Analysis of the Secondary Phases Formed by Corrosion of U3Si2-Al Research Reactor Fuel Elements in the Presence of Chloride Rich Brines


Abstract
:1. Introduction
2. Materials and Methods
2.1. Setup of the Corrosion Experiments and Sample Pre-Treatment
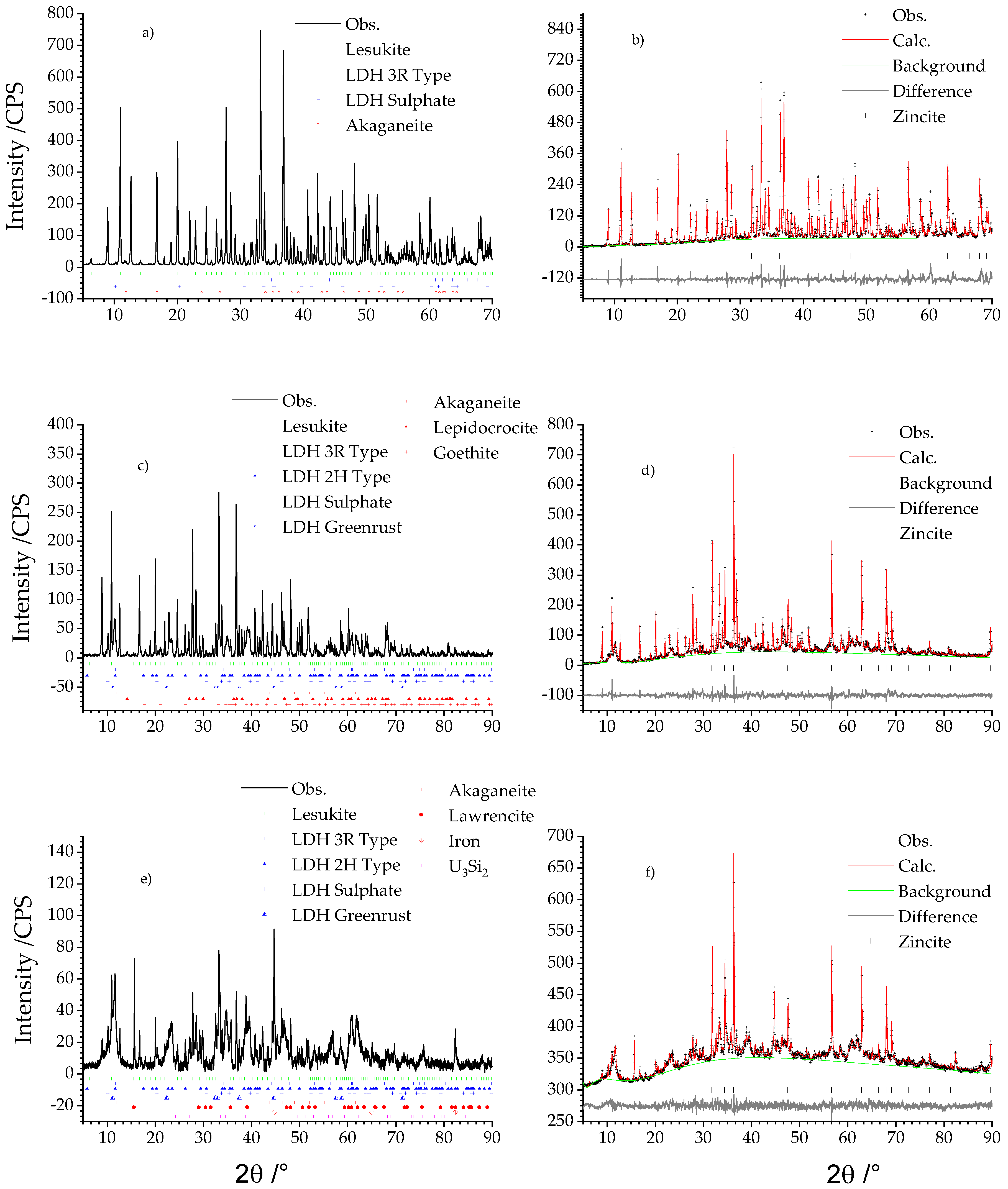
2.2. X-ray Diffraction Analysis
2.3. SEM/EDS Analysis
3. Results and Discussion
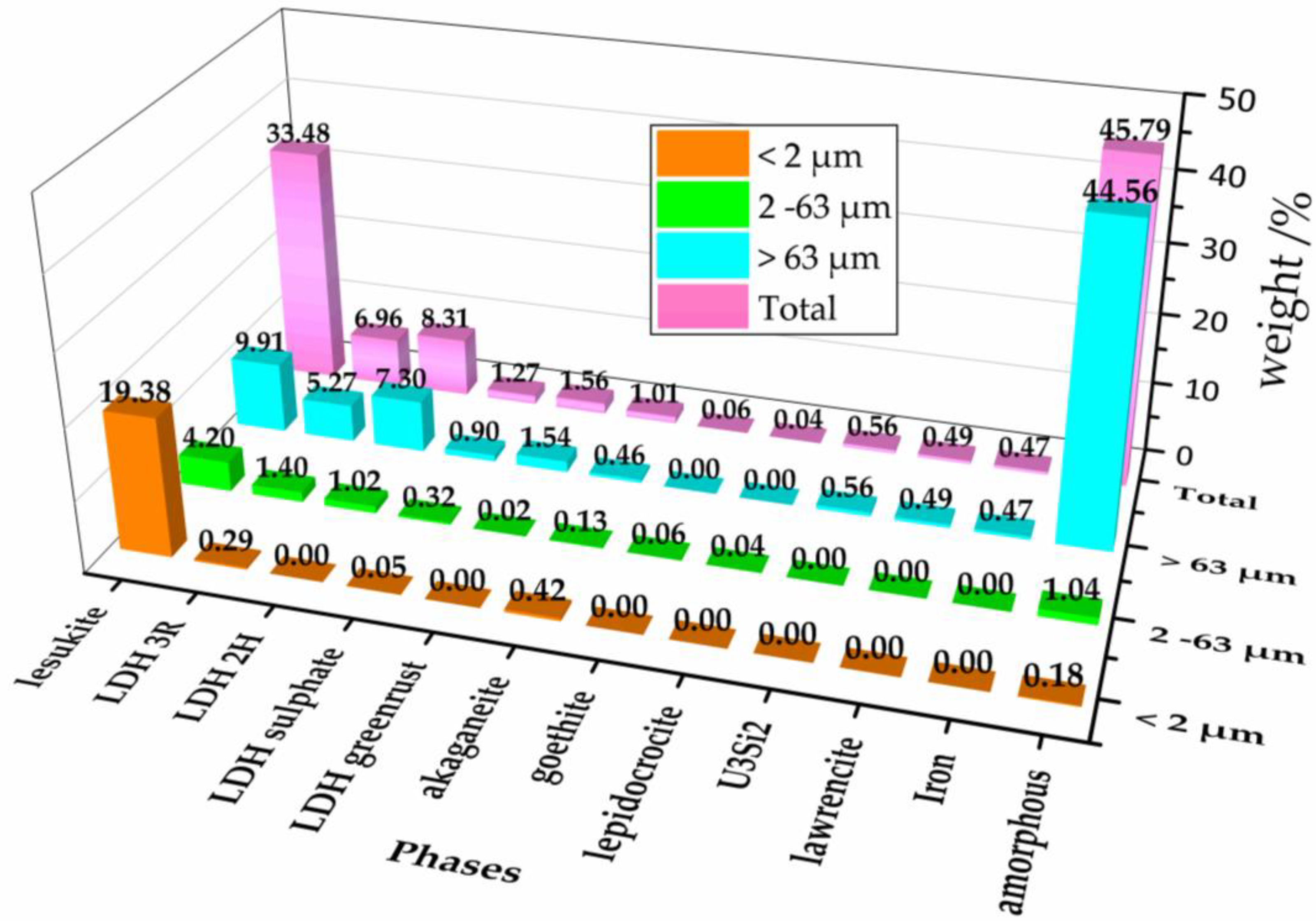
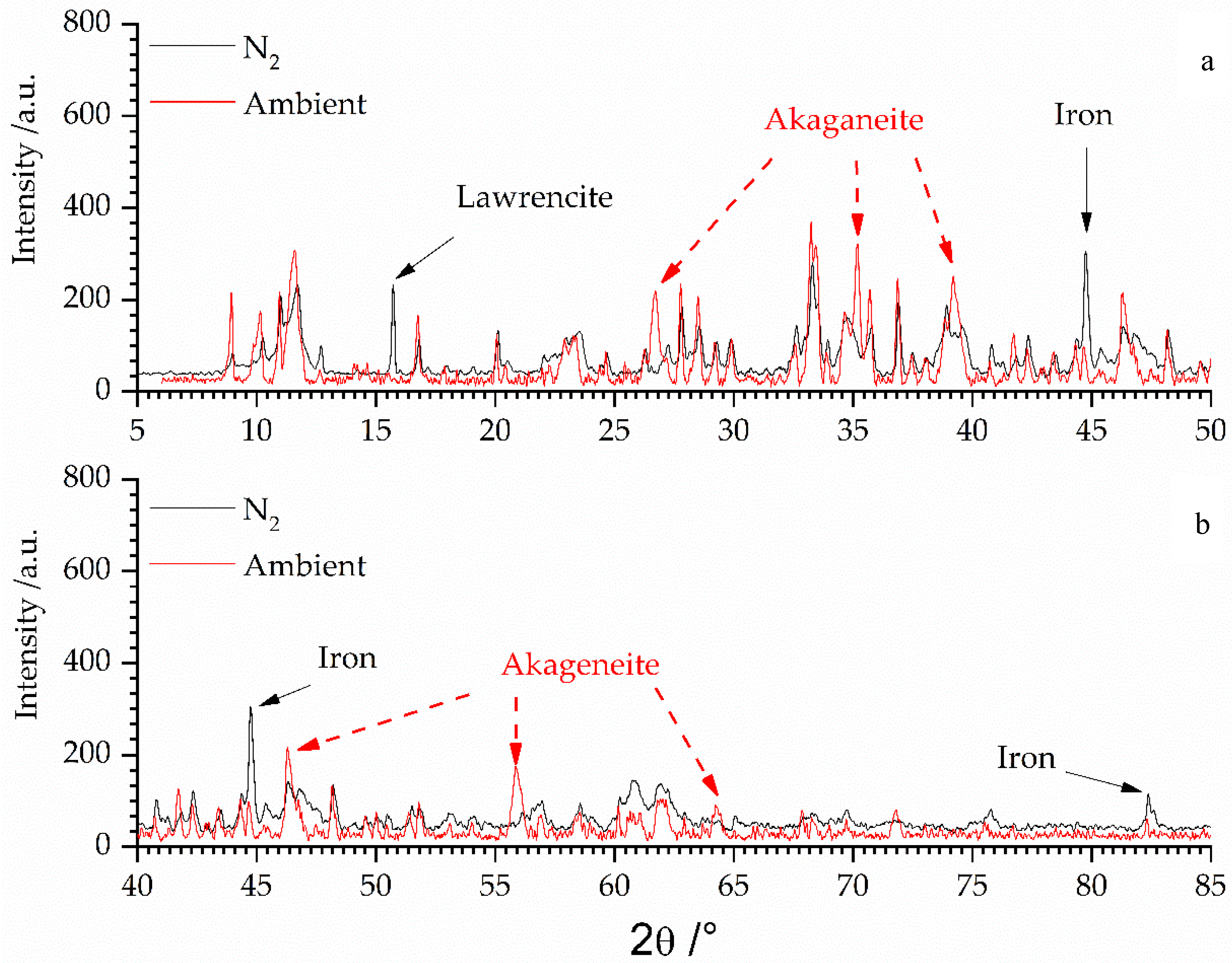
3.1. X-ray Analysis of the Secondary Phases
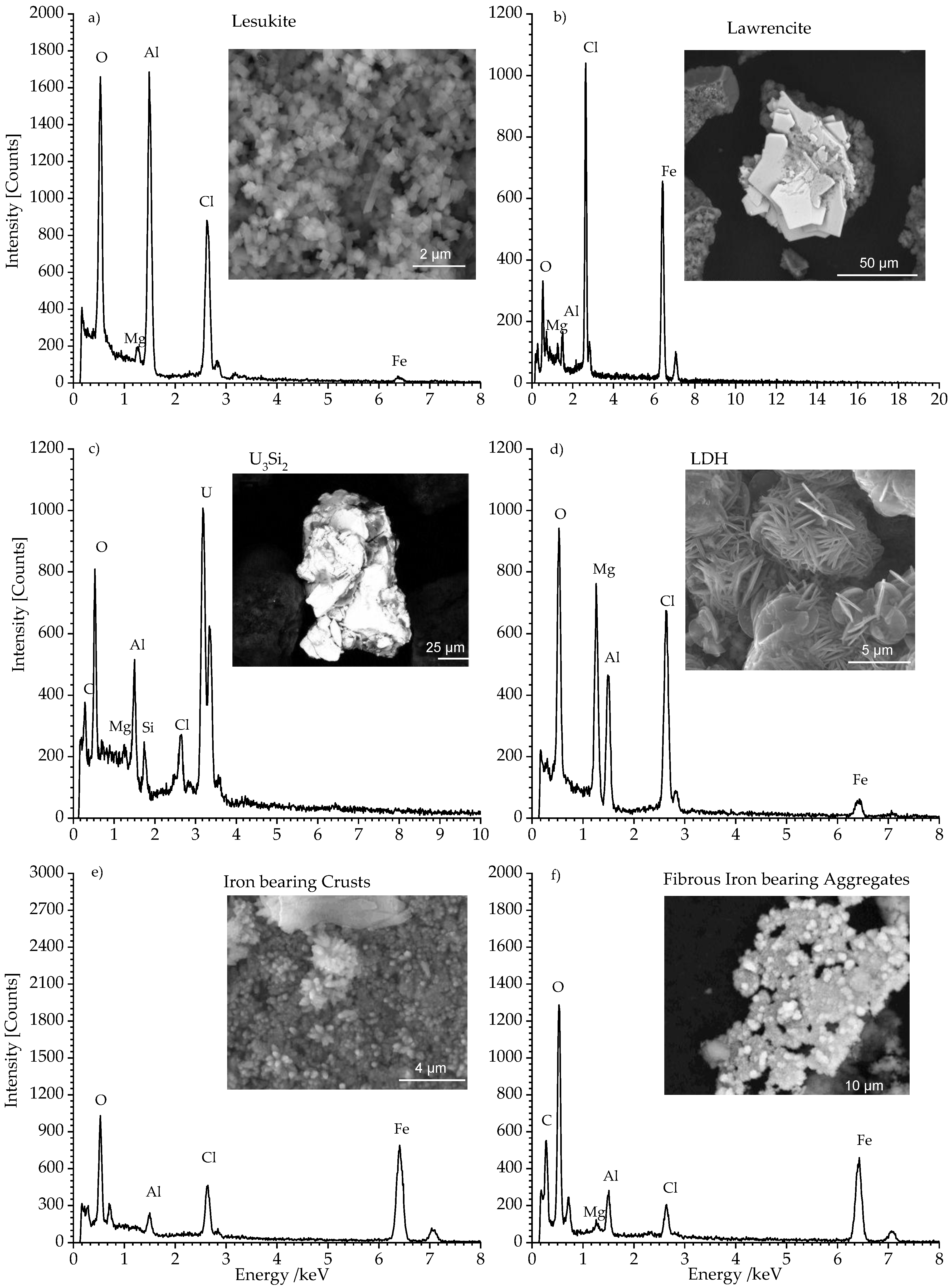
3.2. SEM and EDS Analysis of the Secondary Phases
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Johnson, L.H.; Shoesmith, D.W. Spent fuel. In Radioactive Waste Forms for the Future; Lutze, W., Ewing, R.C., Eds.; Elsevier: Amsterdam, The Netherlands, 1988; p. 635. ISBN 0444871047. [Google Scholar]
- Kienzler, B.; Loida, A. Endlagerrelevante Eigenschaften von Hochradioaktiven Abfallprodukten—Charakterisierung und Bewertung—Empfehlungen des Arbeitskreises HAW-Produkte; Karlsruhe Institute of Technology: Karlsruhe, Germany, 2001. [Google Scholar]
- Fein, E.; Müller-Lyda, I.; Rübel, A. Anhang Langzeitsicherheitsanalyse. In GRS Bericht 247/7—Endlagerung Wärmeentwickelnder Radioaktiver Abfälle in Deutschland; Gesellschaft für Anlagen- und Reaktorsicherheit GRS: Cologne, Germany, 2008; ISBN 978-3-939355-22-9. [Google Scholar]
- Brücher, H.; Curtius, H.; Kaiser, G.; Mazeina, L.; Fachinger, J. Untersuchungen zur Radionuklidfreisetzung und zum Korrosionsverhalten von Bestrahltem Kernbrennstoff aus Forschungsreaktoren unter Endlagerbedingungen; Berichte des Forschungszentrum Jülich 4104: Jülich, Germany, 2001. [Google Scholar]
- Cloke, P.L.; Gottlieb, P.; Lester, D.H.; Fuenties, E.; Benton, H.A. Geochemical Analysis of Degradation Modes of HEU SNF in a Codisposal Waste Package with HLW Canisters; BBA000000-0171740200-0059 REV 01; CRWMS/M&O: Las Vegas, NV, USA, 1998. [Google Scholar]
- Fachinger, J.; Brücher, H.; Rainer, H.; Kaiser, G.; Syuhada, I.; Zschunke, S.; Nau, K. Untersuchungen zur Radionuklidfreisetzung durch Einwirkung konzentrierter Salzlaugen auf Alu-MTR-Brennelemente; Berichte des Forschungszentrums Jülich 4104: Jülich, Germany, 1998. [Google Scholar]
- Curtius, H.; Kaiser, G.; Paparigas, Z.; Ufer, K.; Müller, E.; Enge, E.; Brücher, H. Untersuchungen zum Verhalten von FR-BE in Wirtsgesteinswässern Möglicher Endlager; Berichte des Forschungszentrums Jülich 4237: Jülich, Germany, 2006. [Google Scholar]
- Curtius, H.; Paparigas, Z.; Kaiser, G. Sorption of selenium on Mg-Al and Mg-Al-Eu layered double hydroxides. Radiochim. Acta 2008, 96, 651–655. [Google Scholar] [CrossRef]
- Wiersma, B.J.; Mickalonis, J.I. Preliminary Report on the Dissolution Rate and Degradation of Aluminum Spent Nuclear Fuels in Repository Environments; WSRC-TR-98-00290 (U) Report; U.S. Department of Energy: Washington, DC, USA, 1998. [Google Scholar]
- McClure, J.A.; Davis, J.W.; Gottlieb, P.; Cloke, P.L.; Nitti, D.A.; Benton, H.A. Evaluation of Codisposal Viability for Aluminum-Clad DOE-Owned Spent Fuel: Phase II—Degraded Codisposal Waste; Package Internal Criticality; BBA000000-01717-5705-00017 REV 01; CRWMS/M&O: Las Vegas, NV, USA, 1998. [Google Scholar]
- Kaminski, M.D.; Goldberg, M.M. Corrosion of breached Aluminide Fuel under Potential Repository Conditions. In Proceedings of the International High Level Radioactive Waste Management Conference, Las Vegas, NV, USA, 29 April–3 May 2001. [Google Scholar]
- Kaminski, M.D.; Goldberg, M.M. Aqueous corrosion of aluminum-based nuclear fuel. J. Nucl. Mater. 2002, 304, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, M.D. Aqueous Corrosion of Aluminum-Based Nuclear Fuel; DOE: ANL-CMT-03/1; Argonne National Laboratory: Argonne, IL, USA, 2003. [Google Scholar]
- Kaminski, M.D.; Goldberg, M.M.; Mertz, C.J. Colloids from the aqueous corrosion of aluminium-based nuclear fuel. J. Nucl. Mater. 2005, 347, 88–93. [Google Scholar] [CrossRef]
- Shelton-Davis, C.; Loo, H.; Mackay, N.; Wheatley, P. Review of DOE Spent Nuclear Fuel Release Rate Test Results; DOE/SNF/REP-073; Idaho National Laboratory: Idaho Falls, ID, USA, 2003. [Google Scholar]
- Standortauswahlgesetz—StandAG: Gesetz zur Suche und Auswahl Eines Standortes für ein Endlager für Wärme Entwickelnde Radioaktive Abfälle; Bundesgesetzblatt Teil I Nr. 26, S. 1074-1102: Bonn, Germany, 2017.
- Code of Federal Regulations (Annual Edition)—Titel 10: Energy; Office of the Federal Register: Washington, DC, USA, 2017.
- Dörr, S.; Bollingerfehr, W.; Filbert, W.; Tholen, M. Status quo der Lagerung ausgedienter Brennelemente aus stillgelegten/rückgebauten deutschen Forschungsreaktoren und Strategie (Lösungsansatz) zu deren künftigen Behandlung/Lagerung—LABRADOR; Abschlussbericht FKZ 02 S 8679; DBE Technology GmbH: Peine, Germany, 2011. [Google Scholar]
- Dörr, S.; Bollingerfehr, W.; Filbert, W.; Tholen, M. Quantity and Management of Spent Fuel of Prototype and Research Reactors in Germany. In Proceedings of the German Annual Meeting on Nuclear Technology 2012, Stuttgart, Germany, 22–24 May 2012. [Google Scholar]
- Hilton, B.A. Review of Oxidation Rates of DOE Spent Nuclear Fuel, Part 1: Metallic Fuel; W-31109-ENG-38, ANL-00/24 Report; Argonne National Laboratory: Argonne, IL, USA, 2000. [Google Scholar]
- Curtius, H.; Kaiser, G.; Paparigas, Z.; Hansen, B.; Neumann, A.; Klinkenberg, M.; Müller, E.; Brücher, H.; Bosbach, D. Wechselwirkung Mobilisierter Radionuklide mit Sekundären Phasen in Endlagerrelevanten Formationswässern; Berichte des Forschungszentrums Jülich 4333: Jülich, Germany, 2010. [Google Scholar]
- Loida, A.; Grambow, B.; Geckeis, H. Anoxic corrosion of various high burnup spent fuel samples. J. Nucl. Mater. 1996, 238, 11–22. [Google Scholar] [CrossRef]
- Mazeina, L.; Curtius, H.; Fachinger, J.; Odoj, R. Characterisation of secondary products of uranium-aluminium material test reactor fuel element corrosion in repository-relevant brine. J. Nucl. Mater. 2003, 323, 1–7. [Google Scholar] [CrossRef]
- Mazeina, L. Investigation of the Corrosion Behaviour of U-Al Material Test Reactor Fuel Elements in Repository-Relevant Solutions and Characterisation of the Secondary Phases Formed; Berichte des Forschungszemtrums Jülich 4063: Jülich, Germany, 2003. [Google Scholar]
- Klinkenberg, M.; Neumann, A.; Curtius, H.; Kaiser, G.; Bosbach, D. Research reactor fuel element corrosion under repository relevant conditions: Seperation, identification, and quantification of secondary alteration phases of UAlx-Al in MgCl2 rich brine. Radiochim. Acta 2014, 102, 311–324. [Google Scholar] [CrossRef]
- Vergasova, L.P.; Stepanova, E.L.; Serafimova, E.K.; Filatov, S.K. Lesukite Al2(OH)5Cl·2H2O—A new mineral from volcanic exhalation. Proc. Russ. Miner. Soc. 1997, 126, 104–110. [Google Scholar]
- Witzke, T. A new aluminium chloride mineral from Oelsnitz near Zwickau, Saxony, Germany. Neues Jahrb. Mineral. Monatshefte 1997, 7, 301–308. [Google Scholar]
- Neumann, A. Bildung von Sekundären Phasen bei Tiefengeologischer Endlagerung von Forschungsreaktor-Brennelementen—Struktur-und Phasenanalyse; Schriften des Forschungszentrums Jülich, Reihe Energie & Umwelt, Band 153: Jülich, Germany, 2012. [Google Scholar]
- Neumann, A.; Klinkenberg, M.; Curtius, H. Corrosion of non-irradiated UAlx–Al fuel in the presence of clay pore solution: A quantitative XRD secondary phase analysis applying the DDM method. Radiochim. Acta 2017, 105, 85–94. [Google Scholar] [CrossRef]
- Pearson, F.J. Opalinus Clay Experimental Water: A1 Type, version 980318; PSI Internal Report TM-44-98-07; Paul Scherrer Institut: Villigen, Switzerland, 1998. [Google Scholar]
- Atterberg, A. Die rationelle Klassifikation der Sande und Kiese. Chem.-Zeitung 1905, 29, 195–198. [Google Scholar]
- Rietveld, H.M. Line profiles of neutron powder-diffraction peaks for structure refinement. Acta Cryst. 1967, 22, 151–152. [Google Scholar] [CrossRef] [Green Version]
- Rietveld, H.M. A profile refinement method for nuclear and magnetic structures. J. Appl. Crystallogr. 1969, 2, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Cheary, R.W.; Coelho, A.A. A fundamental parameters approach to X-ray line-profile fitting. J. Appl. Crystallogr. 1992, 25, 109–121. [Google Scholar] [CrossRef] [Green Version]
- Coelho, A.A. Indexing of powder diffraction patterns by iterative use of singular value decomposition. J. Appl. Crystallogr. 2003, 36, 86–95. [Google Scholar] [CrossRef]
- Bergmann, J.; Friedel, P.; Kleeberg, R. BGMN—A new fundamental parameters based Rietveld program for laboratory X-ray sources, its use in quantitative analysis and structure investigations. CPD Newslett. 1998, 20, 5–8. [Google Scholar]
- Hölzer, G.; Fritsch, M.; Deutsch, M.; Härtwig, J.; Förster, E. Kα1,2 and Kβ1,3 X-ray emission lines of the 3d transition metals. Phys. Rev. A 1997, 56, 4554–4568. [Google Scholar] [CrossRef]
- Brindley, G.W. The effect of grain or particle size on X-ray reflections from mixed powder and alloys considered in relation to the quantitative determination of crystalline substances by X-ray methods. Philos. Mag. 1945, 36, 347–369. [Google Scholar] [CrossRef]
- Curtius, H.; Kaiser, G.; Müller, E.; Bosbach, D. Radionuclide release from research reactor spent fuel. J. Nucl. Mater. 2011, 416, 211–215. [Google Scholar] [CrossRef]
- Vettier, C.; Yelon, W.B. The structure of FeCl2 at high pressures. J. Phys. Chem. Solids 1975, 36, 401–405. [Google Scholar] [CrossRef]
- Curtius, H.; Kaiser, G.; Rozov, K.; Neumann, A.; Dardenne, K.; Bosbach, D. Preparation and characterization of Fe-, Co-, and Ni-containing Mg-Al-layered double hydroxides. Clays Clay Miner. 2013, 61, 424–439. [Google Scholar] [CrossRef]
- Rozov, K.; Curtius, H.; Bosbach, D. Preparation, characterization and thermodynamic properties of Zr-containing Cl-bearing layered double hydroxides (LDHs). Radiochim. Acta 2015, 103, 369–378. [Google Scholar] [CrossRef]
- Poonoosamy, J.; Brandt, F.; Stekiel, M.; Kegler, P.; Klinkenberg, M.; Winkler, B.; Vinograd, V.; Bosbach, D.; Deissmann, G. Zr-containing layered double hydroxides: Synthesis, characterization, and evaluation of thermodynamic properties. Appl. Clay Sci. 2018, 151, 54–65. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase (PDF-2 No./ICSD No.) | Weight/% | ||
|---|---|---|---|
| <2 µm | 2–63 µm | >63 µm | |
| Al2Cl(OH)5·2H2O lesukite (00-031-0006/-) | 95.37 ± 0.33 | 51.00 ± 2.75 | 13.87 ± 1.26 |
| (Mg0,67Al0,33(OH)2)·(CO3)0.165·(H2O)0.48 LDH 3R (01-089-0460/86655) | 1.43 ± 0.28 | 17.00 ± 4.08 | 7.37 ± 0.66 |
| Al2Mg4(OH)12 (CO3) (H2O)3 LDH 2H 00-020-0658/82874 | 12.35 ± 1.33 | 10.21 ± 0.96 | |
| ((Zn0,625 Al0,375) (OH)2) (SO4)0.188 LDH sulphate (01-070-6422/91859) | 0.25 ± 0.08 | 3.94 ± 0.28 | 1.26 ± 0.19 |
| (Fe(OH)2) ((OH)0.25 (H2O)0.5) green rust (00-040-0127/159700) | 0.20 ± 0.06 | 2.15 ± 0.21 | |
| Fe8O8(OH)8Cl1.35 akaganeite (00-034-1266/69606) | 2.05 ± 0.16 | 1.55 ± 0.15 | 0.65 ± 0.17 |
| FeO(OH) goethite (01-081-0462/245057) | 0.53 ± 0.08 | ||
| FeO(OH) lepidocrocite (01-070-8045/93948) | 0.73 ± 0.11 | ||
| FeCl2 lawrencite (01-070-1634/64830) | 0.68 ± 0.07 | ||
| U3Si2 uranium silizide (00-005-0628/73695) | 0.78 ± 0.07 | ||
| Fe iron (00-006-0696/84483) | 0.66 ± 0.10 | ||
| Amorphous | 0.90 ± 2.00 | 12.70 ± 5.30 | 62.36 ± 2.74 |
| Total | 100.00 | 100.00 | 100.00 |
| Relative fraction amount (%) | 20.30 | 8.20 | 71.50 |
| Rwp (%) | 12.21 | 8.20 | 0.95 * |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neumann, A.; Klinkenberg, M.; Curtius, H. Analysis of the Secondary Phases Formed by Corrosion of U3Si2-Al Research Reactor Fuel Elements in the Presence of Chloride Rich Brines. Materials 2018, 11, 1121. https://doi.org/10.3390/ma11071121
Neumann A, Klinkenberg M, Curtius H. Analysis of the Secondary Phases Formed by Corrosion of U3Si2-Al Research Reactor Fuel Elements in the Presence of Chloride Rich Brines. Materials. 2018; 11(7):1121. https://doi.org/10.3390/ma11071121
Chicago/Turabian StyleNeumann, Andreas, Martina Klinkenberg, and Hildegard Curtius. 2018. "Analysis of the Secondary Phases Formed by Corrosion of U3Si2-Al Research Reactor Fuel Elements in the Presence of Chloride Rich Brines" Materials 11, no. 7: 1121. https://doi.org/10.3390/ma11071121