Metal Phosphate-Supported Pt Catalysts for CO Oxidation


Abstract
:1. Introduction
2. Results and Discussion
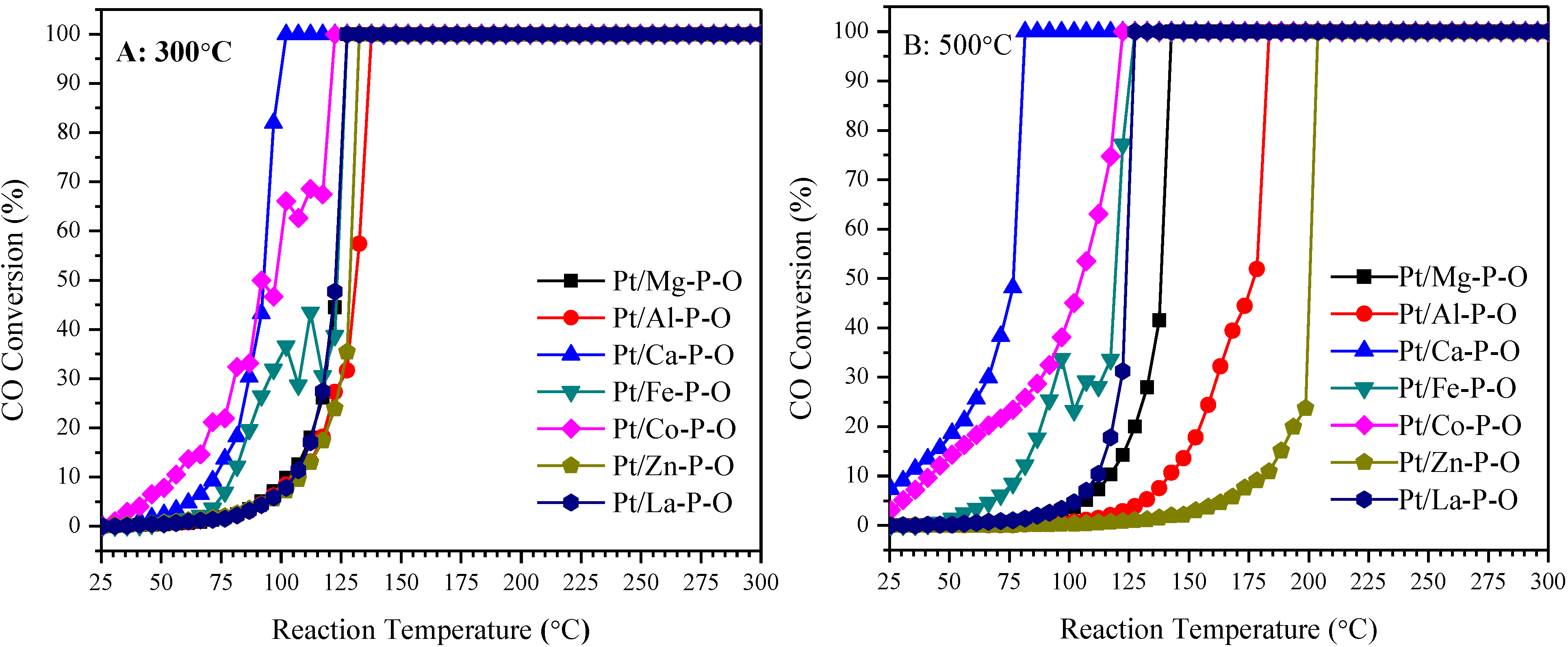
2.1. Catalytic Activity of Pt/M-P-O

2.2. BET Surface Areas and Pt Contents
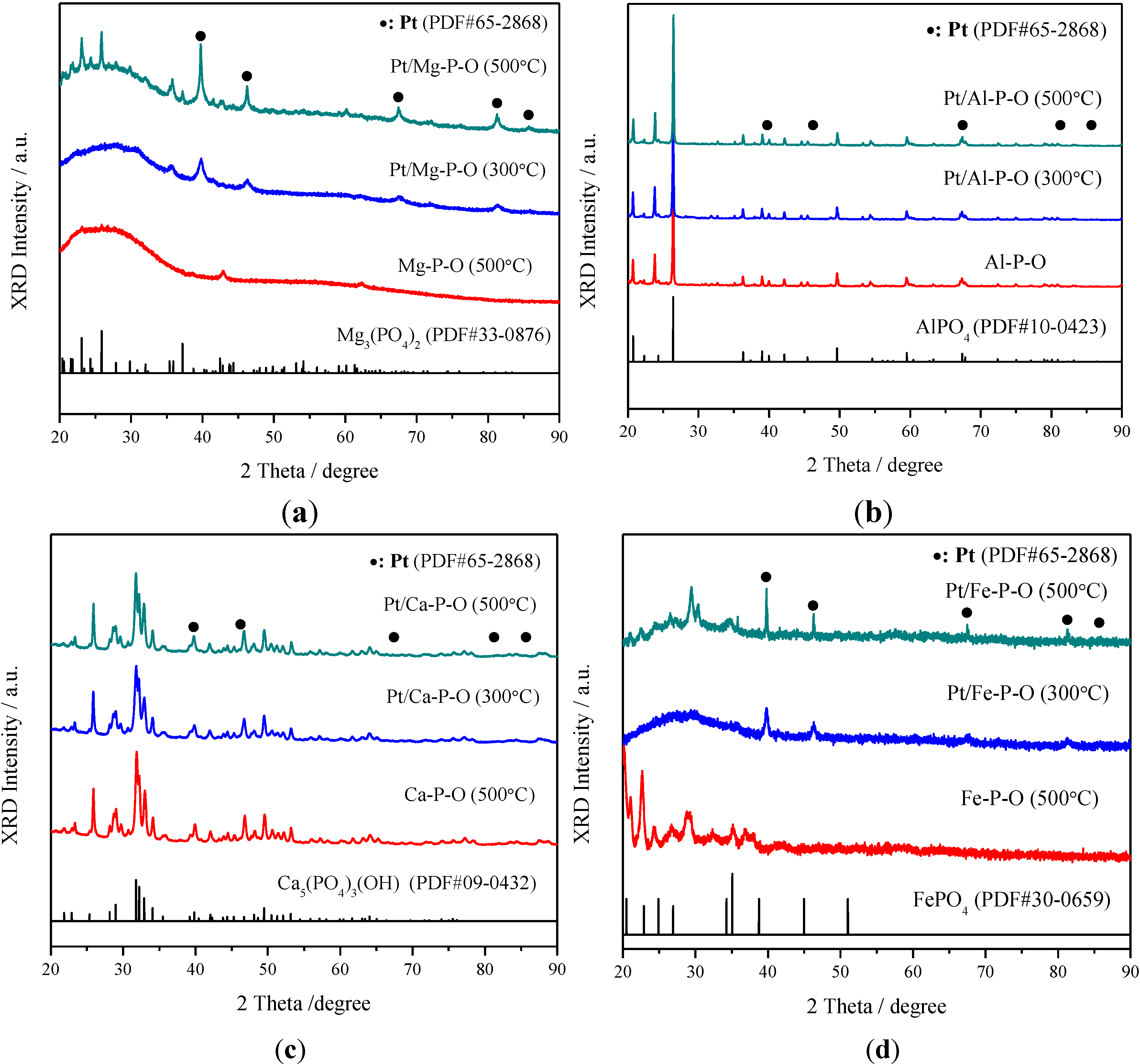
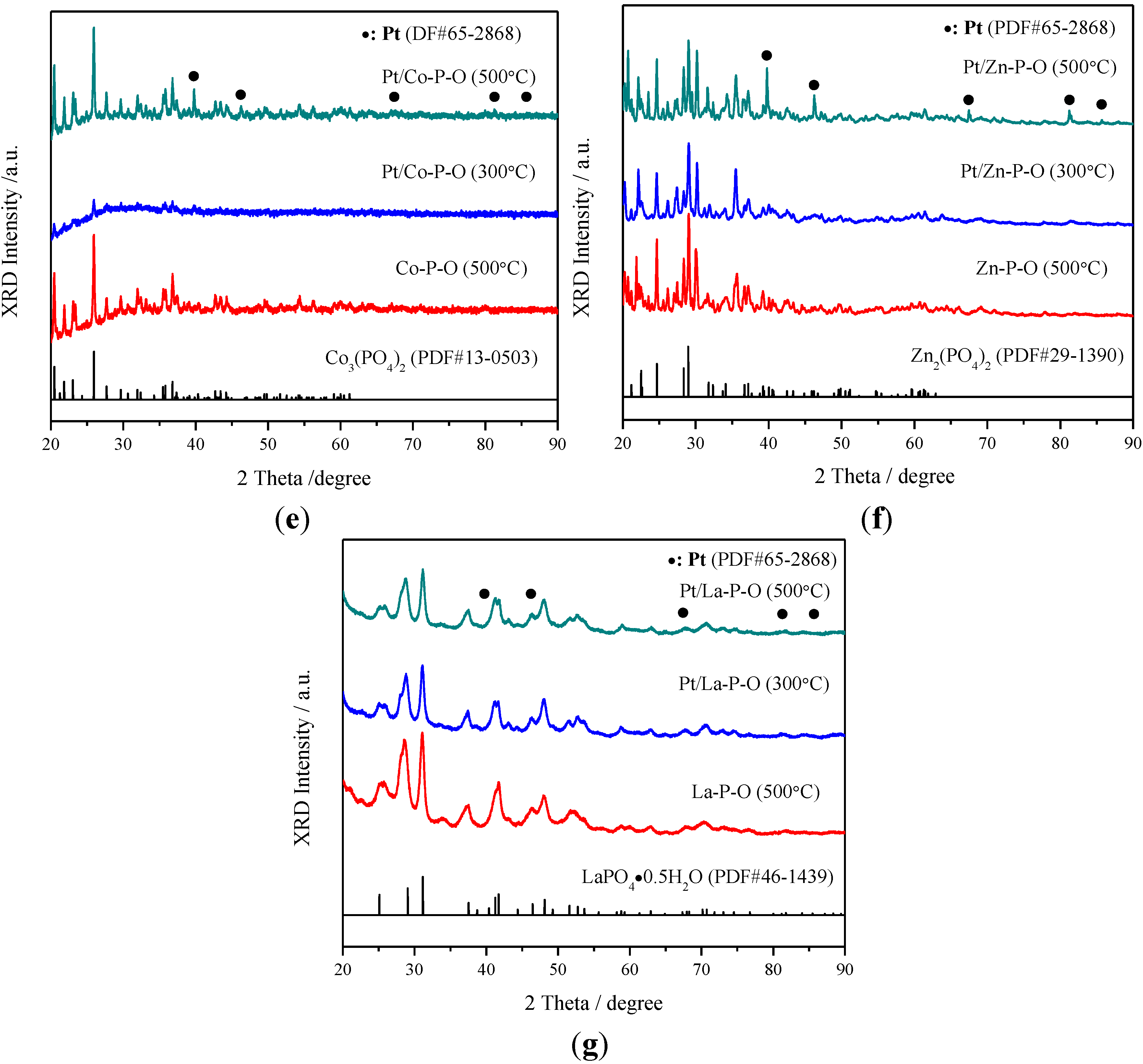
2.3. XRD Characterization


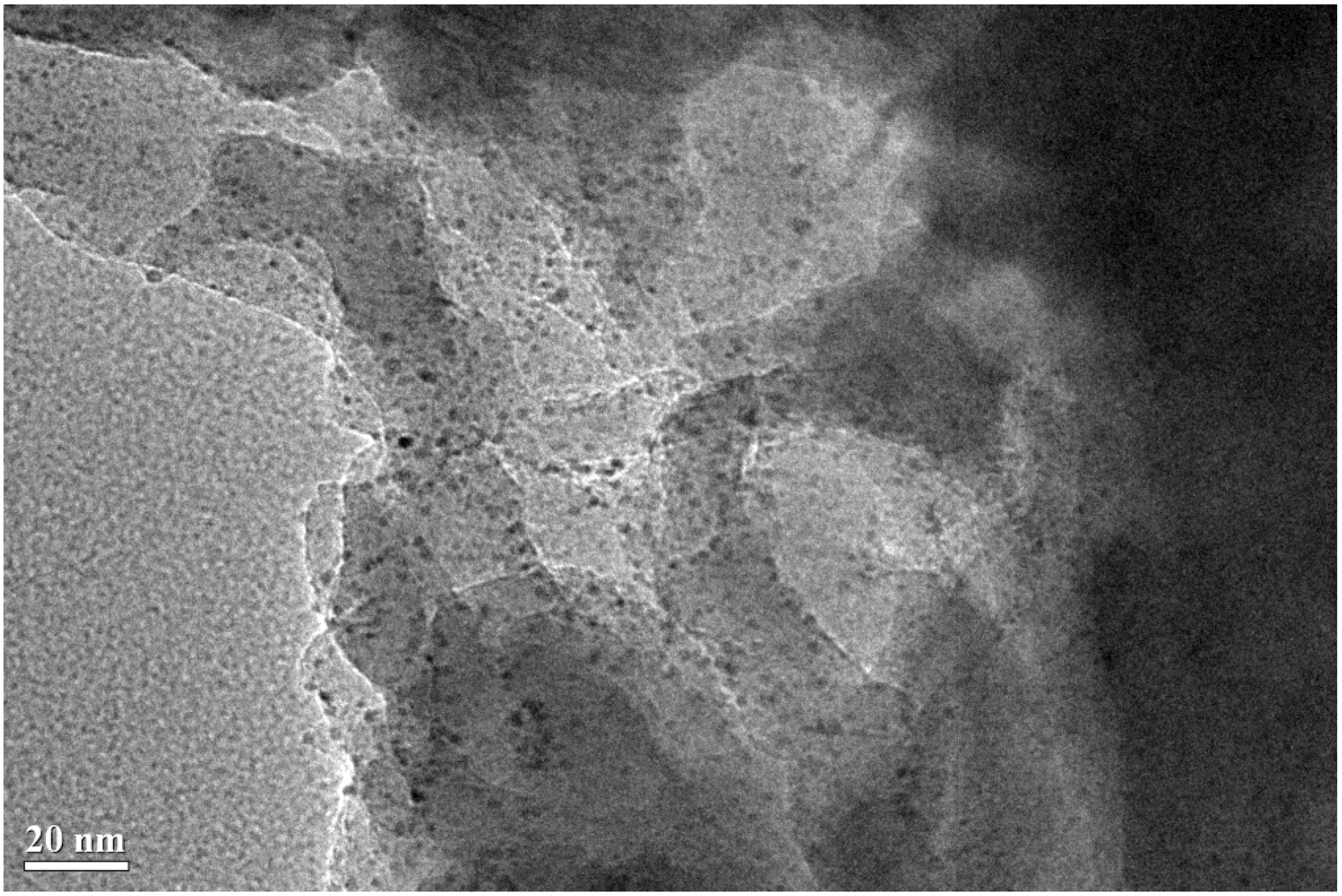
2.4. TEM Characterization



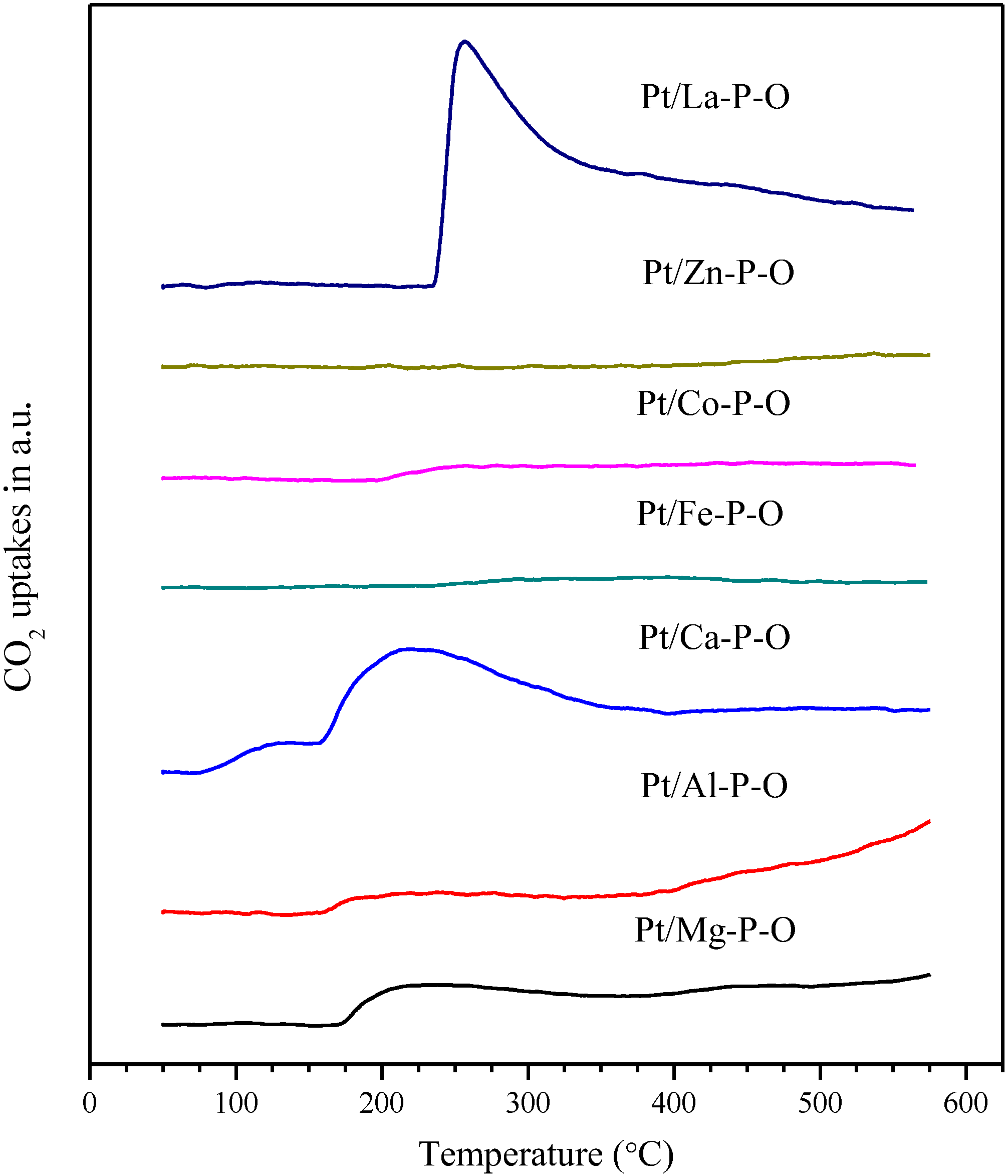
2.5. CO2-TPD



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | T50 (°C) | SBET (m2/g) | Pt content (%) | Average Pt particle size (nm) | Basic sites (μmol/g) | O1s (%) | ||
|---|---|---|---|---|---|---|---|---|
| OB | ONB | OOH | ||||||
| Pt/Mg-P-O | 140 | 24.4 | 0.49 | 2–5, some are very big | 26.6 | 29.8 | 57.0 | 13.2 |
| Pt/Al-P-O | 178 | 5.2 | 1.35 | 2–3 | 9.1 | 30.2 | 61.2 | 8.6 |
| Pt/Ca-P-O | 76 | 47.1 | 0.85 | 2–3 | 76.8 | 16.6 | 45.5 | 37.9 |
| Pt/Fe-P-O | 120 | 8.7 | 1.42 | 2–5, some are big (10 nm) | 0 | 29.6 | 57.1 | 13.3 |
| Pt/Co-P-O | 107 | 7.7 | 1.43 | 2–5, some are big (10 nm) | 1.3 | 35.1 | 52.1 | 12.8 |
| Pt/Zn-P-O | 202 | <1 | 1.23 | very big | 0 | 20.1 | 66.8 | 13.1 |
| Pt/La-P-O | 123 | 71.3 | 1.45 | 2–3 | 147.9 | 32.2 | 59.0 | 8.8 |
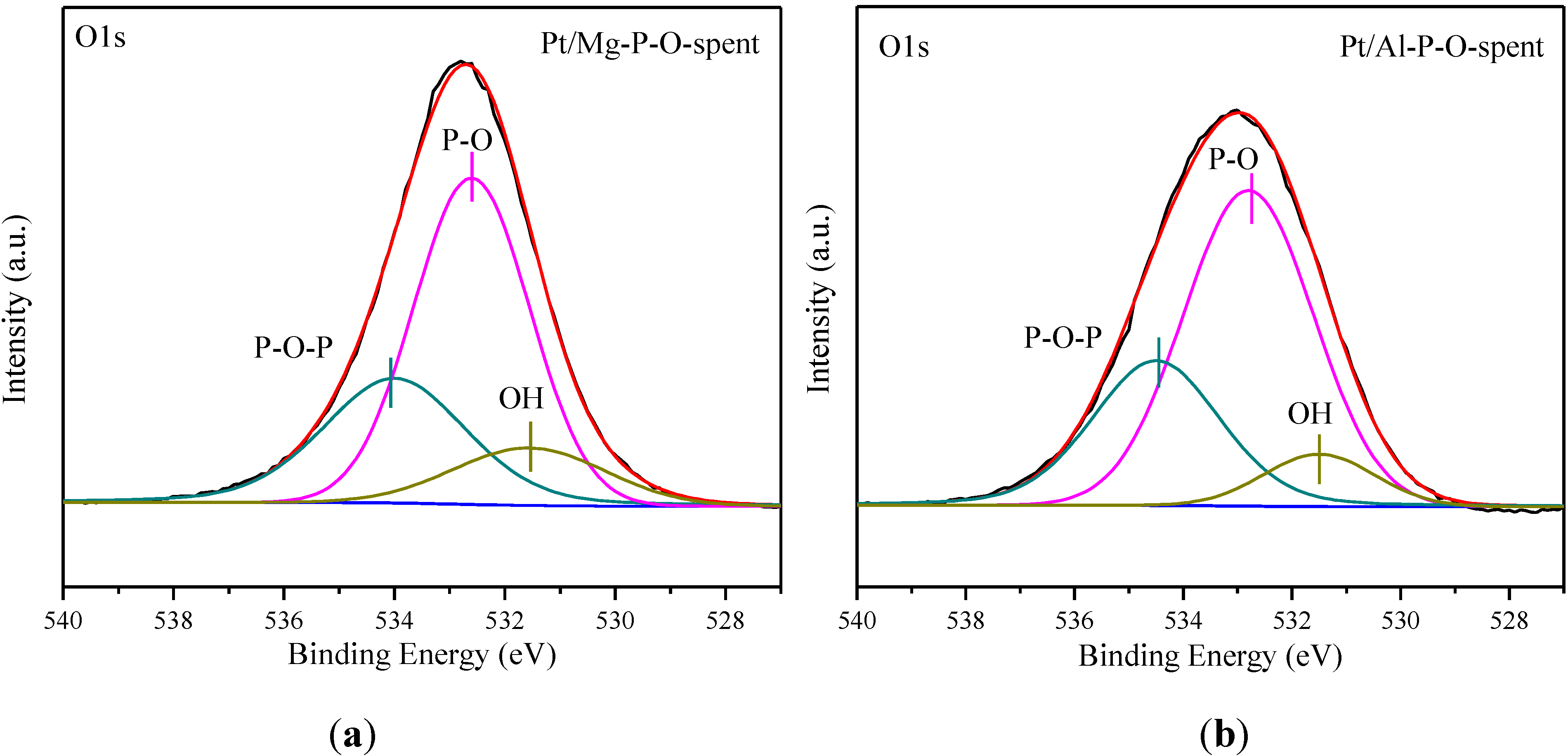
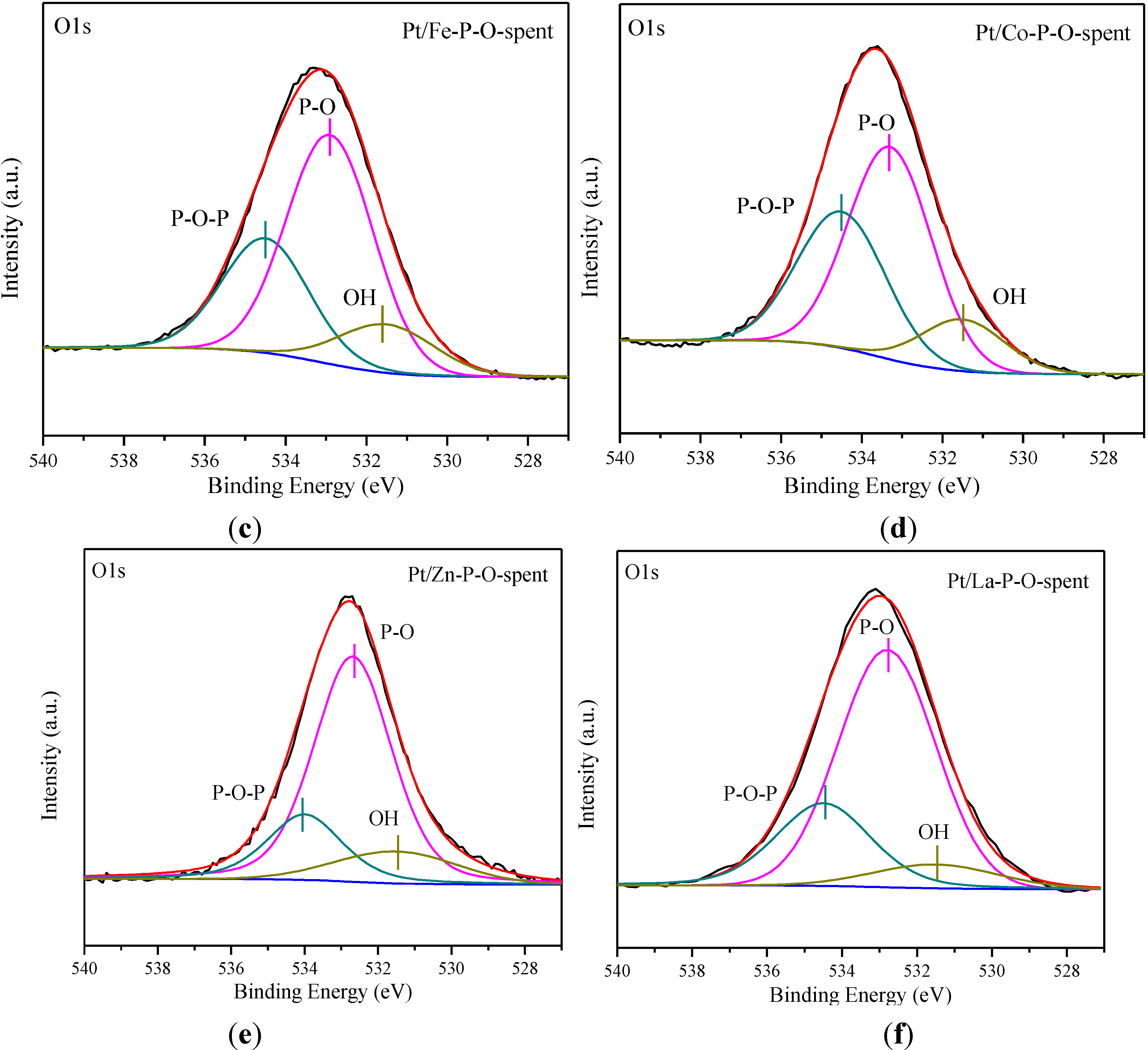
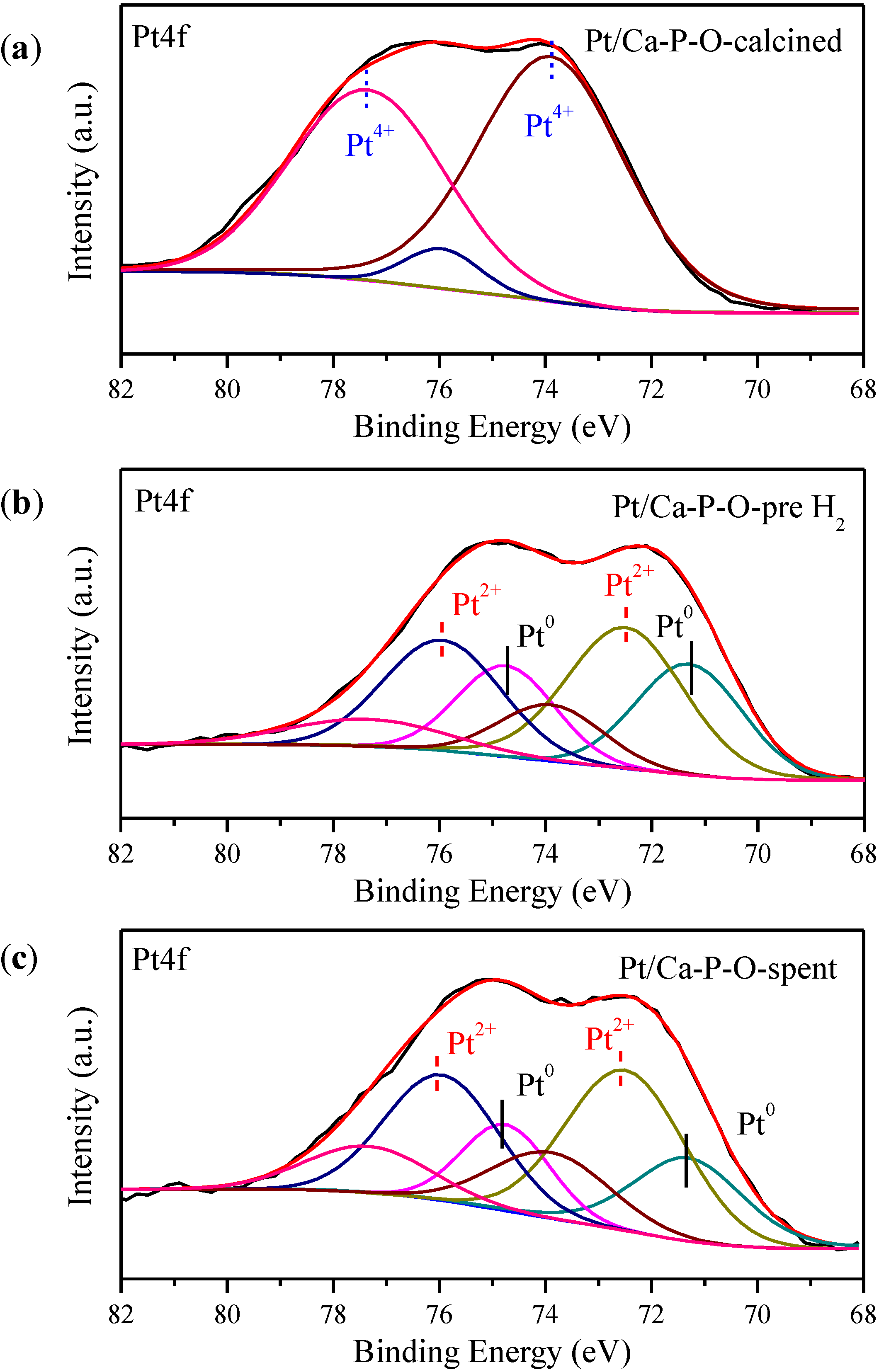
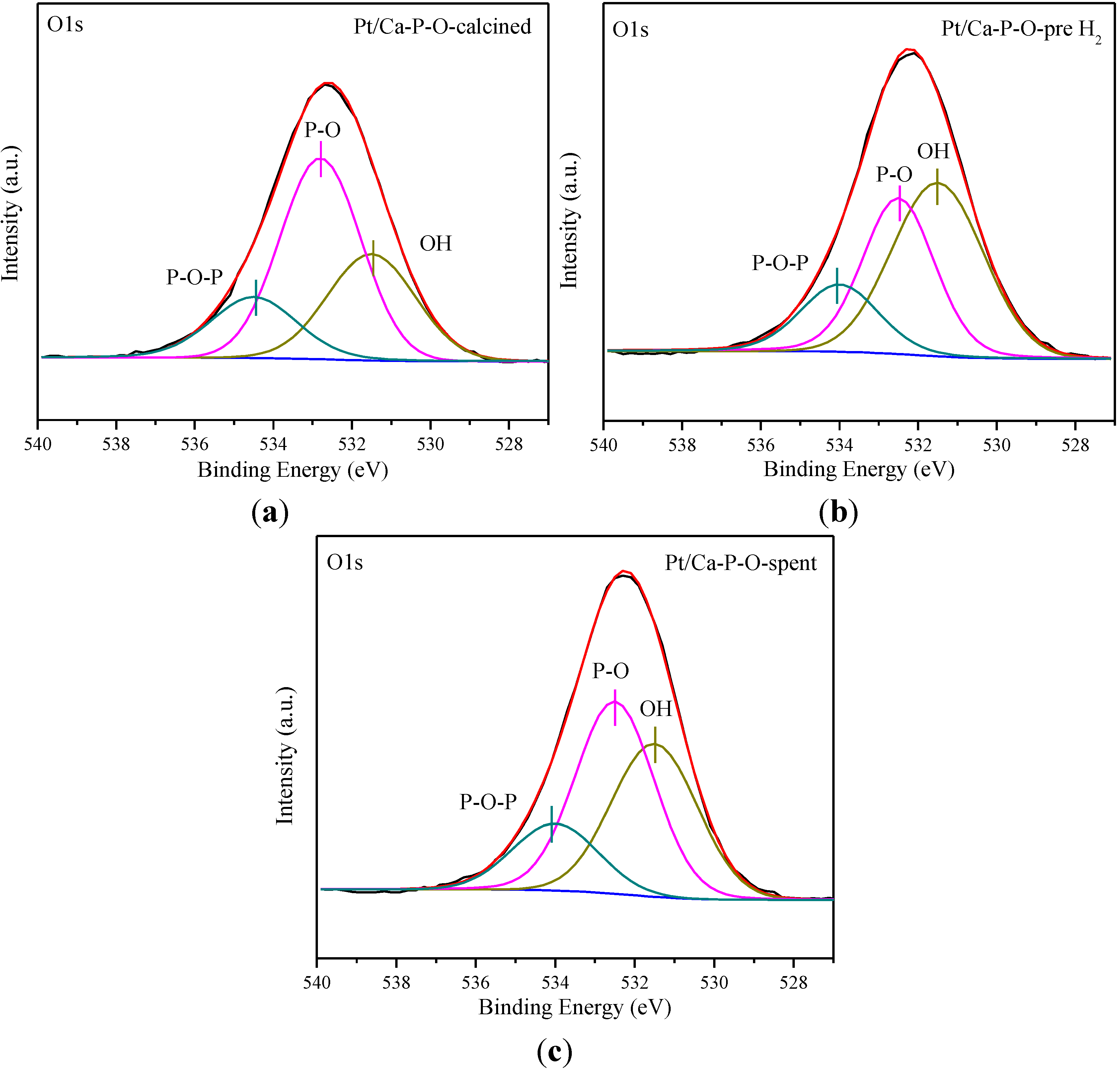
2.6. XPS Characterization



| Sample | Ca/P | Pt0/Pttotal | Pt2+/Pttotal | Pt4+/Pttotal | OOH (%) | Cl/Pt |
|---|---|---|---|---|---|---|
| Pt/Ca-P-O-calcined | 1.29 | 0 | 0.092 | 0.908 | 30.1 | 1.39 |
| Pt/Ca-P-O-pre H2 | 1.29 | 0.338 | 0.495 | 0.167 | 46.1 | 1.26 |
| Pt/Ca-P-O-spent | 1.38 | 0.272 | 0.517 | 0.211 | 37.9 | 1.22 |
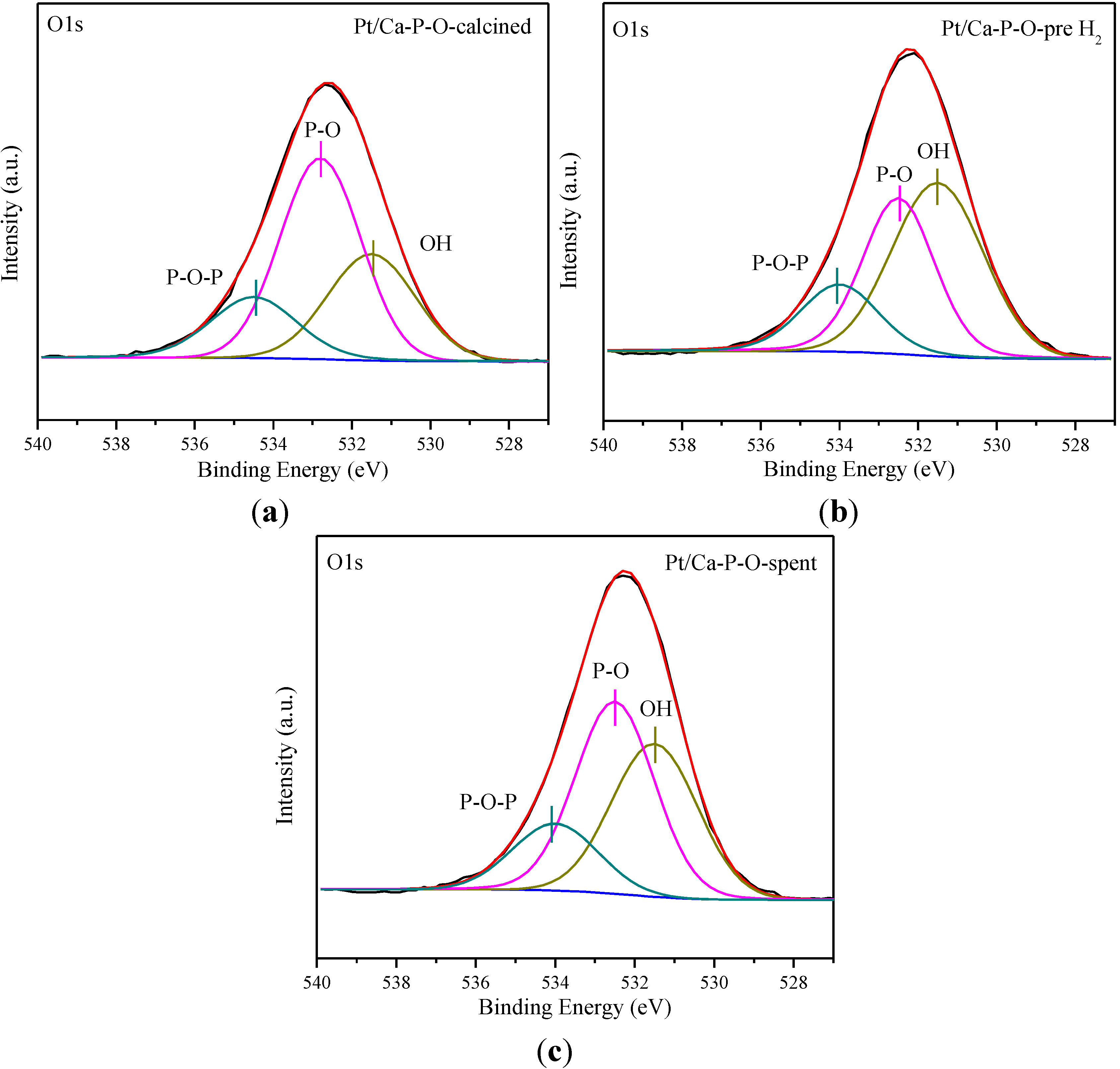
2.7. Discussion
3. Experimental Section
3.1. Synthesis of Catalysts
3.2. Characterization
3.3. Catalytic Testing
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ma, Z.; Zaera, F. Heterogeneous catalysis by metals. In Encyclopedia of Inorganic and Bioinorganic Chemistry; Scott, R.A., Ed.; John Wiley &Sons: Chichester, West Sussex, UK, 2014; eibc0079; pp. 1–16. [Google Scholar]
- Ma, Z.; Tao, F. Metal salt-based gold nanocatalysts. In Metal Nanoparticles for Catalysis: Advances and Applications; Tao, F., Ed.; the Royal Society of Chemistry: London, UK, 2014; pp. 157–171. [Google Scholar]
- Tanabe, K.; Misono, M.; Ono, Y.; Hattori, H. New Acid and Bases; Elsevier: Amsterdam, The Netherlands, 1989. [Google Scholar]
- Takita, Y.; Yamashita, H.; Moritaka, K. Selective partial oxidation of propane over metal phosphate catalysts. Chem. Lett. 1989, 18, 1733–1736. [Google Scholar] [CrossRef]
- Takita, Y.; Sano, K.; Kurosaki, K.; Kawata, N.; Nishiguchi, H.; Ito, M.; Ishihara, T. Oxidative dehydrogenation of iso-butane to iso-butene—I. Metal phosphate catalysts. Appl. Catal. A 1998, 167, 49–56. [Google Scholar] [CrossRef]
- Takita, Y.; Sano, K.; Muraya, T.; Nishiguchi, H.; Kawata, N.; Ito, M.; Akbay, T.; Ishihara, T. Oxidative dehydrogenation of iso-butane to iso-butene—II. Rare earth phosphate catalysts. Appl. Catal. A 1998, 170, 23–31. [Google Scholar] [CrossRef]
- Takita, Y.; Qing, X.; Takami, A.; Nishiguchi, H.; Nagaoka, K. Oxidative dehydrogenation of isobutane to isobutene—III. Reaction mechanism over CePO4. Appl. Catal. A 2005, 296, 63–69. [Google Scholar] [CrossRef]
- Bautista, F.M.; Campelo, J.M.; Luna, D.; Marinas, J.M.; Quirós, R.A.; Romero, A.A. Screening of amorphous metal-phosphate catalysts for the oxidative dehydrogenation of ethylbenzene to styrene. Appl. Catal. B 2007, 70, 611–620. [Google Scholar] [CrossRef]
- Johnstone, R.A.W.; Liu, J.Y.; Whittaker, D. Mechanism of cyclohexanol dehydration catalysed by zirconium phosphate. J. Chem. Soc. Perkin Trans. 1998, 2, 1287–1288. [Google Scholar] [CrossRef]
- Costa, M.C.C.; Hodson, L.F.; Johnstone, R.A.W.; Liu, J.Y.; Whittaker, D. The mechanism of gas-phase dehydration of cyclohexanol and the methylcyclohexanols catalysed by zirconium phosphate. J. Mol. Catal. A 1999, 142, 349–360. [Google Scholar] [CrossRef]
- Johnstone, R.A.W.; Liu, J.Y.; Whittaker, D. Mechanism of heterogeneous gas phase dehydration of 1-methylcyclohexanol catalysed by metal(IV) phosphates. J. Mol. Catal. A 2001, 174, 159–168. [Google Scholar] [CrossRef]
- Al-Qallaf, F.A.H.; Johnstone, R.A.W.; Liu, J.Y.; Lu, L.; Whittaker, D. Metal(IV) phosphate catalyzed retro-Prins reaction involving an oxetane intermediate. J. Chem. Soc. Perkin Trans. 1999, 2, 1421–1423. [Google Scholar] [CrossRef]
- Al-Qallaf, F.A.H.; Hodson, L.F.; Johnstone, R.A.W.; Liu, J.Y.; Lu, L.; Whittaker, D. Heterogeneous liquid phase catalysis by metal (IV) phosphates of cyclic ether formation and a reverse Prins reaction. J. Mol. Catal. A 2000, 152, 187–200. [Google Scholar] [CrossRef]
- Costa, M.C.C.; Johnstone, R.A.W.; Whittaker, D. Catalysis of terpene rearrangements by zirconium phosphates and zirconium organo-substituted phosphonates. J. Mol. Catal. A 1998, 129, 79–89. [Google Scholar] [CrossRef]
- Costa, M.C.C.; Johnstone, R.A.W.; Whittaker, D. Properties of polymeric zirconium phosphates as Friedel-Crafts catalysts. J. Mol. Catal. A 1995, 103, 155–162. [Google Scholar] [CrossRef]
- Li, G.L.; Ishihara, T.; Nishiguchi, H.; Moro-oka, Y.; Takita, Y. Novel catalysts effective for dehydrofluorination of CH3CH3 (HFC143a) into CF2CH2. Chem. Lett. 1996, 507–508. [Google Scholar]
- Li, G.L.; Nishiguchi, H.; Ishihara, T.; Moro-oka, Y.; Takita, Y. Catalytic dehydrofluorination of CF3CH3(HFC143a) into CF2CH2(HFC1132a). Appl. Catal. B 1998, 16, 309–317. [Google Scholar] [CrossRef]
- Takita, Y.; Ninomiya, M.; Matsuzaki, R.; Wakamatsu, H.; Nishiguchi, H.; Ishihara, T. Decomposition of chlorofluorocarbons over metal phosphate catalysts—Part I. Decomposition of CCl2F2 over metal phosphate catalysts. Phys. Chem. Chem. Phys. 1999, 1, 2367–2373. [Google Scholar] [CrossRef]
- Takita, Y.; Wakamatsu, H.; Li, G.L.; Moro-oka, Y.; Nishiguchi, H.; Ishihara, T. Decomposition of chlorofluorocarbons over metal phosphate catalysis—II. Origin of the stability of AlPO4 and the location of Ce as a promoter. J. Mol. Catal. A 2000, 155, 111–119. [Google Scholar] [CrossRef]
- Takita, Y.; Moriyama, J.I.; Yoshinaga, Y.; Nishiguchi, H.; Ishihara, T.; Yasuda, S.; Ueda, Y.; Kubo, M.; Miyamoto, A. Adsorption of water vapor on the AlPO4-based catalysts and reaction mechanism for CFCs decomposition. Appl. Catal. A 2004, 271, 55–60. [Google Scholar] [CrossRef]
- Takita, Y.; Morita, C.; Ninomiya, M.; Wakamatsu, H.; Nishiguchi, H.; Ishihara, T. Catalytic decomposition of CF4 over AlPO4-based catalysts. Chem. Lett. 1999, 417–418. [Google Scholar]
- Takita, Y.; Ninomiya, M.; Miyake, H.; Wakamatsu, H.; Yoshinaga, Y.; Ishihara, T. Catalytic decomposition of perfluorocarbons—Part II. Decomposition of CF4 over AlPO4-rare earth phosphate catalysts. Phys. Chem. Chem. Phys. 1999, 1, 4501–4504. [Google Scholar] [CrossRef]
- Takita, Y.; Tanabe, T.; Ito, M.; Ogura, M.; Muraya, T.; Yasuda, S.; Nishiguchi, H.; Ishihara, T. Decomposition of CH2FCF3 (134a) over metal phosphate catalysts. Ind. Eng. Chem. Res. 2002, 41, 2585–2590. [Google Scholar] [CrossRef]
- Takita, Y.; Sadatomi, Y.; Nishiguchi, H.; Nagaoka, K. Decomposition of chlorobenzene over phosphate and sulphate catalysts properties. Bull. Chem. Soc. Jpn. 2006, 79, 145–148. [Google Scholar] [CrossRef]
- Kashiwagi, D.; Takai, A.; Takubo, T.; Nagaoka, K.; Inoue, T.; Takita, Y. Metal phosphate catalysts efficient for degradation of sulfur hexafluoride. Ind. Eng. Chem. Res. 2009, 48, 632–640. [Google Scholar] [CrossRef]
- Kashiwagi, D.; Takita, A.; Takubo, T.; Yamada, H.; Inoue, T.; Nagaoka, K.; Takita, Y. Catalytic activity of rare earth phosphates for SF6 decomposition and promotion effects of rare earths added in AlPO4. J. Colloid Interface Sci. 2009, 332, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Takubo, T.; Hirose, Y.; Kashiwagi, D.; Inoue, T.; Yamada, H.; Nagaka, K.; Takita, Y. Metal phosphate and fluoride catalysts active for hydrolysis of NF3. Catal. Commun. 2009, 3, 147–150. [Google Scholar] [CrossRef]
- Lisnyak, V.V.; Ischenko, E.V.; Stratiichuk, D.A.; Zaderko, A.N.; Boldyrieva, Q.Yu.; Safonova, V.V.; Yatsymyrskyi, A.V. Pt, Pd supported on niobium phosphates as catalysts for the hydrogen oxidation. Res. J. Chem. Sci. 2013, 3, 30–33. [Google Scholar]
- Johnstone, R.A.W.; Liu, J.Y.; Lu, L.; Whittaker, D. Hydrogenation of alkenes over palladium and platinum metals supported on a variety of metal(IV) phosphates. J. Mol. Catal. A 2003, 191, 289–294. [Google Scholar] [CrossRef]
- Danjo, Y.; Kikuchi, I.; Ino, Y.; Ohshima, M.; Kurokawa, H.; Miura, H. Support effect of Pd/AlPO4 catalyst in hydrogen storage of organic hydride method in the presence of CO. React. Kinet. Mech. Catal. 2012, 105, 381–389. [Google Scholar] [CrossRef]
- Takita, Y.; Ohkuma, T.; Nishiguchi, H.; Nagaoka, K.; Nakajo, T. Novel tough catalyst supports for reactions involving HF. Appl. Catal. A 2005, 283, 47–52. [Google Scholar] [CrossRef]
- Yan, W.F.; Brown, S.; Pan, Z.W.; Mahurin, S.M.; Overbury, S.H.; Dai, S. Ultrastable gold nanocatalyst supported by nanosized non-oxide substrate. Angew. Chem. Int. Ed. 2006, 45, 3614–3618. [Google Scholar]
- Ma, Z.; Yin, H.F.; Overbury, S.H.; Dai, S. Metal phosphates as a new class of supports for gold nanocatalysts. Catal. Lett. 2008, 126, 20–30. [Google Scholar] [CrossRef]
- Ma, Z.; Yin, H.F.; Dai, S. Influence of preparation methods on the performance of metal phosphate-supported gold catalysts in CO oxidation. Catal. Lett. 2010, 138, 40–45. [Google Scholar] [CrossRef]
- Li, M.J.; Wu, Z.L.; Ma, Z.; Schwartz, V.; Mullins, D.R.; Dai, S.; Overbury, S.H. CO oxidation on Au/FePO4 catalyst: Reaction pathways and nature of Au sites. J. Catal. 2009, 266, 98–105. [Google Scholar] [CrossRef]
- Li, M.J.; Wu, Z.L.; Overbury, S.H. CO oxidation on phosphate-supported Au catalysts: Effect of support reducibility on surface reactions. J. Catal. 2011, 278, 133–142. [Google Scholar] [CrossRef]
- Zhang, D.L.; Zhao, H.W.; Zhao, X.Y.; Liu, Y.M.; Chen, H.; Li, X.J. Application of hydroxyapatite as catalyst and catalyst carrier. Prog. Chem. 2011, 23, 687–694. [Google Scholar]
- Tsuchida, T.; Sakuma, S.; Takeguchi, T.; Ueda, W. Direct synthesis of n-butanol from ethanol over nonstoichiometric hydroxyapatite. Ind. Eng. Chem. Res. 2006, 45, 8634–8642. [Google Scholar] [CrossRef]
- Tsuchida, T.; Yoshioka, T.; Sakuma, S.; Takeguchi, T.; Ueda, W. Synthesis of biogasoline from ethanol over hydroxyapatite catalyst. Ind. Eng. Chem. Res. 2008, 47, 1443–1452. [Google Scholar] [CrossRef]
- Tsuchida, T.; Kubo, J.; Yoshioka, T.; Sakuma, S.; Takeguchi, T.; Ueda, W. Reaction of ethanol over hydroxyapatite affected by Ca/P ratio of catalyst. J. Catal. 2008, 259, 183–189. [Google Scholar] [CrossRef]
- Sebti, S.; Tahir, R.; Nazih, R.; Saber, A.; Boulaajai, S. Hydroxyapatite as a new solid support for the Knoevenagel reaction in heterogeneous media without solvent. Appl. Catal. A 2002, 228, 155–159. [Google Scholar] [CrossRef]
- Mallouk, S.; Bougrin, K.; Laghzizil, A.; Benhida, R. Microwave-assisted and efficient solvent-free Knoevenagel condensation. A suitable protocol using porous calcium hydroxyapatite as catalyst. Molecules 2010, 15, 813–823. [Google Scholar] [PubMed]
- Subramanian, M.; Vanangamudi, G.; Thirunarayanan, G. Hydroxyapatite catalyzed aldol condensation: Synthesis, spectral linearity, antimicrobial and inset antifeedant activities of some 2,5-dimethyl-3-furyl chalcones. Spectrochim. Acta A 2013, 110, 116–123. [Google Scholar] [CrossRef]
- Tahir, R.; Banert, K.; Sebti, S. Natural and synthetic phosphates: New and clean heterogeneous catalysts for the synthesis of 5-arylhydantoins. Appl. Catal. A 2006, 298, 261–264. [Google Scholar] [CrossRef]
- Zahouily, M.; Abrouki, Y.; Bahlaouan, B.; Rayadh, A.; Sebti, S. Hydroxyapatite: New efficient catalyst for the Michael addition. Catal. Commun. 2003, 4, 521–524. [Google Scholar] [CrossRef]
- Xu, J.; White, T.; Li, P.; He, C.H.; Han, Y.-F. Hydroxyapatite foam as a catalyst for formaldehyde combustion at room temperatue. J. Am. Chem. Soc. 2010, 132, 13172–13173. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Moffat, J.B.; Sugiyama, S.; Hayashi, H.; Shigemoto, N.; Saltoh, K. Selective oxidative coupling of methane catalyzed over hydroxyapatite ion-exchanged with lead. J. Chem. Soc. Faraday Trans. 1994, 90, 2133–2140. [Google Scholar] [CrossRef]
- Masuyama, Y.; Nakajima, Y.; Okabe, J. Environmentally-benign palladium(II)-exchanged hydroxyapatite-catalyzed allylic alkylation of allyl methyl carbonate in water. Appl. Catal. A 2010, 387, 107–112. [Google Scholar] [CrossRef]
- Masuyama, Y.; Sugioka, Y.; Chonan, S.; Suzuki, N.; Fujita, M.; Hara, K.; Fukuoka, A. Palladium(II)-exchanged hydroxyapatite-catalyzed Suzuki-Miyaura-type cross-coupling reactions with potassium aryltrifluoroborates. J. Mol. Catal. A 2013, 352, 81–85. [Google Scholar] [CrossRef]
- Saha, D.; Chatterjee, T.; Mukherjee, M.; Ranu, B. Copper(I) hydroxyapatite catalyzed Sonogashira reaction of alkynes with styrenyl bromides. Reaction of cis-styrenyl bromides forming unsymmetric diynes. J. Org. Chem. 2012, 77, 9379–9383. [Google Scholar] [CrossRef] [PubMed]
- Maaten, B.; Moussa, J.; Desmarets, C.; Gredin, P.; Beaunier, P.; Kanger, T.; Tõnsuaadu, K.; Villemin, D.; Gruselle, M. Cu-modified hydroxyl-apatite as catalyst for Glaser-Hay C–C homo-coupling reaction of terminal alkynes. J. Mol. Catal. A 2014, 393, 112–116. [Google Scholar] [CrossRef]
- Qu, Z.P.; Sun, Y.H.; Chen, D.; Wang, Y. Possible sites of copper located on hydroxyalatites structure and the identification of active sites for formaldehyde oxidation. J. Mol. Catal. A 2014, 393, 182–190. [Google Scholar] [CrossRef]
- Low, H.R.; Avdeev, M.; Ramesh, K.; White, T.J. Zinc hydroxyapatite catalyst for decomposition of 2-propanol. Adv. Mater. 2012, 24, 4175–4179. [Google Scholar] [CrossRef] [PubMed]
- Neelakandeswari, N.; Sangami, G.; Emayavaramban, P.; Karvembu, R.; Dharmaraj, N.; Kim, H.Y. Mesoporous nickel hydroxyapatite nanocomposite for microwave-assisted Henry reaction. Tetrahedron Lett. 2012, 53, 2980–2984. [Google Scholar] [CrossRef]
- Ogo, S.; Onda, A.; Yanagisawa, K. Hydrothermal synthesis of vanadate-substituted hydroxyapatites, and catalytic properties for conversion of 2-propanol. Appl. Catal. A 2008, 348, 129–135. [Google Scholar] [CrossRef]
- Carniti, P.; Gervasini, A.; Tiozzo, C.; Guidotti, M. Niobium-containing hydroxyapatites as amorphous catalysts: synthesis, properties, and activity. ACS Catal. 2014, 4, 469–479. [Google Scholar] [CrossRef]
- Ogo, S.; Onda, A.; Iwasa, Y.; Hara, K.; Fukuoka, A.; Yanagisawa, K. 1-Butanol synthesis from ethanol over strontium phosphate hydroxyapatite catalysts with various Sr/P ration. J. Catal. 2012, 296, 24–30. [Google Scholar] [CrossRef]
- Venugopal, A.; Scurrell, M.S. Hydroxyapatite as a novel support for gold and ruthenium catalysts—Behavior in the water gas shift reaction. Appl. Catal. A 2003, 245, 137–147. [Google Scholar] [CrossRef]
- Phonthammachai, N.; Zhong, Z.Y.; Guo, J.; Han, Y.F.; White, T.J. Synthesis of high performance hydroxyapatite-gold catalysts for CO oxidation. Gold Bull. 2008, 41, 42–50. [Google Scholar] [CrossRef]
- Domínguez, M.I.; Romero-Sarria, F.; Centeno, M.A.; Odriozola, J.A. Gold/hydroxyapatite catalysts: Synthesis, characterization and catalytic activity to CO oxidation. Appl. Catal. B 2009, 87, 245–251. [Google Scholar] [CrossRef]
- Huang, J.; Wang, L.-C.; Liu, Y.-M.; Cao, Y.; He, H.-Y.; Fan, K.-N. Gold nanoparticles supported on hydroxyapatite as high performance catalysts for low temperature CO oxidation. Appl. Catal. B 2011, 101, 560–569. [Google Scholar] [CrossRef]
- Zhao, K.F.; Qiao, B.T.; Wang, J.H.; Zhang, Y.J.; Zhang, T. A highly active and sintering-resistant Au/FeOx-hydroxyapatite catalyst for CO oxidation. Chem. Commun. 2011, 47, 1779–1781. [Google Scholar] [CrossRef]
- Mikami, Y.; Noujima, A.; Mitsudome, T.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. Highly efficient gold nanoparticle catalyzed deoxygenation of amides, sulfoxides, and pyridine N-oxides. Chem. Eur. J. 2011, 17, 1768–1772. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.M.; Tsunoyama, H.; Akita, T.; Xie, S.H.; Tsukuda, T. Aerobic oxidation of cyclohexane catalyzed by size-controlled Au clusters on hydroxyapatite: Size effect in the sub-2 nm regime. ACS Catal. 2011, 1, 2–6. [Google Scholar] [CrossRef]
- Sun, H.; Su, F.-Z.; Ni, J.; Cao, Y.; He, H.Y.; Fan, K.N. Gold supported on hydroxyapatite as a versatile multifunctional catalyst for the direct tandem synthesis of imines and oximes. Angew. Chem. Int. Ed. 2009, 48, 4390–4393. [Google Scholar] [CrossRef]
- Han, Y.-F.; Phonthammachai, N.; Ramesh, K.; Zhong, Z.Y.; White, T. Removing organic compounds from aqueous medium via wet peroxidation by gold catalysts. Environ. Sci. Technol. 2008, 42, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Chambyal, O.S.; Paul, S.; Shamim, T.; Gupta, M.; Gupta, R.; Loupy, A. HAP-Pd(0): A highly efficient recyclable heterogeneous catalyst for the selective reduction of carbon-carbon double bond in α,-β-unsaturated ketones. Synth. Commun. 2013, 43, 656–667. [Google Scholar] [CrossRef]
- Wuyts, S.; De Vos, D.E.; Verpoort, F.; Depla, D.; De Gryse, R.; Jacobs, P.A. A heterogeneous Ru-hydroxyapatite catalyst for mild racemization of alcohols. J. Catal. 2003, 219, 417–424. [Google Scholar] [CrossRef]
- Ho, C.-M.; Yu, W.-J.; Che, C.-M. Ruthenium nanoparticles supported on hydroxyapatite as an efficient and recyclable catalyst for cis-dihydroxylation and oxidative cleavage of alkenes. Angew. Chem. Int. Ed. 2003, 43, 3303–3307. [Google Scholar] [CrossRef] [Green Version]
- Durak, H.; Gulcan, M.; Zahmakiran, M.; Ozkar, S.; Kaya, M. Hydroxyapatite-nanosphere supported ruthenium(0) nanoparticle catalyst for hydrogen generation from ammonia-borane solution: Kinetic studies for nanoparticle formation and hydrogen evolution. RSC Adv. 2014, 4, 28947–28955. [Google Scholar] [CrossRef]
- Mitsudome, T.; Arita, S.; Mori, H.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. Supported silver-nanoparticle-catalyzed highly efficient aqueous oxidation of phenylsilanes to silanols. Angew. Chem. Int. Ed. 2008, 47, 7938–7940. [Google Scholar] [CrossRef]
- Kumar, P.A.; Reddy, M.P.; Ju, L.K.; Phil, H.H. Novel silver loaded hydroxyapatite catalyst for the selective catalytic reduction of NOx by propene. Catal. Lett. 2008, 126, 78–83. [Google Scholar] [CrossRef]
- Haneda, M.; Watanabe, T.; Kamiuchi, N.; Ozawa, M. Effect of platinum dispersion on the catalytic activity of Pt/Al2O3 for the oxidation of carbon monoxide and propene. Appl. Catal. B 2013, 142–143, 8–14. [Google Scholar] [CrossRef]
- Li, N.; Chen, Q.Y.; Luo, L.F.; Huang, W.X.; Luo, M.F.; Hu, G.S.; Lu, J.Q. Kinetic study and the effect of particle size on low temperature CO oxidation over Pt/TiO2 catalysts. Appl. Catal. B 2013, 142–143, 523–532. [Google Scholar] [CrossRef]
- Kim, M.Y.; Choi, J.S.; Toops, T.J.; Jeong, E.S.; Han, S.W.; Schwartz, V.; Chen, J.H. Coating SiO2 support with TiO2 or ZrO2 and effects on structure and CO oxidation performance of Pt catalysts. Catalysts 2013, 3, 88–103. [Google Scholar] [CrossRef]
- Kamiuchi, N.; Haneda, M.; Ozawa, M. CO oxidation over Pt/Ce-Zr oxide catalysts with low content of platinum and cerium components. Catal. Today 2013, 201, 79–84. [Google Scholar] [CrossRef]
- Shen, M.Q.; Lv, L.F.; Wang, J.Q.; Zhu, J.X.; Huang, Y.; Wang, J. Study of Pt dispersion on Ce based supports and the influence on the CO oxidation reaction. Chem. Eng. J. 2014, 255, 40–48. [Google Scholar] [CrossRef]
- Chua, Y.P.G.; Gunasooriya, G.T.K.K.; Saeys, M.; Seebauer, E.G. Controlling the CO oxidation rate over Pt/TiO2 catalysts by defect engineering of the TiO2 support. J. Catal. 2014, 311, 306–313. [Google Scholar] [CrossRef]
- Jung, C.H.; Yun, J.; Qadir, K.; Naik, B.; Yun, J.Y.; Park, J.Y. Catalytic activity of Pt/SiO2 nanocatalysts synthesized via ultrasonic spray pyrolysis process under CO oxidation. Appl. Catal. B 2014, 154–155, 171–176. [Google Scholar] [CrossRef]
- An, N.H.; Yuan, X.L.; Pan, B.; Li, Q.L.; Li, S.Y.; Zhang, W.X. Design of a highly active Pt/Al2O3 catalyst for low-temperature CO oxidation. RSC Adv. 2014, 4, 38250–38257. [Google Scholar] [CrossRef]
- Oh, Y.; Kang, J.; Nam, S.; Byun, S.; Park, B. Pt/AlPO4 nanocomposite thin-film electrodes for ethanol electrooxiadtion. Mater. Chem. Phys. 2012, 135, 188–192. [Google Scholar] [CrossRef]
- Park, J.; Oh, Y.; Park, Y.; Nam, S.; Moon, J.; Kang, J.; Jung, D.R.; Byun, S.; Park, B. Methanol oxidation in nanostructured platinum/cerium-phosphate thin films. Curr. App. Phys. 2011, 11, S2–S5. [Google Scholar] [CrossRef]
- Bouwman, P.J.; Dmowski, W.; Stanley, J.; Cotton, G.B.; Swider-Lyons, K.E. Platinum-iron phosphate electrocatalysts for oxygen reduction in PEMFCs. J. Electrochem. Soc. 2004, 151, A1989–A1998. [Google Scholar] [CrossRef]
- Lee, B.; Kim, C.; Park, Y.; Kim, T.G.; Park, B. Nanostructured platinum/iron phosphate thin film electrodes for methanol oxidation. Electrochem. Solid-State Lett. 2006, 9, E27–E30. [Google Scholar] [CrossRef]
- Gardner, S.D.; Hoflund, G.B.; Schryer, D.R.; Upchurch, B.T. Characterization study of silica-supported platinized tin oxide catalysts used for low-temperature CO oxidation: Effect of pretreatment temperature. J. Phys. Chem. 1991, 95, 835–838. [Google Scholar] [CrossRef]
- Lieske, H.; Lietz, G.; Spindler, H.; Völter, J. Reactions of platinum in oxygen- and hydrogen-treated Pt/γ-Al2O3 catalysts: I. Temperature-programmed reduction, adsorption, and redispersion of platinum. J. Catal. 1983, 81, 8–16. [Google Scholar] [CrossRef]
- Gracia, F.J.; Miller, J.T.; Kropf, A.J.; Wolf, E.E. Kinetics, FTIR, and controlled atmosphere EXAFS study of the effect of chlorine on Pt-supported catalysts during oxidation reactions. J. Catal. 2002, 209, 341–354. [Google Scholar] [CrossRef]
- Ma, Z.; Zaera, F. Characterization of heterogeneous catalysts. In Surface and Nanomolecular Catalysis; Richards, R., Ed.; Taylor & Fransic: Boca Raton, FL, USA, 2006; pp. 1–37. [Google Scholar]
- Chen, G.X.; Zhao, Y.; Fu, G.; Duchesne, P.N.; Gu, L.; Zheng, Y.P.; Weng, X.F.; Chen, M.S.; Zhang, P.; Pao, C.W.; Lee, J.F.; Zheng, N.F. Interfacial effects in iron-nickel hydroxide-platinum nanoparticles enhance catalytic oxidation. Science 2014, 344, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Brow, R.K. An XPS study of oxygen bonding in zinc phosphate and zinc borophosphate glasses. J. Non-Cryst. Solids 1996, 194, 267–273. [Google Scholar] [CrossRef]
- Grosseau-Poussard, J.L.; Panicaud, B.; Pedraza, F.; Renault, P.O.; Silvain, J.F. Iron oxidation under the influence of phosphate thin films. J. Appl. Phys. 2003, 94, 784–788. [Google Scholar] [CrossRef]
- Mercado, D.F.; Magnacca, G.; Malandrino, M.; Rubert, A.; Montoneri, E.; Celi, L.; Prevot, A.B.; Gonzalez, M.C. Paramagnetic iron-doped hydroxyapatite nanoparticles with improved metal sorption properties. A bioorganic substrates-mediated synthesis. ACS Appl. Mater. Interfaces 2004, 6, 3937–3946. [Google Scholar] [CrossRef] [Green Version]
- Satterfield, C.N. Heterogeneous Catalysis in Industrial Practice, 2nd ed.; Krieger Publishing: Malabar, FL, USA, 1996; pp. 358–364. [Google Scholar]
- Schubert, M.M.; Hackenberg, S.; van Veen, A.C.; Muhler, M.; Plzak, V.; Behm, R.J. CO oxidation over supported gold catalysts—“Inert” and “active” support materials and their role for the oxygen supply during reaction. J. Catal. 2001, 197, 113–122. [Google Scholar] [CrossRef]
- Weiher, N.; Bus, E.; Delannoy, L.; Louis, C.; Ramaker, D.E.; Miller, J.T.; van Bokhoven, J.A. Structure and oxidation state of gold on different supports under various CO oxidation conditions. J. Catal. 2006, 240, 100–107. [Google Scholar] [CrossRef]
- Lee, C.H.; Chen, Y.W. Effect of basic additives on Pt/Al2O3 for CO and propylene oxidation under oxygen deficient conditions. Ind. Eng. Chem. Res. 1997, 36, 1498–1506. [Google Scholar] [CrossRef]
- Liu, X.J.; Wang, R.; Song, L.Y.; He, H.; Zhang, G.Z.; Zi, X.H.; Qiu, W.G. The oxidation of carbon monoxide over the palladium nanocube catalysts: Effect of the basic-property of the support. Catal. Commun. 2014, 46, 213–218. [Google Scholar] [CrossRef]
- Wu, Z.L.; Zhou, S.H.; Zhu, H.G.; Dai, S.; Overbury, S.H. DRIFTS-QMS study of room temperature CO oxidation on Au/SiO2 catalyst: Nature and role of different Au species. J. Phys. Chem. C 2009, 113, 3726–3734. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, X.; Qin, H.; Meng, T.; Lin, Y.; Ma, Z. Metal Phosphate-Supported Pt Catalysts for CO Oxidation. Materials 2014, 7, 8105-8130. https://doi.org/10.3390/ma7128105
Qian X, Qin H, Meng T, Lin Y, Ma Z. Metal Phosphate-Supported Pt Catalysts for CO Oxidation. Materials. 2014; 7(12):8105-8130. https://doi.org/10.3390/ma7128105
Chicago/Turabian StyleQian, Xiaoshuang, Hongmei Qin, Tao Meng, Yi Lin, and Zhen Ma. 2014. "Metal Phosphate-Supported Pt Catalysts for CO Oxidation" Materials 7, no. 12: 8105-8130. https://doi.org/10.3390/ma7128105