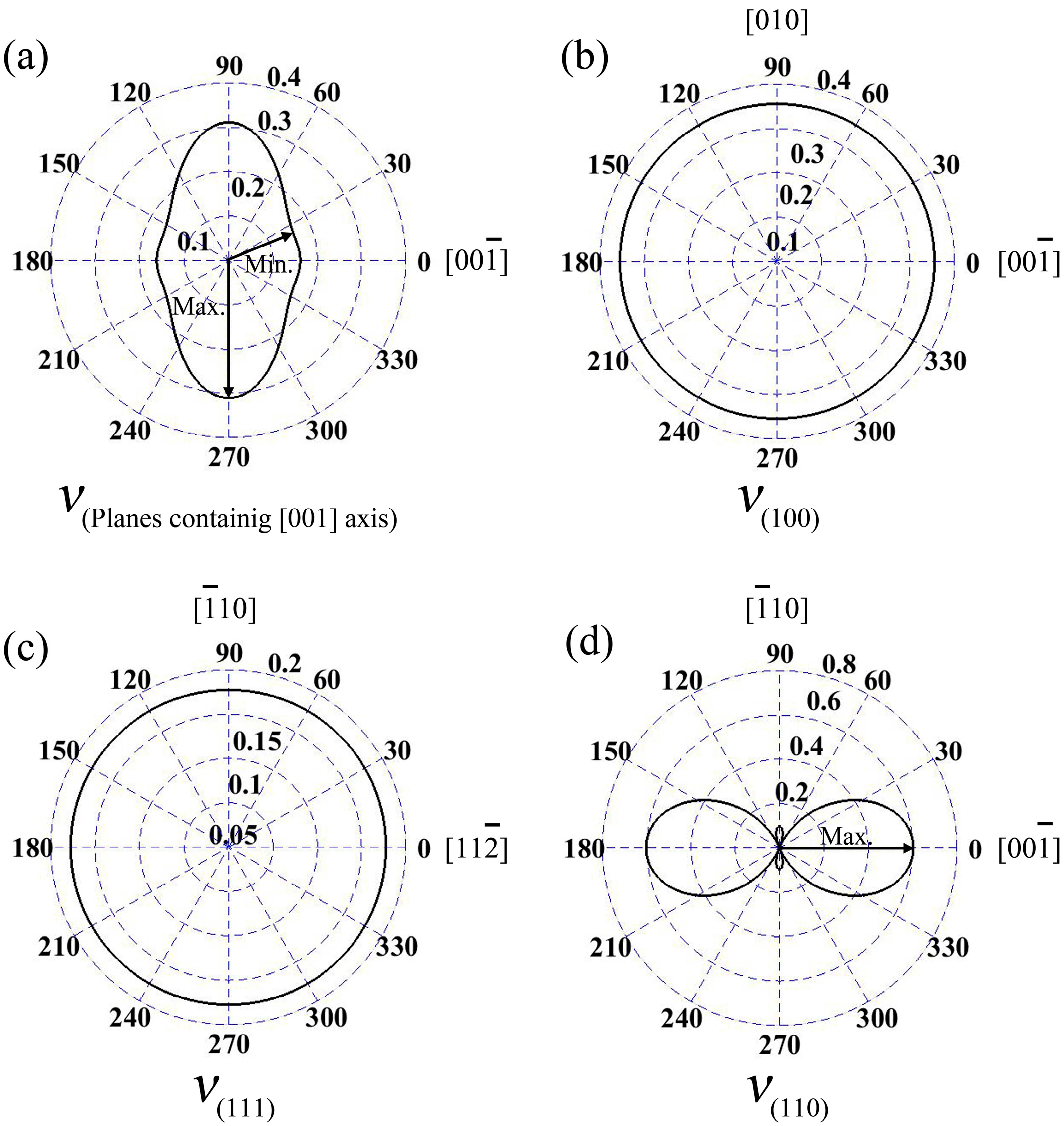
In the wurtzite GaN, Poisson’s ratios at the planes containing [001] axis are illustrated in
Figure 4a; the maximum value is 0.31, and it is located at the directions vertical to [001] axis. Meanwhile, the minimum
ν is 0.158, and the angle between the direction and [001] axis is 19.8°. In addition,
ν along [001] axis of wurtzite GaN is 0.162. Additionally, for zinc-blende GaN, Poisson’s ratios at planes (100) and (111) are isotropic, as shown in
Figure 4b,c, and the values are 0.35 and 0.18, respectively. However,
ν at plane (110) exhibits obvious anisotropic behavior (see
Figure 4d), where the maximum value 0.60 is located at [001] and
directions, while the minimum value is 0.10 (actually, −0.10) and it is in the orientations
or
.
3.4.Thermodynamic Properties
The calculation results of phonon spectra can be used to compute lattice heat capacity (
CV) as functions of temperature [
60]. The temperature dependence of the energy can be calculated as follows:
where
Etot is the total static energy at 0 K, which can be calculated by first principles,
Ezp is the zero-point vibrational energy,
ω is the frequency,
k is the Boltzmann’s constant,
T is the Kelvin temperature,
ħ is Planck’s constant and
F(
ω) is the phonon density of states.
Ezp can be expressed as follows:
The lattice contribution to the heat capacity,
CV, is calculated as follows:
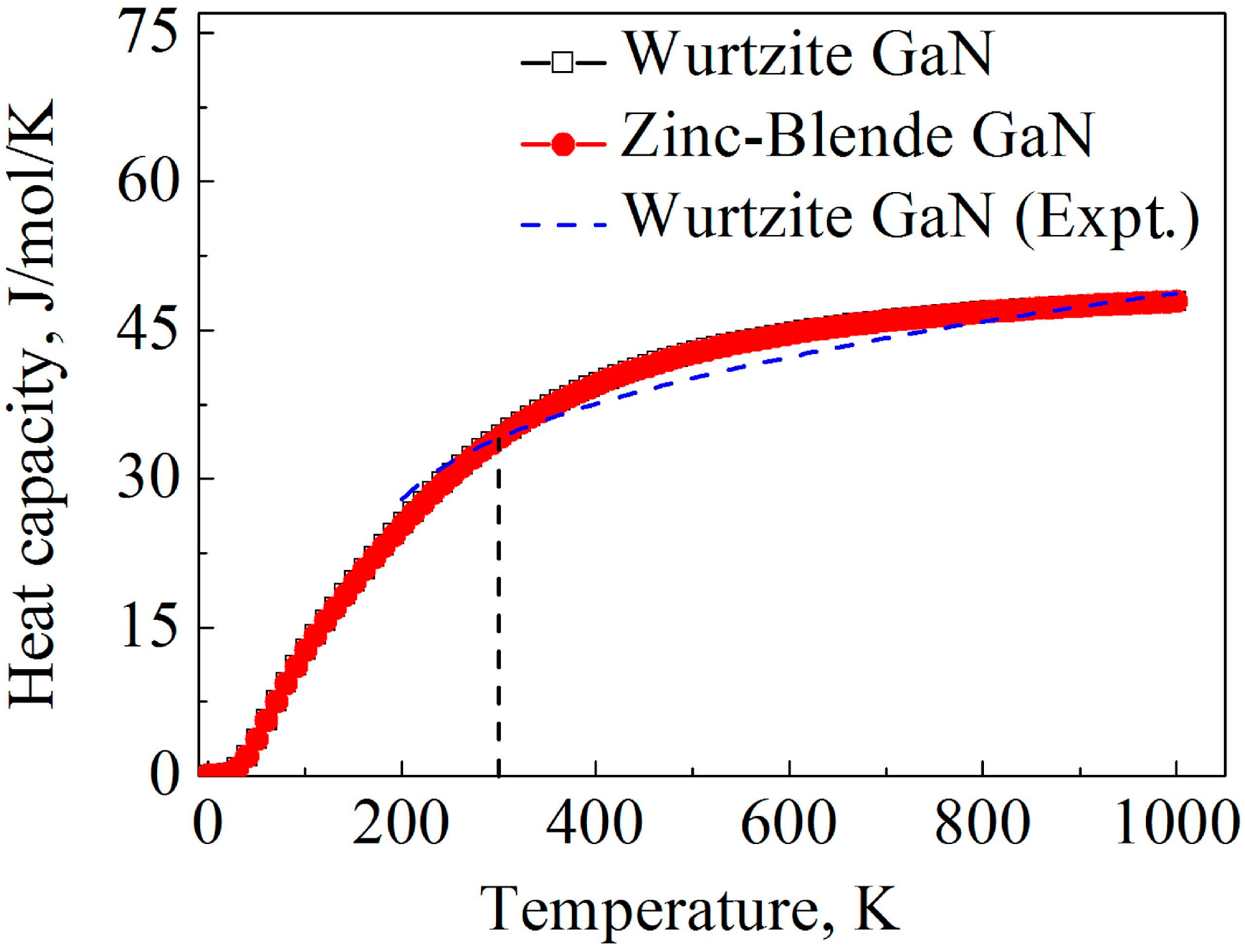
The heat capacity calculated by the LDA method is illustrated in
Figure 5. At low temperatures, power-law temperature dependence is clearly observed for the heat capacity, and the predicted power-law exponent is approximately 3 for the two GaN crystals according to the calculation result, which is consistent with the Debye heat capacity theory. Moreover, at 300 K, the calculated heat capacities of wurtzite and zinc-blende are 34.4 and 34.3 J mol
−1 K
−1, respectively, which are close to the experimental value of wurtzite GaN at 300 K (34.1 J mol
−1 K
−1) [
61]. At high temperatures, the heat capacity of the two GaN crystals tends to converge at 49.4 mol
−1 K
−1, which is the Dulong–Petit limit. Obviously, heat capacities of the two GaN crystals are quite close as the temperature is increased from 0 to 1000 K, and the effect of crystal structures on the heat capacity of GaN is very limited.
Further, Debye temperature is one of important basic physical parameters, which has connections with many mechanical and physical properties such as elastic constants, heat capacity, bond strength, and so on [
62]. By employing elastic constants and average sound velocity
νm, Debye temperature Θ
D can be approximately calculated by
where
n is the number of atoms in the cell,
NA is Avogadro constant,
M is the molecular weight,
ρ is the density, and
νt and
νl are longitudinal and transverse sound velocities, respectively, which can be obtained from Equations (27) and (28), respectively.
The thermodynamic properties of GaN crystals calculated by LDA method are listed in
Table 6. Obviously, the densities of the two structure GaN crystals are extremely close, and wurtzite GaN has higher
νt,
νl and
νm. Besides, the measured Debye temperatures of GaN are scattered, and previous study reported that the experimental measured Debye temperatures of wurtzite GaN range from 586 K to 898 K [
63]. In this study, the calculated Debye temperatures of wurtzite and zinc-blende GaN are 641.8 and 620.2 K, respectively. Compared with zinc-blende GaN, wurtzite GaN has the slightly higher Debye temperature, indicating stronger Ga–N ionic bonds in the wurtzite GaN. Therefore, wurtzite GaN exhibits slightly higher stiffness and melt point to a small degree.
3.5. Electronic Properties
Band gap is the energy required for a valence electron to become a conduction electron, which moves freely and serves as a charge carrier. According to previous experimental results, the band gaps of wurtzite and zinc-blende GaN crystals are 3.5 eV [
55] and 3.1 eV [
64], respectively. However, Band gaps of wurtzite and zinc-blende GaN crystals calculated by LDA are 1.66 eV and 1.48 eV, respectively, and they are 2.01 and 1.91 eV by GGA. Here, HSE06 scheme [
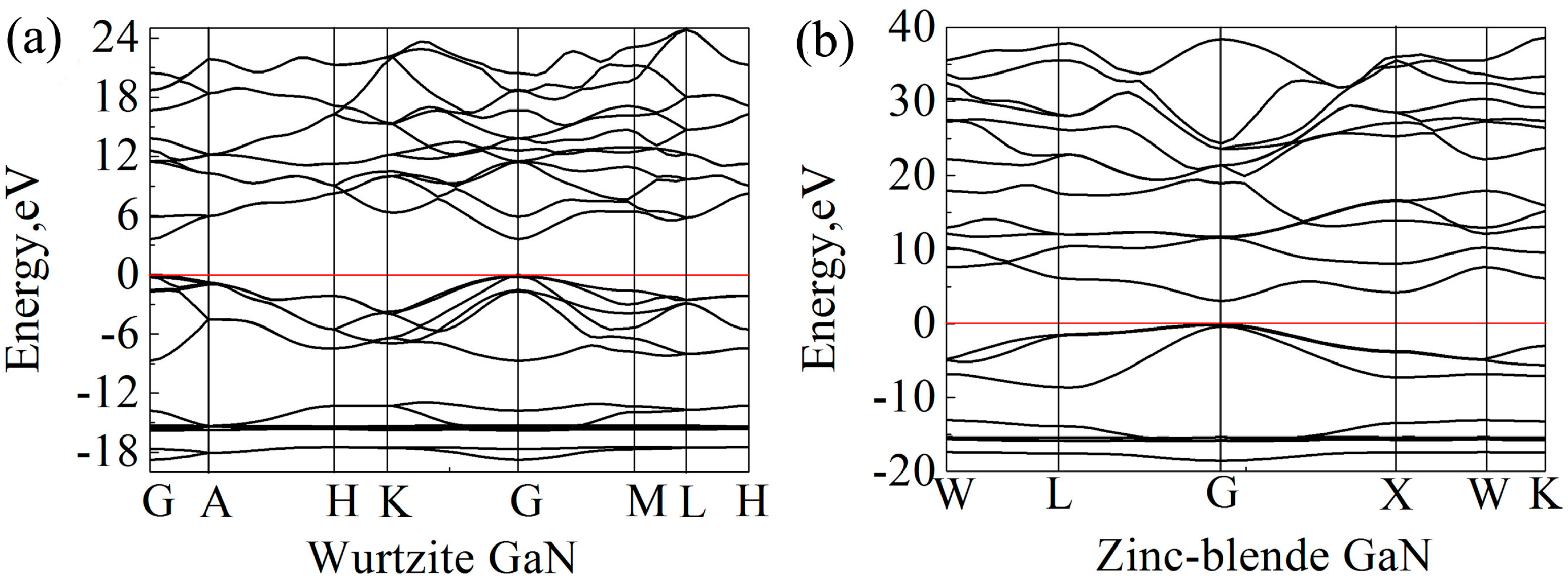
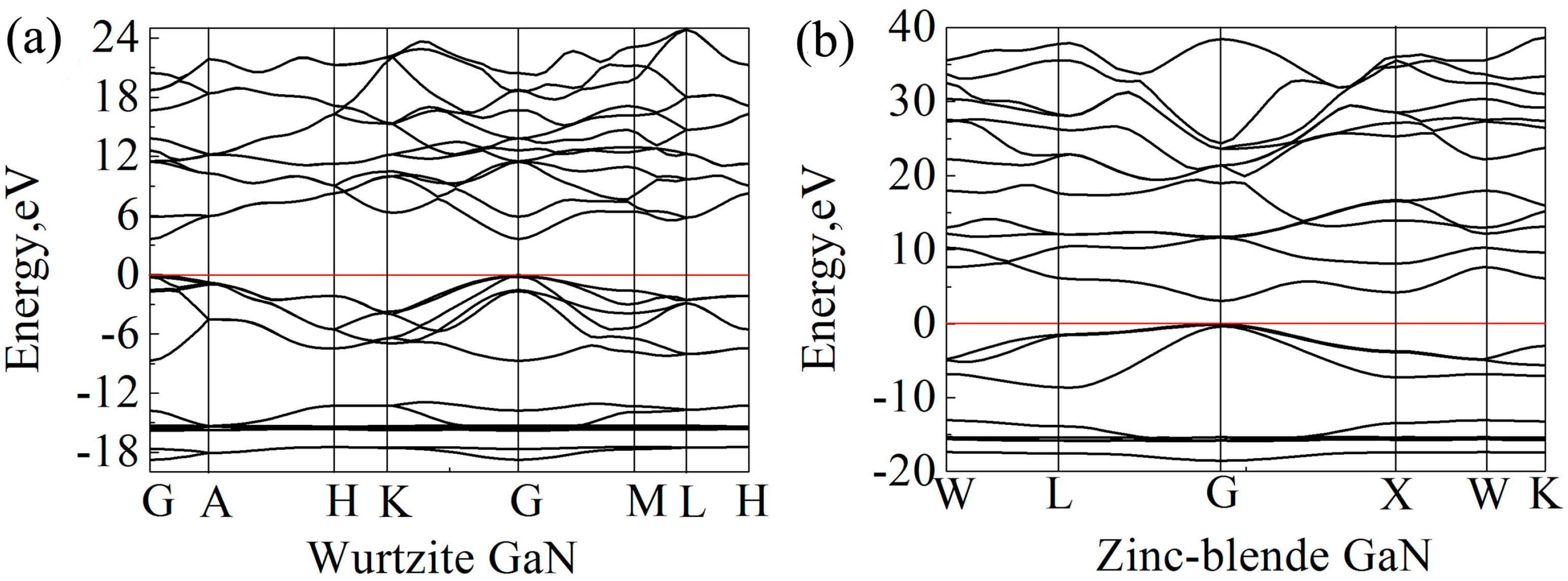
39] was applied for band gap calculations, and the calculated band gaps of wurtzite and zinc-blende GaN are 3.62 eV and 3.06 eV, respectively, which are close to the experimental values. Clearly, the results also show that the band gap of wurtzite GaN is larger than that of zinc-blende GaN. In previous studies, it is reported that the band gap of wurtzite GaN is located at the Γ (G) point [
65,
66]. In the present study, the band structures of wurtzite and zinc-blende GaN crystals are shown in
Figure 6. It is also found that the maximum value of valence band and the minimum value of conduction band are located at the G point for the two crystals, indicating that all of them are direct gap semiconductors. At the G point, the lowest energy of conduction band in the wurtzite GaN is larger than that in the zinc-blende GaN, which results in a wider band gap for the wurtzite GaN.
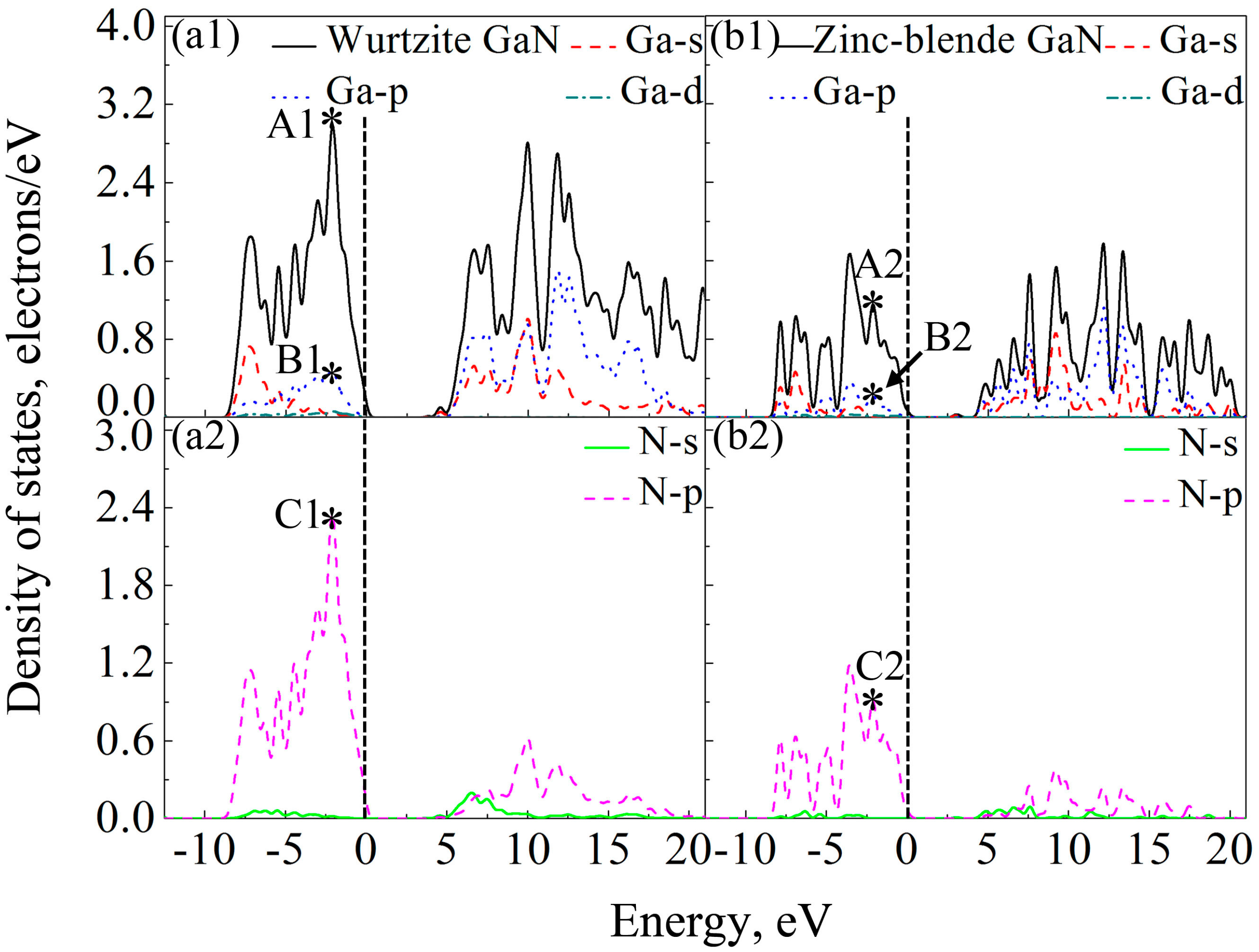
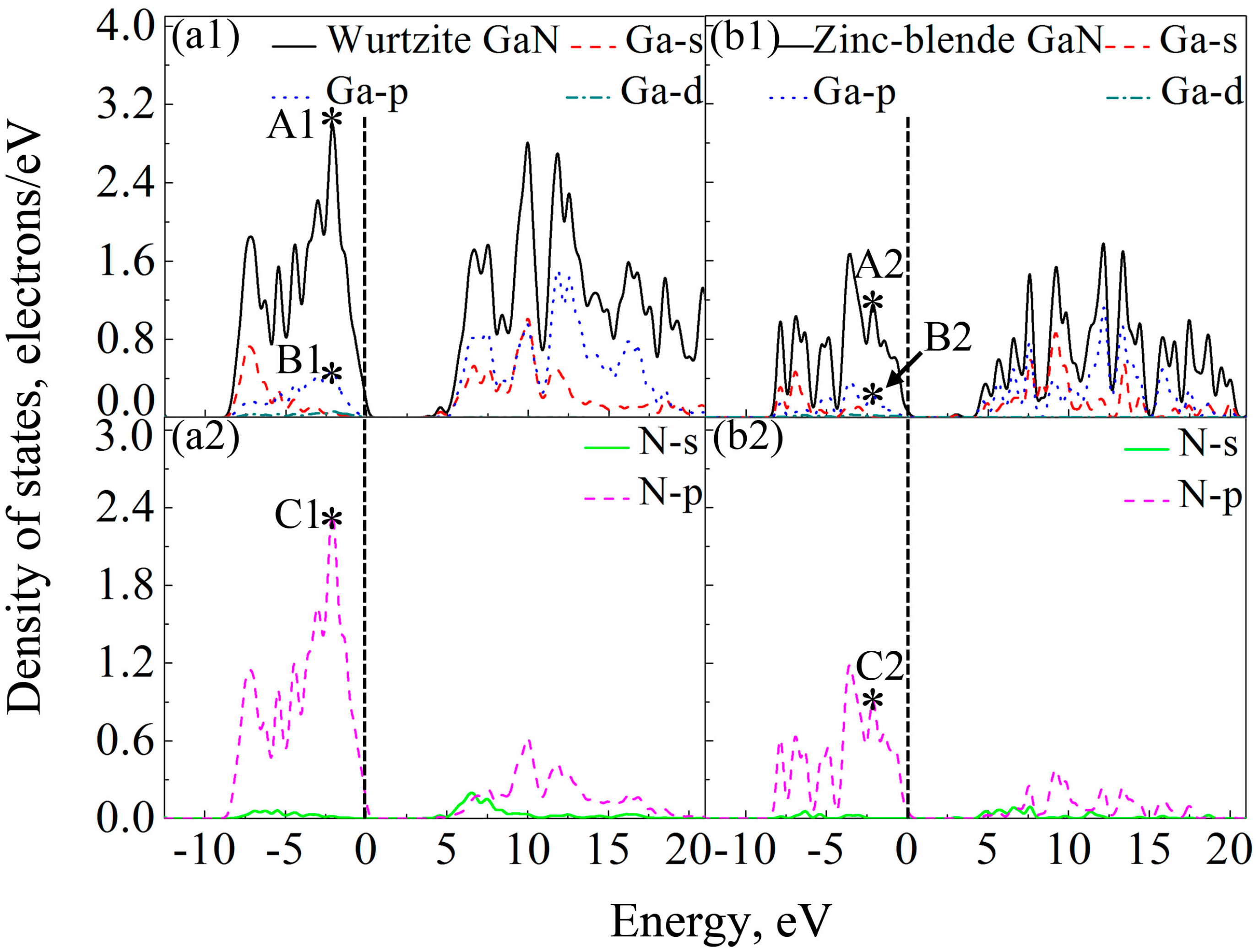
Figure 7 shows DOSs of the GaN crystals around band gaps. According to the distribution characteristics of DOSs of GaN crystals, the two crystals exhibit similar atomic bonding and hybrid behavior. In the energy band with the energy ranging from −10 to 0 eV, DOSs of the two GaN crystals are primarily contributed by p orbits of N atoms (N-p). However, for the energy band in the conduction band, which is close to the band gap, e.g., the energies ranging from 3.62 eV to 20 eV for wurtzite GaN, DOSs of GaN crystals are mainly due to the hybridization between N-p and s and p orbits of Ga (Ga-s and Ga-p). Meanwhile, although the two crystals exhibit similar atomic bonding and hybridization behavior, the DOSs of Ga atoms, N atoms and wurtzite GaN are much larger than those of zinc-blende GaN, as observed in
Figure 7. For example, the values of A1 (peak of DOS of GaN), B1 (peak of Ga-p) and C1 (peak of N-p) at −2.1 eV are 2.98, 0.48 and 2.36 electrons/eV, respectively, while A2, B2 and C2 at −2.1 eV for zinc-blende GaN are 1.15, 0.22 and 0.91 electrons/eV, respectively. Obviously, the values of A1, B1 and C1 are much larger than those of A2, B2 and C2, respectively. Therefore, it can be inferred that more electrons in corresponding atomic orbitals participate in forming Ga-N ionic bonds in the wurtzite GaN, which is why the wurtzite GaN has larger elastic modulus, higher Debye temperature, smaller Poisson’s ratio and shows more evident characteristic of brittleness.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}