On the Stability of c-BN-Reinforcing Particles in Ceramic Matrix Materials



Abstract
:
1. Introduction
2. Materials and Methods
3. Results
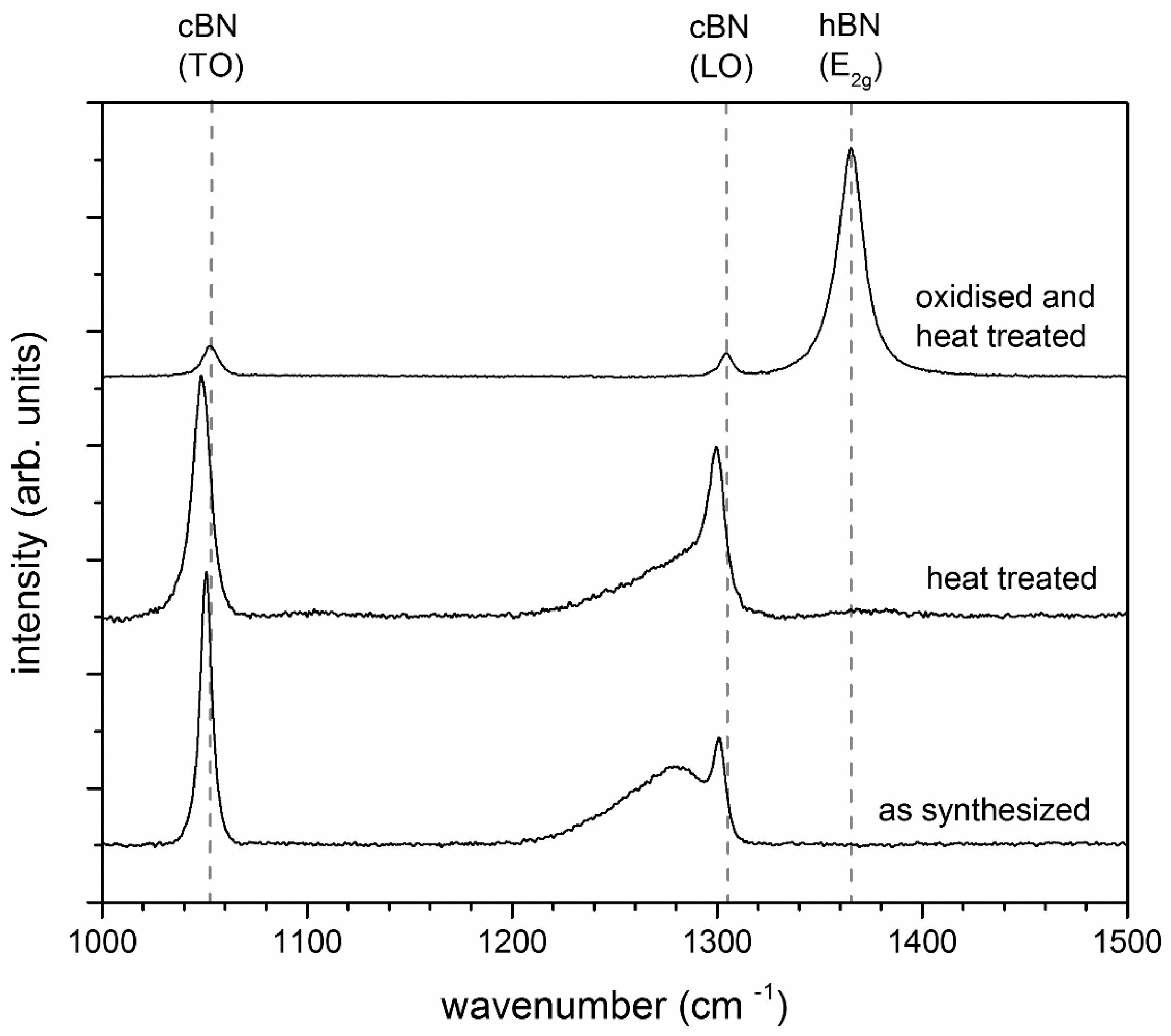
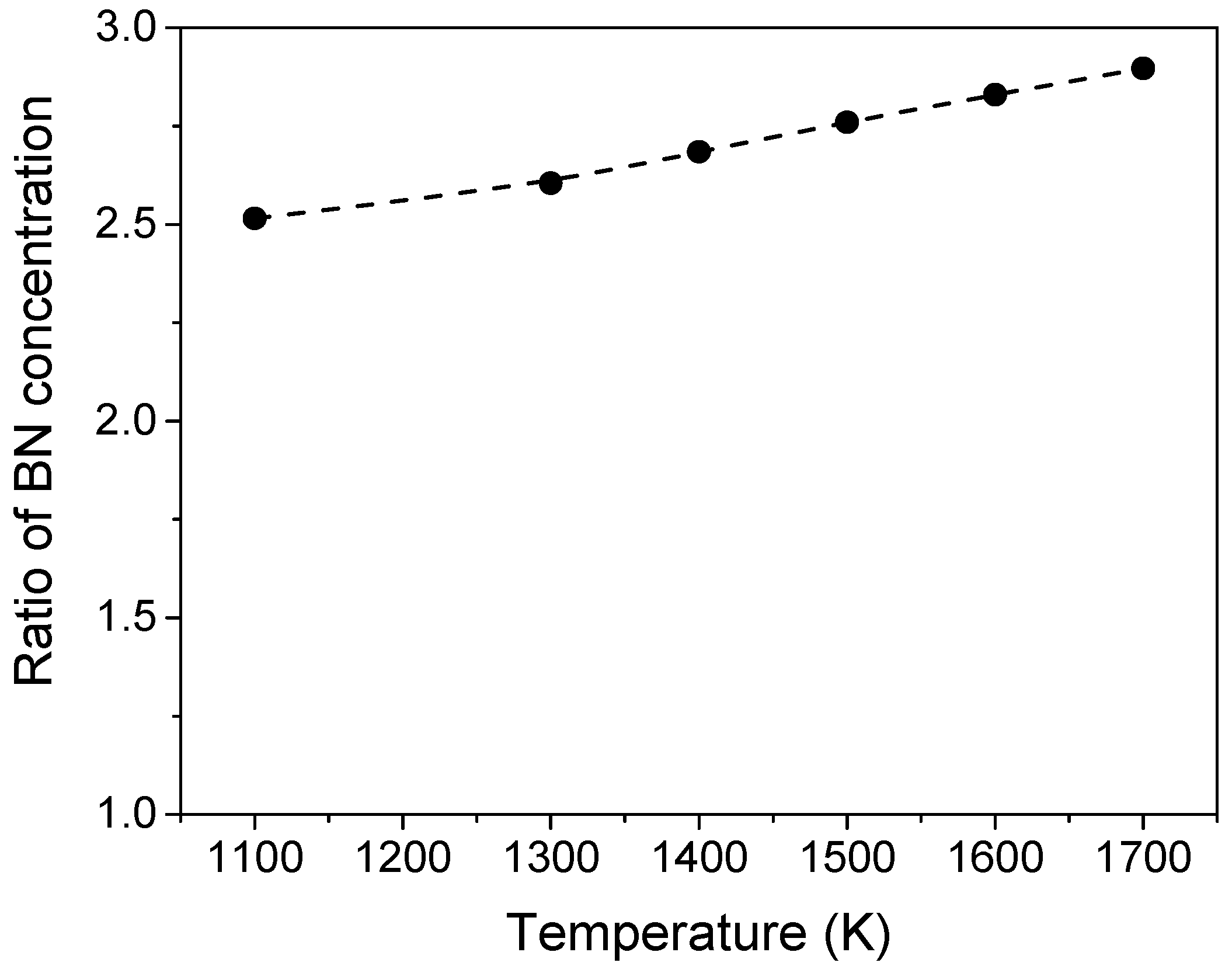
3.1. Heat Treatment of the Pure and Oxidized c-BN Powders
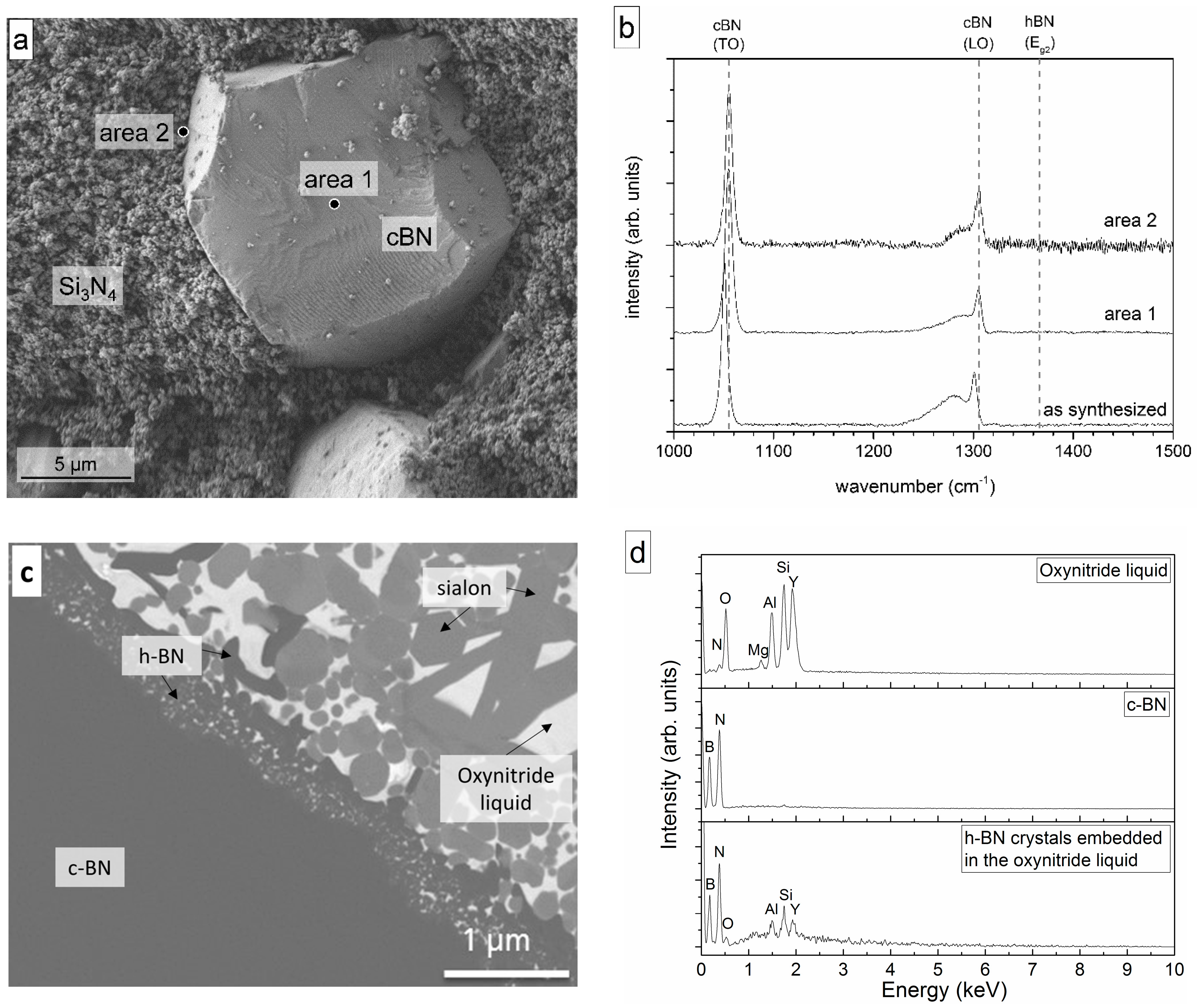
3.2. Heat Treatment of the c-BN–Si3N4 Composite with and without a Liquid Phase
3.3. Heat Treatment of c-BN–Glass Composites
4. Discussion
- Heat treatment of pure c-BN in an argon atmosphere yielded no transformation. In contrast, c-BN showed pronounced transformation to h-BN in the presence of B2O3 under the same conditions (Section 3.1).
- c-BN showed no conversion in pure Si3N4 powder at 1575 °C in the absence of a liquid phase, but transformation took place if an oxynitride liquid was formed in the presence of sintering additives (Section 3.2). The latter was also observed in previous experiments performed by the present authors [12,13,14].
- Heat treatment resulted in a much higher c-BN to h-BN transformation rate for mixtures of c-BN and oxide glasses of different compositions than for pure BN, even at 1400 °C and below. This transformation was influenced by the glass composition (Section 3.3).
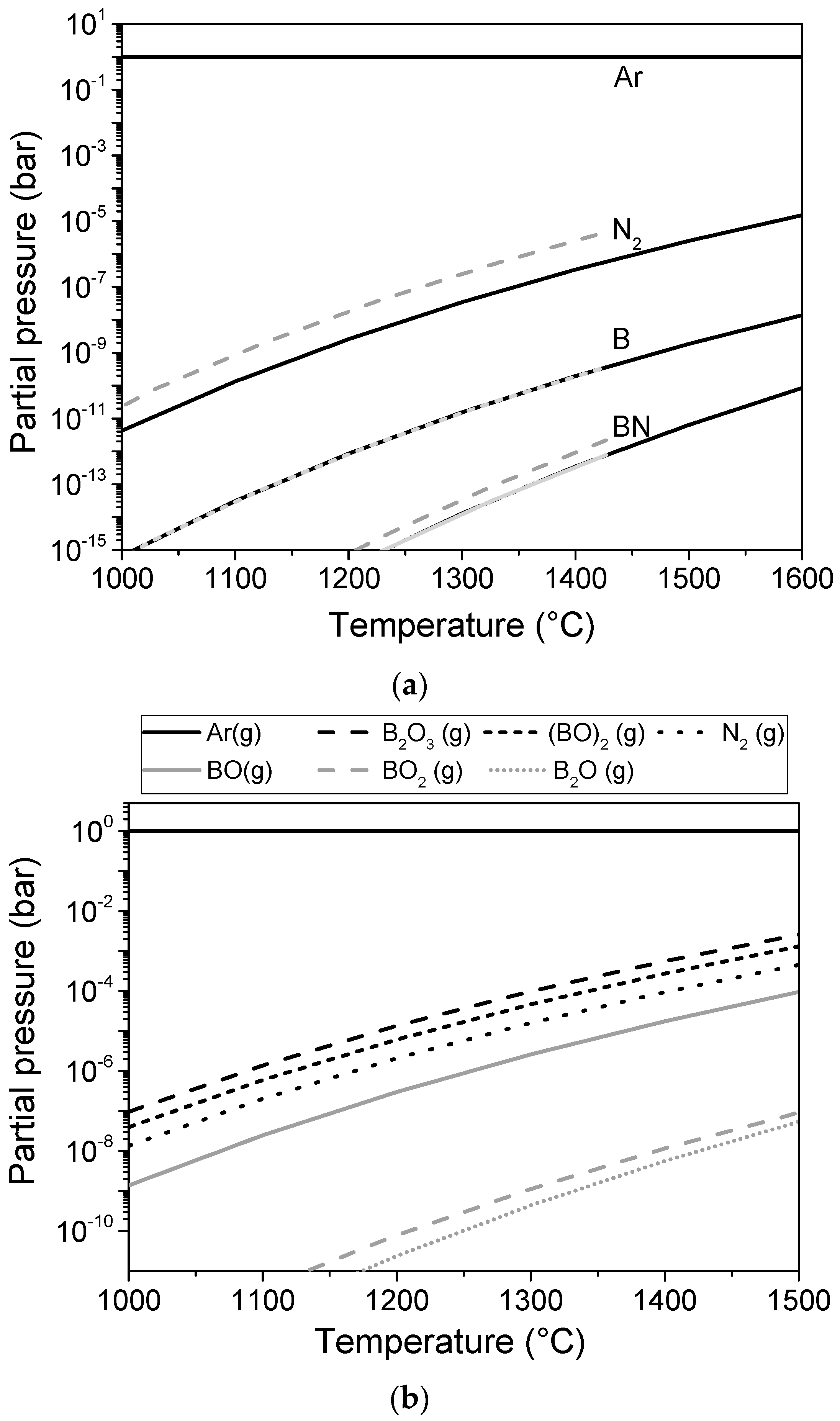
4.1. Gas Phase Mechanism
4.2. Liquid Phase Mechanism
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A

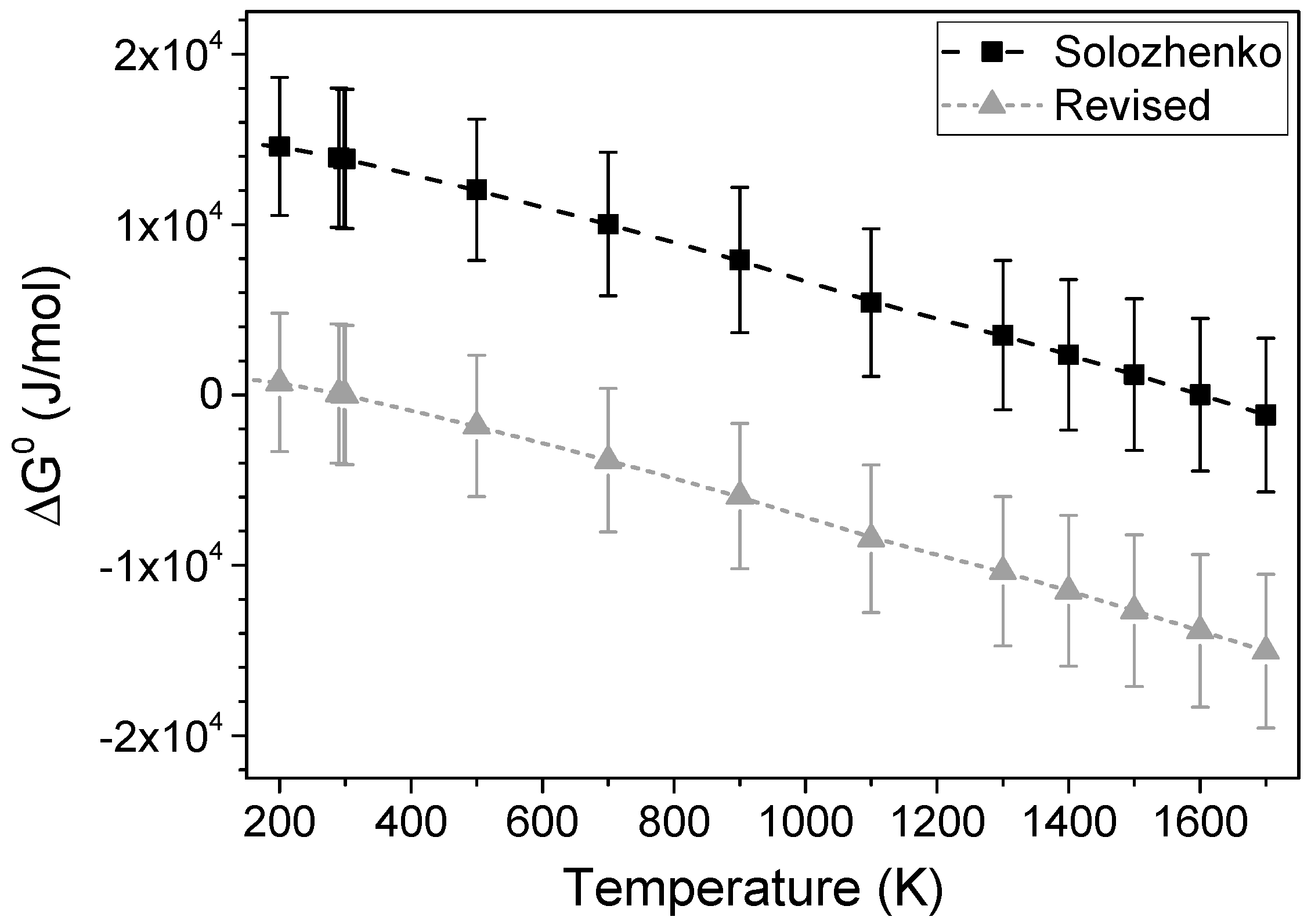
Solubility of c-BN/h-BN

References
- Deutsche Keramische Gesellschaft (DKG). Expertenstudie. In Zukunftspotentiale von Hochleistungskeramiken; DKG: Köln, Germany, 2014; ISBN 978-3-00-045777-7. Available online: www.expertenstudie-hlk.dkg.de (accessed on 1 February 2018).
- Zhang, J.; Tu, R.; Goto, T. Cubic boron nitride-containing ceramic matrix composites for cutting tools. In Advances in Ceramic Matrix Composites; Low, I.M., Ed.; Woodhead Publishing Limited: Oxford, UK, 2014; pp. 570–586. [Google Scholar] [CrossRef]
- McKie, A.; Winzer, J.; Sigalas, I.; Herrmann, M.; Weiler, L.; Rödel, J.; Can, N. Mechanical properties of cBN–Al composite materials. Ceram. Int. 2011, 37, 1–8. [Google Scholar] [CrossRef]
- Herrmann, M.; Matthey, B.; Kunze, S.; Petasch, U. SiC-diamond materials: Wear-resistant and versatile. Ceram. Forum Int. CFI 2014, 91, E39–E43. [Google Scholar]
- Herrmann, M.; Matthey, B.; Höhn, S.; Kinski, I.; Rafaja, D.; Michaelis, A. Diamond-ceramics composites—New materials for a wide range of challenging applications. J. Eur. Ceram. Soc. 2012, 32, 1915–1923. [Google Scholar] [CrossRef]
- Matthée, T.; Schrüfer, A. CVD-Diamantinnovation setzt sich durch. Diam. Bus. 2011, 36, 6–9. [Google Scholar]
- Klimczyk, P.; Figiel, P.; Petrusha, I.; Olszyna, A. Cubic boron nitride based composites for cutting applications. J. Achiev. Mater. Manuf. Eng. 2011, 44, 198–204. [Google Scholar]
- Angseryd, J.; Elfwing, M.; Olsson, E.; Andrén, H.-O. Detailed microstructure of a cBN based cutting tool material. Int. J. Refract. Metals Hard Mater. 2009, 27, 249–255. [Google Scholar] [CrossRef]
- Fukunaga, O. The equilibrium phase boundary between hexagonal and cubic boron nitride. Diam. Relat. Mater. 2000, 9, 7–12. [Google Scholar] [CrossRef]
- Solozhenko, V.L.; Turkevich, V.Z.; Holzapfel, W.B. Refined phase diagram of boron nitride. J. Phys. Chem. B 1999, 103, 2903–2905. [Google Scholar] [CrossRef]
- Zhang, J.; Tu, R.; Goto, T. Densification, microstructure and mechanical properties of SiO2-cBN composites by spark plasma sintering. Ceram. Int. 2012, 38, 351–356. [Google Scholar] [CrossRef]
- Garrett, J.C.; Sigalas, I.; Wolfrum, A.K.; Herrmann, M. Effect of cubic boron nitride grain size in the reinforcing of α-Sialon ceramics sintered via SPS. J. Eur. Ceram. Soc. 2015, 35, 451–462. [Google Scholar] [CrossRef]
- Garrett, J.C.; Sigalas, I.; Herrmann, M. TEM investigation of the interface formation in cubic boron nitride containing α-Sialon composites. Ceram. Int. 2014, 40 Pt B, 16169–16175. [Google Scholar] [CrossRef]
- Garrett, J.C.; Sigalas, I.; Herrmann, M.; Olivier, E.J.; O’Connell, J.H. cBN reinforced Y-α-Sialon composites. J. Eur. Ceram. Soc. 2013, 33, 2191–2198. [Google Scholar] [CrossRef]
- Ye, F.; Hou, Z.; Zhang, H.; Liu, L.; Zhou, Y. Spark plasma sintering of cBN/β-Sialon composites. Mater. Sci. Eng. A 2010, 527, 4723–4726. [Google Scholar] [CrossRef]
- Hotta, M.; Goto, T. Densification and phase transformation of β-Sialon-cubic boron nitride composites prepared by spark plasma sintering. J. Am. Ceram. Soc. 2009, 92, 1684–1690. [Google Scholar] [CrossRef]
- Hotta, M.; Goto, T. Effect of time on microstructure and hardness of β-Sialon-cubic boron nitride composites during spark plasma sintering. Ceram. Int. 2011, 37, 521–524. [Google Scholar] [CrossRef]
- Klimczyk, P.; Cura, M.E.; Vlaicu, A.M.; Mercioniu, I.; Wyzga, P.; Jaworska, L.; Hannula, S.P. Al2O3-cBN composites sintered by SPS and HPHT methods. J. Eur. Ceram. Soc. 2016, 36, 1783–1789. [Google Scholar] [CrossRef]
- Wolfrum, A.-K.; Herrmann, M.; Michaelis, A. Sialon ceramics reinforced with coated cubic boron nitride prepared via FAST/SPS. In Proceedings of the 8th International Symposium on Nitrides—ISNT, Wildbad Kreuth, Germany, 31 August–5 September 2014. [Google Scholar]
- Zhang, J.; Tu, R.; Goto, T. Densification of SiO2-cBN composites by using Ni nanoparticle and SiO2 nanolayer coated cBN powder. Ceram. Int. 2012, 38, 4961–4966. [Google Scholar] [CrossRef]
- Ahmed, B.A.; Hakeem, A.S.; Laoui, T.; Khan, R.M.A.; Malki, M.M.A.; Hamid, A.U.; Khalid, F.A.; Bakhsh, N. Effect of precoursor size on the structure and mechanical properties of calcium-stabilized sialon/cubic boron nitride nanocomposites. J. Alloys Compd. 2017, 728, 836–843. [Google Scholar] [CrossRef]
- Sachdev, H.; Haubner, R.; Noth, H.; Lux, B. Investigation of the c-BN/h-BN phase transformation at normal pressure. Diam. Relat. Mater. 1997, 6, 286–292. [Google Scholar] [CrossRef]
- Sachdev, H.; Tang, X.; Haubner, R.; Lux, B. Patwhwways to c-BN from melts at atmospheric pressure. Int. J. Refract. Metals Hard Mater. 1998, 16, 243–251. [Google Scholar] [CrossRef]
- Höhn, H.; Sempf, K.; Herrmann, M. Artefact-free preparation and characterization of ceramic materials and interfaces. Ceram. Forum Int. 2011, 88, 16–20. [Google Scholar]
- Pfeiffer, T. Viscosities and electrical conductivities of oxidic glass-forming melts. Solid State Ion. 1998, 105, 277–287. [Google Scholar] [CrossRef]
- Rost, A.; Schilm, J.; Kusnezoff, M.; Michaelis, A. Degradation of Sealing Glasses for SOFC under Electrical Load and Dual Atmosphere. J. Ceram. Sci. Technol. 2012, 3, 69–80. [Google Scholar]
- Herrmann, M.; Lippmann, W.; Hurtado, A. Y2O3–Al2O3–SiO2-based glass-ceramic fillers for the laser-supported joining of SiC. J. Eur. Ceram. Soc. 2014, 34, 1935–1948. [Google Scholar] [CrossRef]
- Scarlett, N.V.Y.; Madsen, I.C. Quantification of phases with partial or no known crystal structures. Powder Diffr. 2006, 278–284. [Google Scholar] [CrossRef]
- Sachdev, H. Influence of impurities on the morphology and Raman spectra of cubic boron nitride. Diam. Relat. Mater. 2003, 12, 1275–1286. [Google Scholar] [CrossRef]
- Reich, S.; Ferrari, A.C.; Arenal, R.; Loiseau, A.; Bello, I.; Robertson, J. Resonant Raman scattering in cubic and hexagonal boron nitride. Phys. Rev. B 2005, 71, 205201. [Google Scholar] [CrossRef]
- Mandal, H. New developments in α-SiAlON ceramics. J. Eur. Ceram. Soc. 1999, 19, 2349–2357. [Google Scholar] [CrossRef]
- Ekstrom, T.; Nygren, M. SiAION ceramics. J. Am. Ceram. Soc. 1992, 75, 259–276. [Google Scholar] [CrossRef]
- Rohbeck, N.; Menna, M.; Somers, J.; Couland, M.; Herrmann, M.; Lippmann, W. Chemical interaction of yttrium aluminosilicate glass with CeO2, UO2 and PuO2 at high temperatures. Nucl. Eng. Des. 2011, 241, 2775–2779. [Google Scholar] [CrossRef]
- FactSage. FACTSAGE 7.0; FACT PS Database; Centre de Recherche en Calcul Thermochimique: Montreal, QC, Canada; GTT-Technologies: Aachen, Germany, 2015. [Google Scholar]
- Eustathopoulus, N. Wettability at High Temperature; Pergamon: Amsterdam, The Netherland, 1999. [Google Scholar]
- Solozhenko, V.L.; Turkevich, V.Z.; Kurakevych, O.O.; Turkevich, D.V.; Taniguchi, T. Phase Equilibria in the B–BN–B2O3 System at 5 GPa. J. Phys. Chem. C 2013, 117, 18642–18647. [Google Scholar] [CrossRef]
- Choi, J.-Y.; Kang, S.-J.L.; Fukunaga, O.; Park, J.-K.; Eun, K.Y. Effect of B2O3 and hBN Crystallinity on cBN Synthesis. J. Am. Ceram. Soc. 1993, 76, 2525–2528. [Google Scholar] [CrossRef]
- Sato, T.; Hiraoka, H.; Endo, T.; Fukunaga, O.; Iwata, M. Effect of oxygen on the growth of cubic boron nitride using Mg3N2 as catalyst. J. Mater. Sci. 1981, 16, 1829–1834. [Google Scholar] [CrossRef]
- Singhal, S.K.; Park, J.K. Synthesis of cubic boron nitride from amorphous boron nitride containing impurity using Mg-Al alloy catalyst solvent. J. Cryst. Growth 2004, 260, 217–222. [Google Scholar] [CrossRef]
- Gladkaya, I.S.; Kremkova, G.N.; Bendeliani, N.A. The phase transition of hBN-cBN in the B-N-H-O system. Diam. Relat. Mater. 1996, 5, 1440–1443. [Google Scholar] [CrossRef]
- Wakasugi, T.; Tsukihashi, F.; Sano, N. Thermodynamics of Nitrogen in B2O3, B2O3-SiO2, and B2O3-CaO Systems. J. Am. Ceram. Soc. 1991, 74, 1650–1653. [Google Scholar] [CrossRef]
- Wakasugi, T.; Tsukihashi, F.; Sano, N. The solubilities of BN in B2O3 bearing melts. J. Non-Cryst. Solids 1991, 135, 139–145. [Google Scholar] [CrossRef]
- Frischat, G.; Krause, W.; Hübenthal, H. Preparation and Properties of Nitrogen-Containing Na2O-B203 Glasses. Commun. Am. Ceram. Soc. 1984, 67, C10–C12. [Google Scholar]
- Fu, Z.; Koc, R. Ultrafine TiB2-TiNiFeCrCoAl high-entropy alloy composite with enhanced mechanical properties. Mater. Sci. Eng. A 2017, 702, 184–188. [Google Scholar] [CrossRef]
- German, R.M.; Suri, P.; Park, S.J. Liquid phase sintering. J. Mater. Sci. 2009, 44, 1–39. [Google Scholar] [CrossRef]
- Solozhenko, V.I.; Gavrichev, K.S. Thermodynamik Properties of Boron Nitride. In Wide Band Gab Electronic Materials; Prelas, M.A., Gielisse, P., Popovici, G., Spitsyn, B.V., Stacy, T., Eds.; Kluver Academic Publishers: Dordrecht, The Netherlands, 1995; pp. 377–392. [Google Scholar]
- Solozhenko, V.L. Boron nitride phase diagram. State of the Art. High Press. Res. 1995, 13, 199–214. [Google Scholar] [CrossRef]
- Will, G.; Nover, G.; von der Gonna, J. New Experimental Results on the Phase Diagram of Boron Nitride. J. Solid State Chem. 2000, 154, 280–285. [Google Scholar] [CrossRef]
- Jacobson, N.; Farmer, S.; Moore, A.; Sayir, H. High-Temperature Oxidation of Boron Nitride: I, Monolithic Boron Nitride. J. Am. Ceram. Soc. 1999, 82, 393–398. [Google Scholar] [CrossRef]
- Herrmann, M.; Thiele, M.; Jaenicker-Roessler, K.; Freemantle, C.S.; Sigalas, I. Oxidation resistance of B6O-materials with different additives. J. Eur. Ceram. Soc. 2011, 32, 1771–1777. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Glass Composition (wt %) | Tg (°C) | ||||||
|---|---|---|---|---|---|---|---|---|
| SiO2 | B2O3 | Al2O3 | MgO | CaO | BaO | Y2O3 | ||
| G1 1 | 19.9 | 13.2 | 24.1 | - | - | - | 42.8 | 740 |
| G2 2 | 15.6 | - | 20.5 | - | - | - | 63.9 | 890 |
| G3 3 | 59.6 | 5.3 | 15.6 | 2.5 | 7.7 | 9.4 | - | 725 |
| Temperature (°C) | Holding Time (h) | Phase Composition | ||
|---|---|---|---|---|
| G1-BN | G2-BN | G3-BN | ||
| 1400 | 1 | Amorphous | Mullite α-Y2Si2O7 γ-Y2Si2O7 | Amorphous |
| 1300 | 1 | Y(Al)BO3 (α-Al2O3) Al9BSi2O19 | Mullite Y2Si2O7 YAG SiO2 | Amorphous |
| 1300 | 10 | YBO3 (α-Al2O3) Al9BSi2O19 | Mullite Y2Si2O7 YAG SiO2 | Amorphous |
| 1200 | 10 | YBO3 Al9BSi2O19 α-Y2Si2O7 γ-Y2Si2O7 | Mullite, YAG Y2Si2O7, SiO2 α-Y2Si2O7 γ-Y2Si2O7 | Amorphous |
| 1100 | 10 | YBO3 α-Y2Si2O7 γ-Y2Si2O7 | Not investigated | SiO2 |
| Temperature (°C) | Holding Time (h) | Content of h-BN/(h-BN+c-BN) (wt %) | ||
|---|---|---|---|---|
| G1-BN | G2-BN | G3-BN | ||
| 1400 | 1 | h-BN (24.6%) | h-BN (13.7%) | h-BN (12.2%) |
| 1300 | 1 | No h-BN | No h-BN | No h-BN |
| 1300 | 10 | h-BN (10.2%) | No h-BN | h-BN (6.7%) |
| 1200 | 10 | h-BN (0.6%) | No h-BN | h-BN (0.6%) |
| 1100 | 10 | No h-BN | Not investigated | No h-BN |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wolfrum, A.-K.; Matthey, B.; Michaelis, A.; Herrmann, M. On the Stability of c-BN-Reinforcing Particles in Ceramic Matrix Materials. Materials 2018, 11, 255. https://doi.org/10.3390/ma11020255
Wolfrum A-K, Matthey B, Michaelis A, Herrmann M. On the Stability of c-BN-Reinforcing Particles in Ceramic Matrix Materials. Materials. 2018; 11(2):255. https://doi.org/10.3390/ma11020255
Chicago/Turabian StyleWolfrum, Anne-Kathrin, Björn Matthey, Alexander Michaelis, and Mathias Herrmann. 2018. "On the Stability of c-BN-Reinforcing Particles in Ceramic Matrix Materials" Materials 11, no. 2: 255. https://doi.org/10.3390/ma11020255
APA StyleWolfrum, A.-K., Matthey, B., Michaelis, A., & Herrmann, M. (2018). On the Stability of c-BN-Reinforcing Particles in Ceramic Matrix Materials. Materials, 11(2), 255. https://doi.org/10.3390/ma11020255