Preparation of Poly-(Methyl vinyl ether-co-maleic Anhydride) Nanoparticles by Solution-Enhanced Dispersion by Supercritical CO2


Abstract
:1. Introduction
2. Results and Discussion
2.1. Particle Size and Particle Size Distribution of PVM/MA Particles
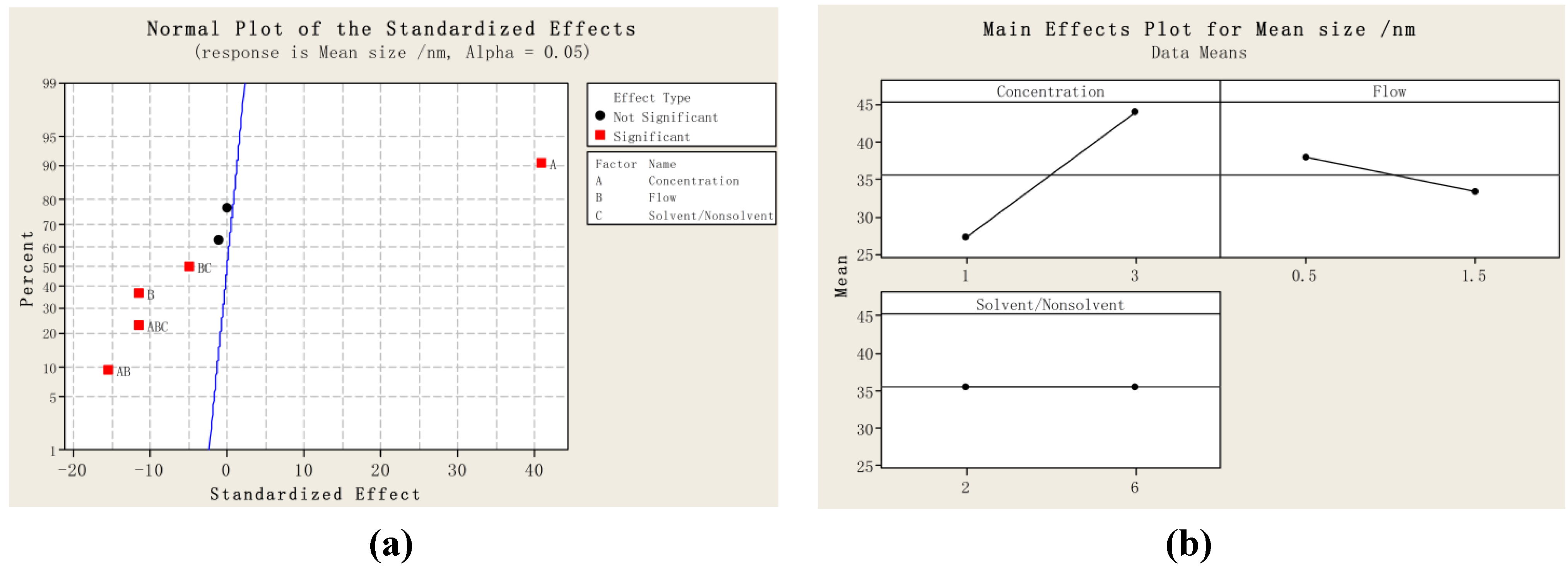
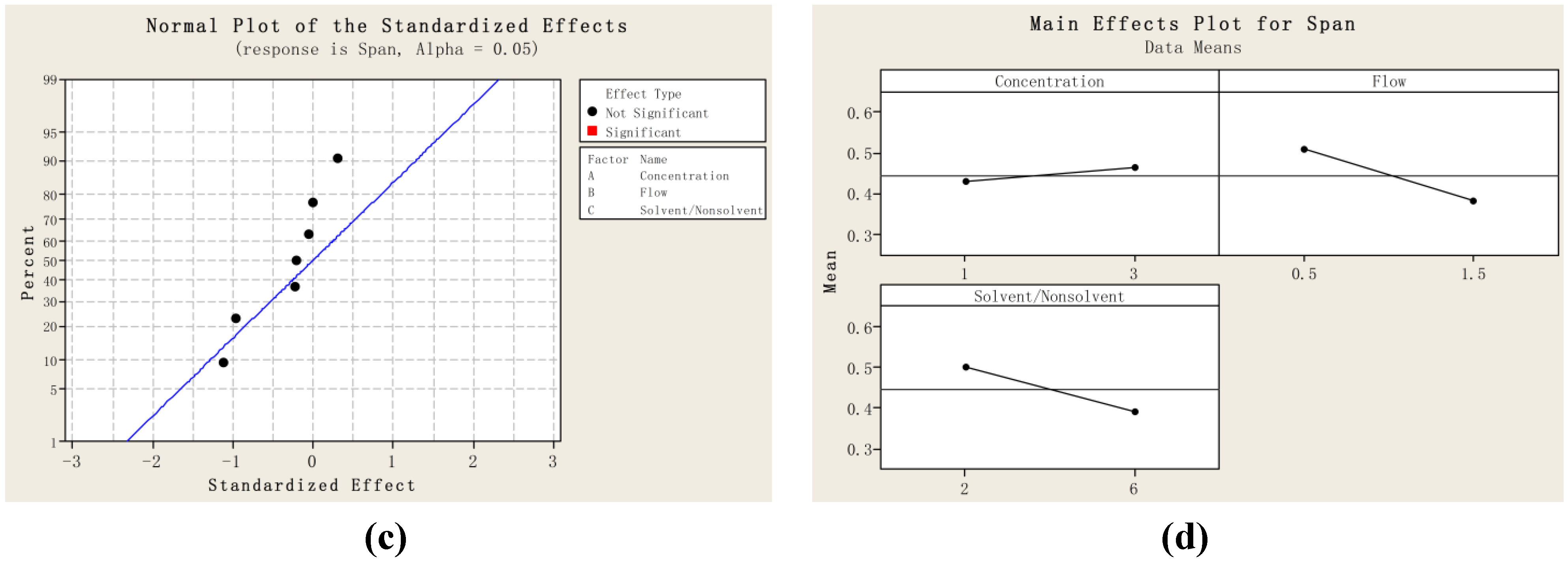

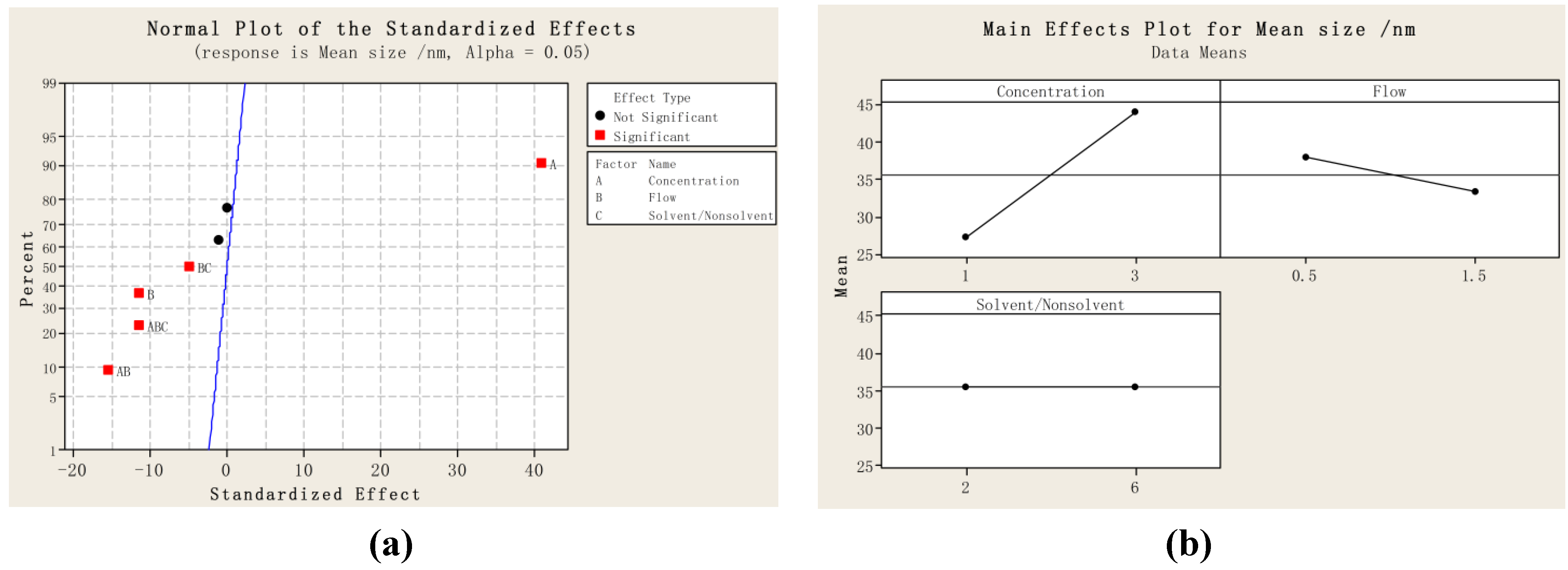{kind=link}
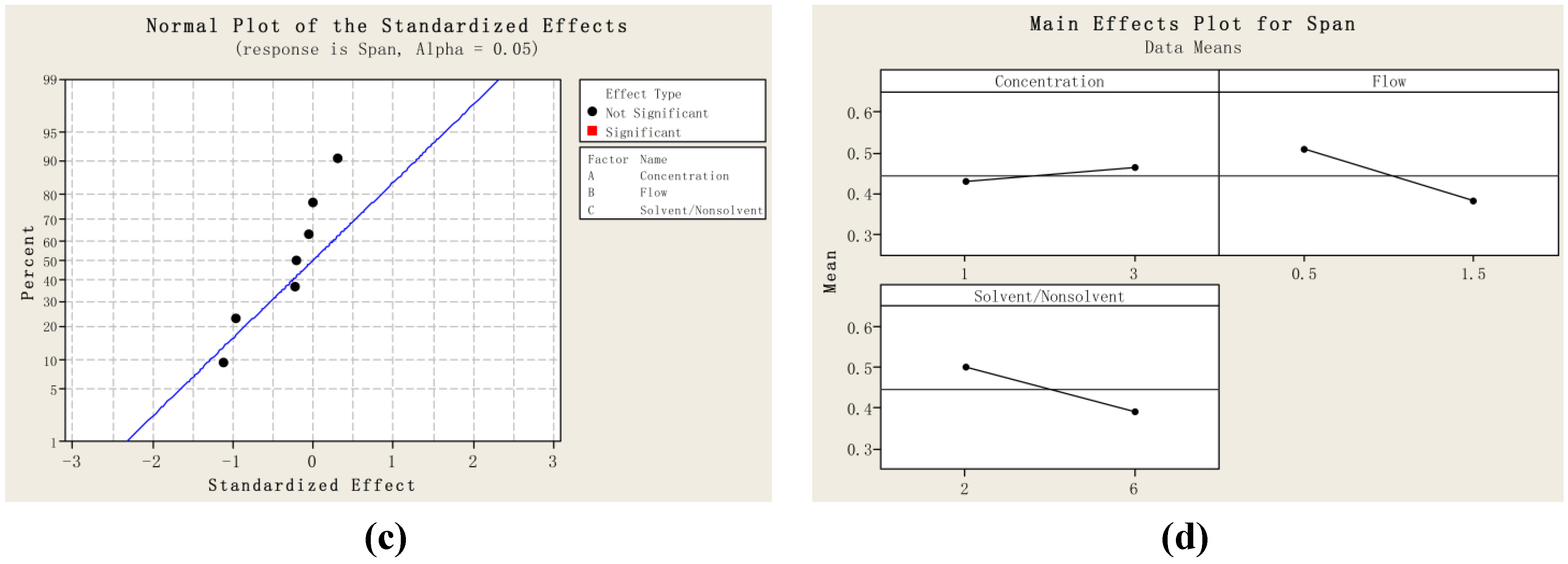{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run order | Mean size/nm | D10 | D50 | D90 | SPAN |
|---|---|---|---|---|---|
| 1 | 27.6 | 21.3 | 27.0 | 36.0 | 0.544 |
| 2 | 46.3 | 34.1 | 44.8 | 60.2 | 0.582 |
| 3 | 26.6 | 21.3 | 26.1 | 32.1 | 0.414 |
| 4 | 42.0 | 32.5 | 42.5 | 52.0 | 0.459 |
| 5 | 25.4 | 20.1 | 25.1 | 30.5 | 0.414 |
| 6 | 52.4 | 38.1 | 52.6 | 64.0 | 0.492 |
| 7 | 29.7 | 24.6 | 29.6 | 34.6 | 0.337 |
| 8 | 34.9 | 29.0 | 35.3 | 40.2 | 0.317 |
| 9 | 35.3 | 29.0 | 35.1 | 43.4 | 0.418 |
| 10 | 36.4 | 28.3 | 36.5 | 45.2 | 0.463 |
| 11 | 35.1 | 22.6 | 35.0 | 48.3 | 0.734 |
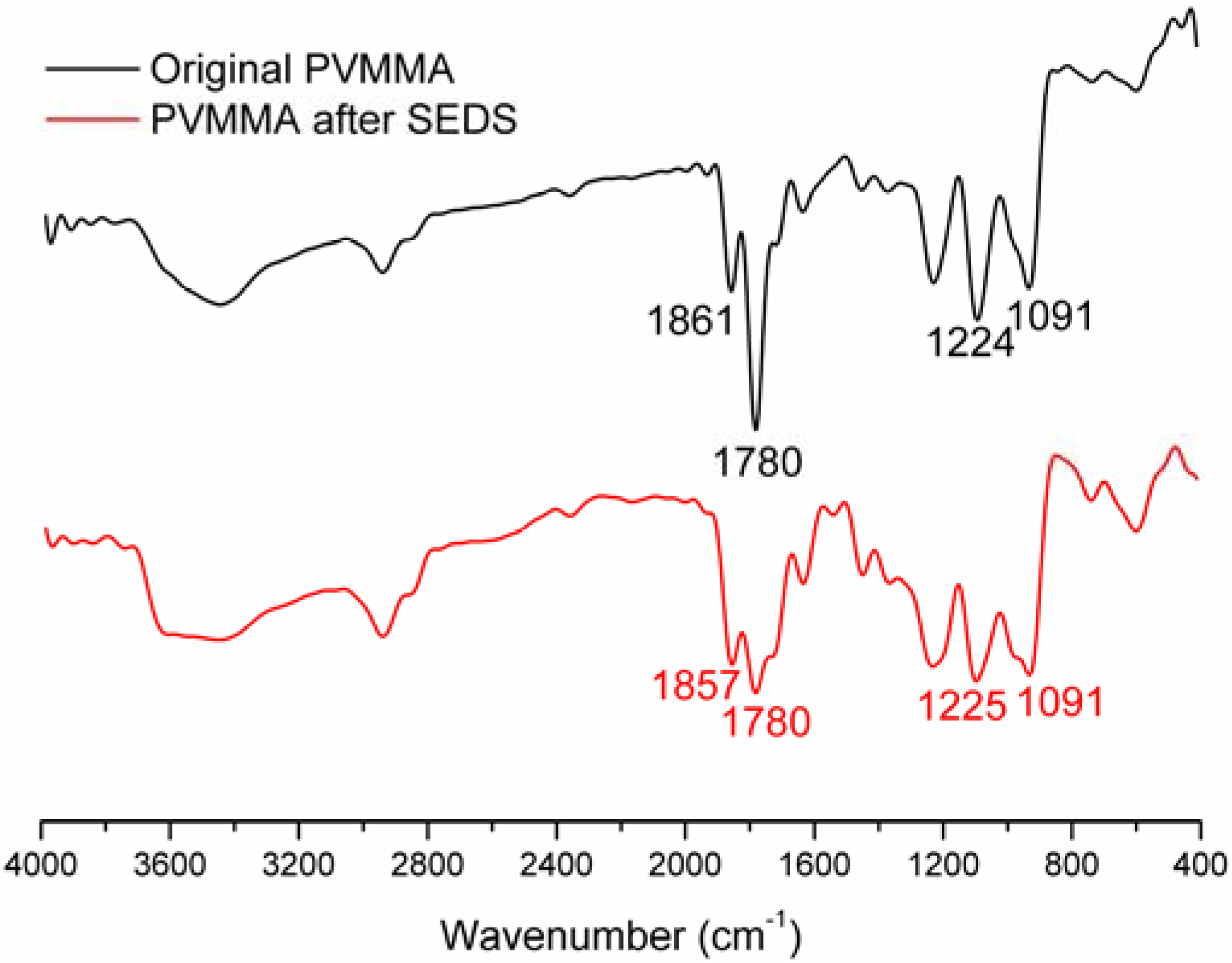
2.2. Physicochemical Characterization of PVM/MA Nanoparticles

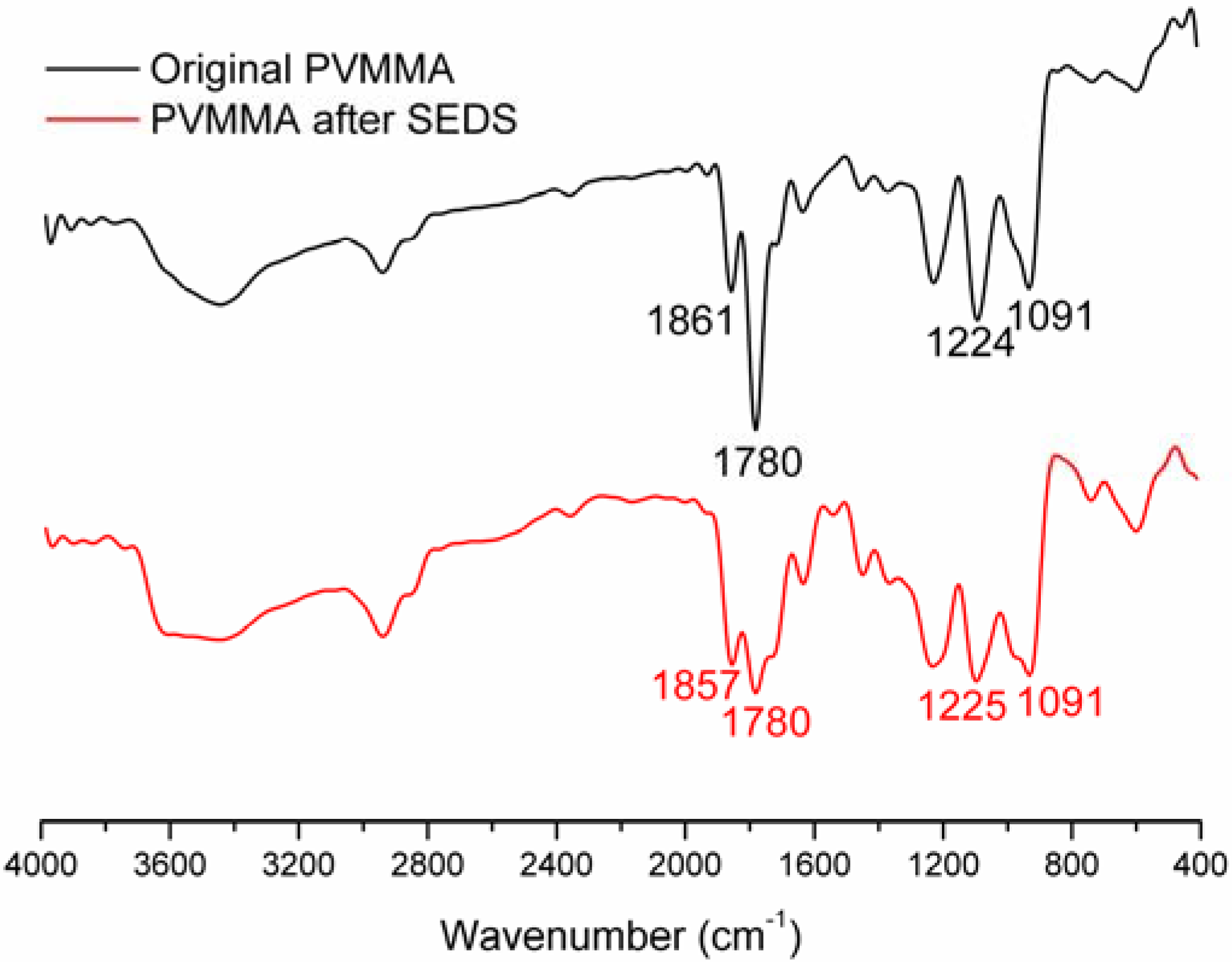
3. Experimental Section
3.1. Materials
3.2. Methods
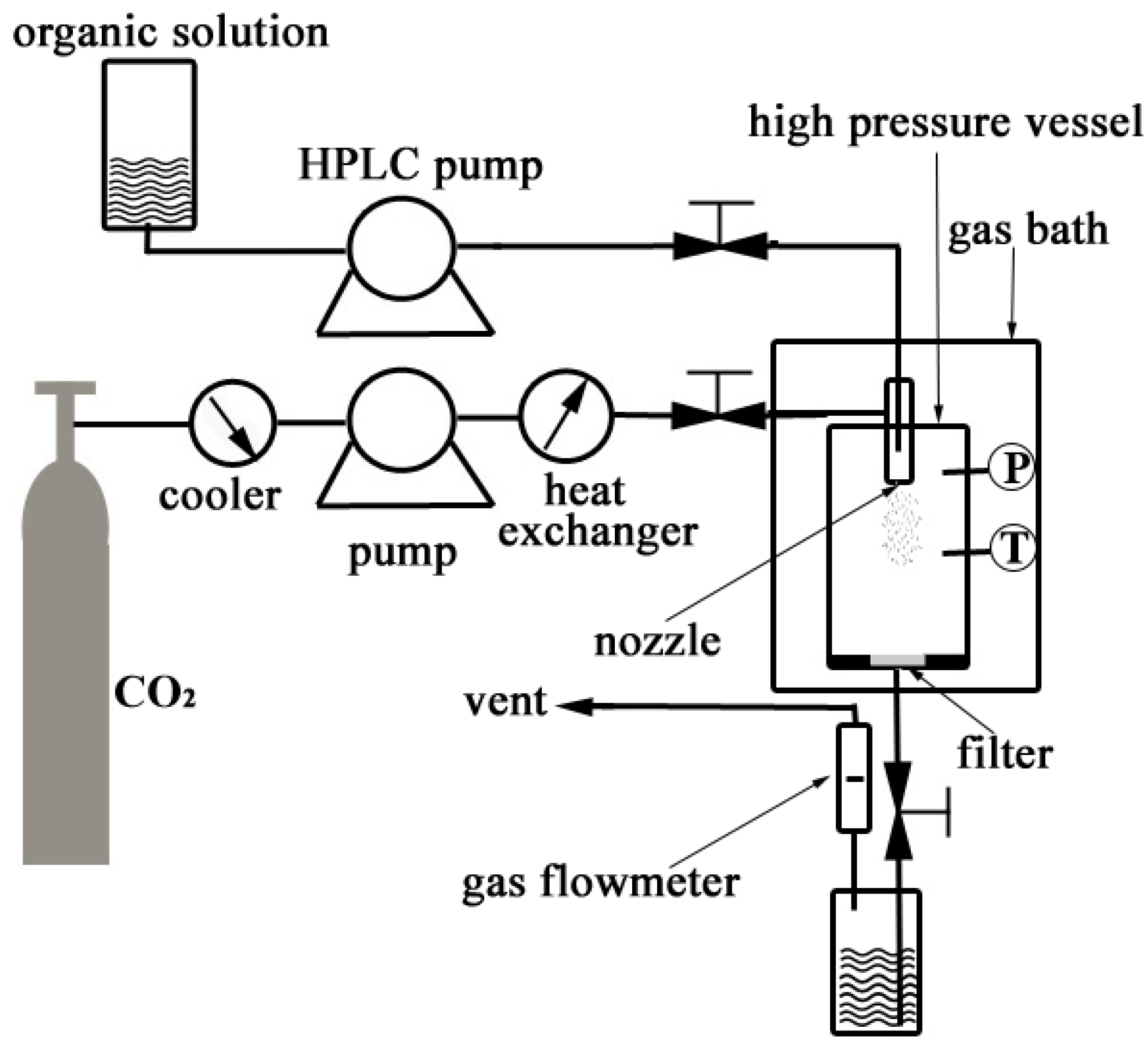
3.2.1. Nanonization of PVM/MA by the SEDS Process
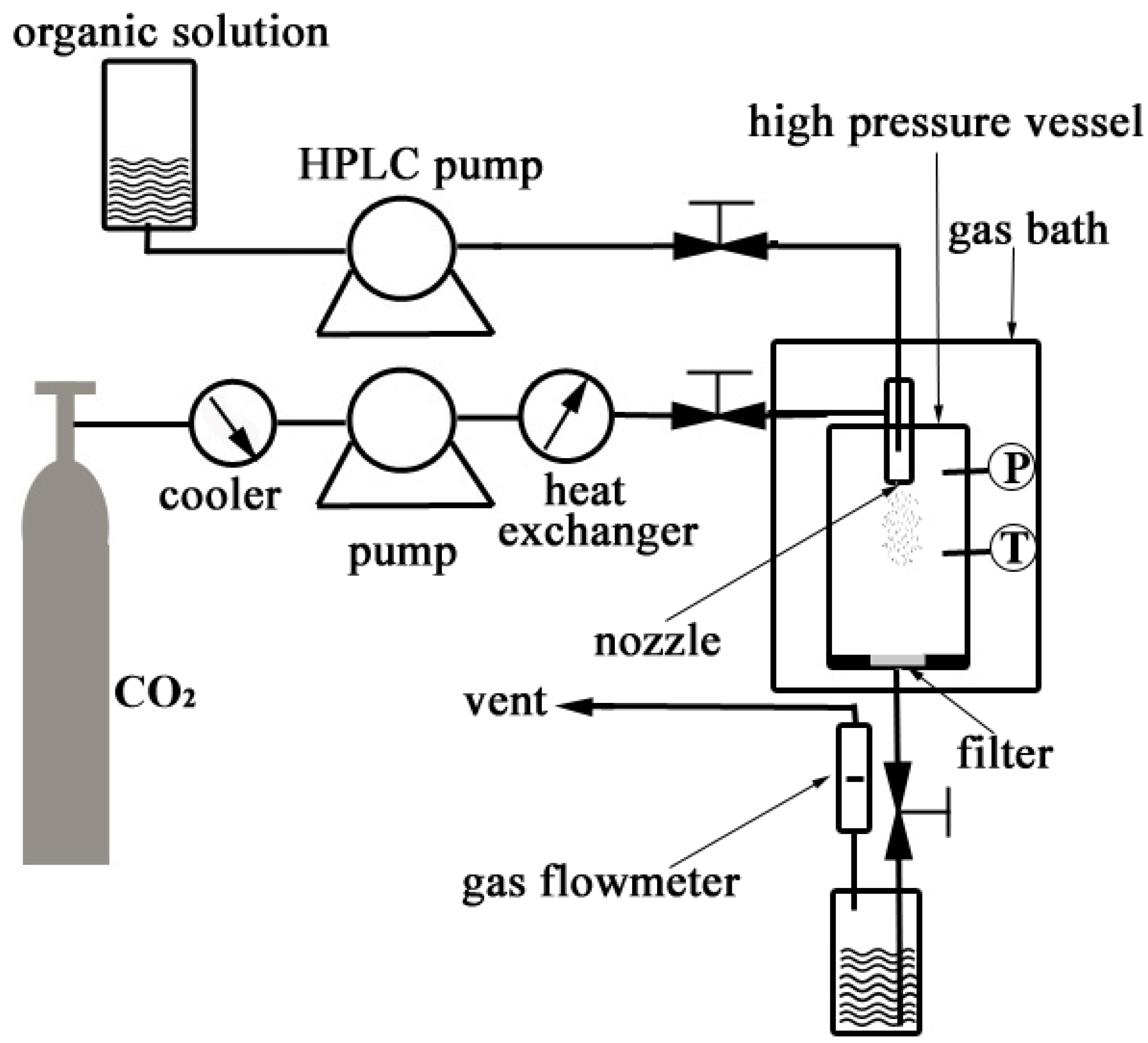
3.2.2. Experimental Design
| Run order | Center point | Blocks | Factor A concentration/% wt/v | Factor B Flow/mL min−1 | Factor C Solvent/nonsolvent |
|---|---|---|---|---|---|
| 1 | 1 | 1 | 1 | 0.5 | 2 |
| 2 | 1 | 1 | 3 | 0.5 | 2 |
| 3 | 1 | 1 | 1 | 1.5 | 2 |
| 4 | 1 | 1 | 3 | 1.5 | 2 |
| 5 | 1 | 1 | 1 | 0.5 | 6 |
| 6 | 1 | 1 | 3 | 0.5 | 6 |
| 7 | 1 | 1 | 1 | 1.5 | 6 |
| 8 | 1 | 1 | 3 | 1.5 | 6 |
| 9 | 1 | 1 | 2 | 1.0 | 4 |
| 10 | 1 | 1 | 2 | 1.0 | 4 |
| 11 | 1 | 1 | 2 | 1.0 | 4 |
3.2.3. Physicochemical Characterization of PVM/MA Nanoparticles
4. Conclusions
Acknowledgments
References
- Nguyen, D.; Hui, A.; Weeks, A.; Heynen, M.; Joyce, E.; Sheardown, H.; Jones, L. Release of ciprofloxacin-HCl and dexamethasone phosphate by hyaluronic acid containing silicone polymers. Materials 2012, 5, 684–698. [Google Scholar] [CrossRef]
- Saralidze, K.; Koole, L.H.; Knetsch, M.L.W. Polymeric microspheres for medical applications. Materials 2010, 3, 3537–3564. [Google Scholar] [CrossRef]
- Solaro, R.; Chiellini, F.; Battisti, A. Targeted delivery of protein drugs by nanocarriers. Materials 2010, 3, 1928–1980. [Google Scholar] [CrossRef]
- Finne, U.; Kyyrönen, K.; Urtti, A. Drug release from monoisopropyl ester of poly(vinyl methyl ether-maleic anhydride) can be modified by basic salts in the polymer matrix. J. Control. Release 1989, 10, 189–194. [Google Scholar] [CrossRef]
- Lendlein, A.; Langer, R. Biodegradable, elastic shape-memory polymers for potential biomedical applications. Science 2002, 296, 1673–1676. [Google Scholar] [CrossRef]
- Paluszkiewicz, C.; Stodolak-Zych, E.; Kwiatek, W.; Jelen, P. Bioactivity of a chitosan based nanocomposite. J. Biomim. Biomater. Tissue Eng. 2011, 10, 95–106. [Google Scholar] [CrossRef]
- Yan, H.; Jiang, W.; Zhang, Y.; Liu, Y.; Wang, B.; Yang, L.; Deng, L.; Singh, G.K.; Pan, J. Novel multi-biotin grafted poly(lactic acid) and its self-assembling nanoparticles capable of binding to streptavidin. Int. J. Nanomed. 2012, 7, 457–465. [Google Scholar]
- Hajiali, H.; Shahgasempour, S.; Naimi-Jamal, M.R.; Peirovi, H. Electrospun PGA/gelatin nanofibrous scaffolds and their potential application in vascular tissue engineering. Int. J. Nanomed. 2011, 6, 2133–2141. [Google Scholar] [CrossRef]
- Lanao, R.P.F.; Leeuwenburgh, S.C.G.; Wolke, J.G.C.; Jansen, J.A. Bone response to fast-degrading, injectable calcium phosphate cements containing PLGA microparticles. Biomaterials 2011, 32, 8839–8847. [Google Scholar] [CrossRef]
- Leon-Rodriguez, L.; Leiro-Vidal, J.; Blanco-Mendez, J.; Luzardo-Alvarez, A. Incorporation of PVMMA to PLGA MS enhances lectin grafting and their in vitro activity in macrophages. Int. J. Pharm. 2010, 402, 165–174. [Google Scholar] [CrossRef]
- Goetz, L.; Foston, M.; Mathew, A.P.; Oksman, K.; Ragauskas, A.J. Poly(methyl vinyl ether-co-maleic acid)-polyethylene glycol nanocomposites cross-linked in situ with cellulose nanowhiskers. Biomacromolecules 2010, 11, 2660–2666. [Google Scholar] [CrossRef]
- Garland, M.J.; Singh, T.R.R.; Woolfson, A.D.; Donnelly, R.F. Electrically enhanced solute permeation across poly(ethylene glycol)-crosslinked poly(methyl vinyl ether-co-maleic acid) hydrogels: Effect of hydrogel crosslink density and ionic conductivity. Int. J. Pharm. 2011, 406, 91–98. [Google Scholar] [CrossRef]
- Elizondo, E.; Cordoba, A.; Sala, S.; Ventosa, N.; Veciana, J. Preparation of biodegradable poly(methyl vinyl ether-co-maleic anhydride) nanostructured microparticles by precipitation with a compressed antisolvent. J. Supercrit. Fluid. 2010, 53, 108–114. [Google Scholar] [CrossRef]
- Cerchiara, T.; Luppi, B.; Chidichimo, G.; Bigucci, F.; Zecchi, V. Chitosan and poly(methyl vinyl ether-co-maleic anhydride) microparticles as nasal sustained delivery systems. Eur. J. Pharm. Biopharm. 2005, 61, 195–200. [Google Scholar] [CrossRef]
- Yoncheva, K.; Lizarraga, E.; Irache, J.M. Pegylated nanoparticles based on poly(methyl vinyl ether-co-maleic anhydride): Preparation and evaluation of their bioadhesive properties. Eur. J. Pharm. Sci. 2005, 24, 411–419. [Google Scholar] [CrossRef]
- Wong, T.W.; Wahab, S.; Anthony, Y. Drug release responses of zinc ion crosslinked poly(methyl vinyl ether-co-maleic acid) matrix towards microwave. Int. J. Pharm. 2008, 357, 154–163. [Google Scholar] [CrossRef]
- Singh, T.R.R.; Woolfson, A.D.; Donnelly, R.F. Swelling, network parameters and permeability of poly(ethylene glycol)-crosslinked poly(methyl vinyl ether-co-maleic anhydride) hydrogels containing pore-forming agent. J. Pharm. Pharmacol. 2010, 62, 1305–1306. [Google Scholar] [CrossRef]
- Beck-Broichsitter, M.; Schweiger, C.; Schmehl, T.; Gessler, T.; Seeger, W.; Kissel, T. Characterization of novel spray-dried polymeric particles for controlled pulmonary drug delivery. J. Control. Release 2012, 158, 329–335. [Google Scholar] [CrossRef]
- Jelvehgari, M.; Barar, J.; Valizadeh, H.; Shadrou, S.; Nokhodchi, A. Formulation, characterization and in vitro evaluation of theophylline-loaded Eudragit RS 100 microspheres prepared by an emulsion-solvent diffusion/evaporation technique. Pharm. Dev. Technol. 2011, 16, 637–644. [Google Scholar] [CrossRef]
- Chen, A.Z.; Li, Y.; Chen, D.; Hu, J.Y. Development of core-shell microcapsules by a novel supercritical CO2 process. J. Mater. Sci. Mater. Med. 2009, 20, 751–758. [Google Scholar] [CrossRef]
- Chen, A.Z.; Zhao, Z.; Wang, S.B.; Li, Y.; Zhao, C.; Liu, Y.G. A continuous RESS process to prepare PLA-PEG-PLA microparticles. J. Supercrit. Fluid. 2011, 59, 92–97. [Google Scholar] [CrossRef]
- Kalani, M.; Yunus, R.; Abdullah, N. Optimizing supercritical antisolvent process parameters to minimize the particle size of paracetamol nanoencapsulated in L-polylactide. Int. J. Nanomed. 2011, 6, 1101–1105. [Google Scholar] [CrossRef]
- Chen, A.Z.; Li, Y.; Chau, F.T.; Lau, T.Y.; Hu, J.Y.; Zhao, Z.; Mok, D.K.W. Microencapsulation of puerarin nanoparticles by poly(L-lactide) in a supercritical CO2 process. Acta Biomater. 2009, 5, 2913–2919. [Google Scholar] [CrossRef]
- Torino, E.; de Marco, I.; Reverchon, E. Organic nanoparticles recovery in supercritical antisolvent precipitation. J. Supercrit. Fluid. 2010, 55, 300–306. [Google Scholar] [CrossRef]
- Chen, A.Z.; Pu, X.M.; Kang, Y.Q.; Liao, L.; Yao, Y.D.; Yin, G.F. Preparation of 5-fluorouracil-poly(L-lactide) microparticles using solution-enhanced dispersion by supercritical CO2. Macromol. Rapid Commun. 2006, 27, 1254–1259. [Google Scholar] [CrossRef]
- Reverchon, E. Supercritical antisolvent precipitation of micro- and nano-particles. J. Supercrit. Fluid. 1999, 15, 1–21. [Google Scholar] [CrossRef]
- Reverchon, E.; de Marco, I. Mechanisms controlling supercritical antisolvent precipitate morphology. Chem. Eng. J. 2011, 169, 358–370. [Google Scholar] [CrossRef]
- Kalani, M.; Yunus, R. Application of supercritical antisolvent method in drug encapsulation: A review. Int. J. Nanomed. 2011, 6, 1429–1442. [Google Scholar] [CrossRef]
- De Diego, Y.P.; Pellikaan, H.C.; Wubbolts, F.E.; Borchard, G.; Witkamp, G.J.; Jansens, P.J. Opening new operating windows for polymer and protein micronisation using the PCA process. J. Supercrit. Fluid. 2006, 36, 216–224. [Google Scholar] [CrossRef]
- Rantakylä, M.; Jäntti, M.; Aaltonen, O.; Hurme, M. The effect of initial drop size on particle size in the supercritical antisolvent precipitation (SAS) technique. J. Supercrit. Fluid. 2002, 24, 251–263. [Google Scholar] [CrossRef]
- Chen, A.Z.; Li, Y.; Chau, F.T.; Lau, T.Y.; Hu, J.Y.; Zhao, Z.; Mok, D.K.W. Application of organic nonsolvent in the process of solution-enhanced dispersion by supercritical CO2 to prepare puerarin fine particles. J. Supercrit. Fluid. 2009, 49, 394–402. [Google Scholar] [CrossRef]
- Knez, Z.; Skerget, M.; Ilic, L.; Lutge, C. Vapor-liquid equilibrium of binary CO2-organic solvent systems (ethanol, tetrahydrofuran, ortho-xylene, meta-xylene, para-xylene). J. Supercrit. Fluid. 2008, 43, 383–389. [Google Scholar] [CrossRef]
- Stievano, M.; Elvassore, N. High-pressure density and vapor-liquid equilibrium for the binary systems carbon dioxide-ethanol, carbon dioxide-acetone and carbon dioxide–dichloromethane. J. Supercrit. Fluid. 2005, 33, 7–14. [Google Scholar] [CrossRef]
- Idrissi, A.; Vyalov, I.; Kiselev, M.; Jedlovszky, P. Assessment of the potential models of acetone/CO2 and ethanol/CO2 mixtures by computer simulation and thermodynamic integration in liquid and supercritical states. Phys. Chem. Chem. Phys. 2011, 13, 16272–16281. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Day, C.Y.; Ko, C.M.; Chiu, K.L. Densities and P-x-y diagrams for carbon dioxide dissolution in methanol, ethanol, and acetone mixtures. Fluid Phase Equilibr. 1997, 131, 243–258. [Google Scholar] [CrossRef]
© 2012 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, A.-Z.; Wang, G.-Y.; Wang, S.-B.; Feng, J.-G.; Liu, Y.-G.; Kang, Y.-Q. Preparation of Poly-(Methyl vinyl ether-co-maleic Anhydride) Nanoparticles by Solution-Enhanced Dispersion by Supercritical CO2. Materials 2012, 5, 1841-1852. https://doi.org/10.3390/ma5101841
Chen A-Z, Wang G-Y, Wang S-B, Feng J-G, Liu Y-G, Kang Y-Q. Preparation of Poly-(Methyl vinyl ether-co-maleic Anhydride) Nanoparticles by Solution-Enhanced Dispersion by Supercritical CO2. Materials. 2012; 5(10):1841-1852. https://doi.org/10.3390/ma5101841
Chicago/Turabian StyleChen, Ai-Zheng, Guang-Ya Wang, Shi-Bin Wang, Jian-Gang Feng, Yuan-Gang Liu, and Yong-Qiang Kang. 2012. "Preparation of Poly-(Methyl vinyl ether-co-maleic Anhydride) Nanoparticles by Solution-Enhanced Dispersion by Supercritical CO2" Materials 5, no. 10: 1841-1852. https://doi.org/10.3390/ma5101841
APA StyleChen, A.-Z., Wang, G.-Y., Wang, S.-B., Feng, J.-G., Liu, Y.-G., & Kang, Y.-Q. (2012). Preparation of Poly-(Methyl vinyl ether-co-maleic Anhydride) Nanoparticles by Solution-Enhanced Dispersion by Supercritical CO2. Materials, 5(10), 1841-1852. https://doi.org/10.3390/ma5101841