
Ferrocene Orientation Determined Intramolecular Interactions Using Energy Decomposition Analysis

Abstract
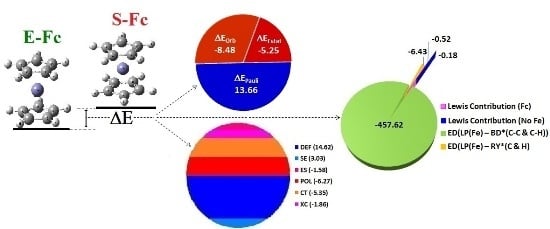
:
1. Introduction
2. Computational Methods and Details
3. Results and Discussion
3.1. ETS: EDA Dependence on the Fragmentation Channels of Fc

{kind=link}
| Frag Channel | M + 2Cp | M2+ + 2Cp− | M2+ + 2Cp− | M2+ + 2Cp− | M2+ + 2Cp− | M2+ + 2Cp− | M2+ + Cp22− | M2+ + Cp22− | MCp+ + Cp− |
|---|---|---|---|---|---|---|---|---|---|
| Model Basis Set | BP86 TZP [21] | BP86 TZP [21] | BP86 TZP [22] | BP86 TZ2P [36] | B_PW91 TZ2P a [39] | OPBE TZP [37] | PW91 TZ2P [38] | PW91 TZ2P [38] | BP86 TZP [20] |
| Conformer | D5d | D5d | D5d | D5d | D5h | D5h | D5h | D5d | D5d |
| ΔEPauli | 409.6 | 279.9 | 272.2 | 282.0 | – | 345.7 | 399.23 | 398.85 | 172.4 |
| ΔEelstat | −307.5 | −599.9 | −598.0 | −600.5 | – | −619.1 | −229.05 | −229.36 | −238.5 |
| ΔEorb | −376.3 | −573.9 | −567.5 | −577.6 | – | −634.5 | −626.35 | −624.17 | −171.5 |
| ΔEint | −274.2 | −893.9 | −893.3 | −896.0 | −919 | −907.9 | −456.17 | −454.68 | −237.6 |
3.2. ETS: EDA Dependence on Quantum Mechanical Models Employed
| Energy Frag 1,2 | BP86/TZP | BP86/TZ2P | BP86/TZ2P+ | ||||||
|---|---|---|---|---|---|---|---|---|---|
| D5h | D5d | ΔΔEi 3 | D5h | D5d | ΔΔEi 3 | D5h | D5d | ΔΔEi 3 | |
| ΔEEstat | −2794.94 | −2789.68 | −5.26 | −2794.94 | −2789.68 | −5.26 | −2793.10 | −2787.85 | −5.25 |
| ΔEPauli | 12,679.83 | 12,666.40 | 13.43 | 12,679.83 | 12,666.40 | 13.43 | 12,675.76 | 12,662.35 | 13.41 |
| ΔEOrb | −12,942.17 | −12,933.04 | −9.13 | −12,954.25 | −12,945.16 | −9.09 | −12,952.85 | −12,943.81 | −9.04 |
| ΔEInt | −3057.29 | −3056.32 | −0.97 | −3069.36 | −3068.45 | −0.91 | −3070.20 | −3069.31 | −0.89 |
| ΔESter | 9884.88 | 9876.71 | 8.17 | 9884.89 | 9876.71 | 8.18 | 9882.66 | 9874.50 | 8.16 |
| Energy Frag 1,2 | HF/TZ2P 4 | PW91/TZ2P 5 | |||||||
| D5h | D5d | ΔΔEi 3 | D5h | D5d | ΔΔEi 3 | ||||
| ΔEEstat | −3332.24 | −3325.18 | −7.06 | −229.05 | −229.36 | 0.31 | |||
| ΔEPauli | 14,486.28 | 14,470.29 | 15.99 | −399.23 | −398.85 | −0.38 | |||
| ΔEOrb | −16,465.98 | −16,457.79 | −8.19 | −626.35 | −624.17 | −2.18 | |||
| ΔEInt | −5311.94 | −5312.68 | 0.74 | −456.17 | −454.68 | −1.49 | |||
| ΔESter | 11,154.04 | 11,145.11 | 8.93 | −628.28 | −628.21 | −0.07 | |||
| Energy Terms 1,2 | HF/TZ2P+ | M06-2X/TZ2P+ | ||||
| D5h | D5d | ΔΔEi 3 | D5h | D5d | ΔΔEi 3 | |
| ΔEEstat | −2793.10 | −2787.85 | −5.25 | −2793.10 | −2787.85 | −5.25 |
| ΔEPauli | −49,351.36 | −49,365.98 | 14.62 | −54,025.19 | −54,039.14 | 13.95 |
| ΔEOrb | −16,840.51 | −16,832.75 | −7.76 | −14,568.75 | −14,560.82 | −7.93 |
| ΔEInt | −68,984.98 | −68,986.58 | 1.60 | −71,387.05 | −71,387.81 | 0.76 |
| ΔESter | −52,144.47 | −52,153.83 | 9.36 | −56,818.3 | −56,826.99 | 8.69 |
| Energy Terms 1,2 | B3LYP/TZ2P+ | BP86/TZ2P+ | ||||
| D5h | D5d | ΔΔEi 3 | D5h | D5d | ΔΔEi 3 | |
| ΔEEstat | −2793.10 | −2787.85 | −5.25 | −2793.10 | −2787.85 | −5.25 |
| ΔEPauli | 222.39 | 208.73 | 13.66 | 12,675.76 | 12,662.35 | 13.41 |
| ΔEOrb | −13,630.90 | −13,622.42 | −8.48 | −12,952.85 | −12,943.81 | −9.04 |
| ΔEInt | −16,201.62 | −16,201.53 | −0.09 | −3070.20 | −3069.31 | −0.89 |
| ΔESter | −2570.72 | −2579.12 | 8.40 | 9882.66 | 9874.50 | 8.16 |
3.3. NBO-NEDA Analysis of Ferrocene Conformers
| Energy Component c | D5h | D5d | ΔE d |
|---|---|---|---|
| EL (ES + POL + SE) | −1011.05 | −1006.23 | −4.82 |
| ES | −676.89 | −675.31 | −1.58 |
| POL | −671.73 | −665.46 | −6.27 |
| SE (Fe) | 35.89 | 35.84 | 0.05 |
| SE (Cp) | 150.84 | 149.35 | 1.49 |
| SE (Cp) | 150.84 | 149.35 | 1.49 |
| Tot SE | 337.57 | 334.54 | 3.03 |
| Charge Transfer (CT) | −634.02 | −628.67 | −5.35 |
| Core (XC + DEF − SE) | 880.64 | 870.91 | 9.73 |
| XC | −200.44 | −198.58 | −1.86 |
| DEF (Fe) | 395.28 | 392.47 | 2.81 |
| DEF (Cp) | 511.69 | 505.78 | 5.91 |
| DEF (Cp) | 511.68 | 505.78 | 5.90 |
| Tot DEF | 1418.65 | 1404.03 | 14.62 |
| Etot (EL + CT + Core) | −764.43 | −763.99 | −0.44 |
| Energy Decomposition | E(D5h)/Eh | E(D5d)/Eh | ΔE a/ kcal·mol−1 |
|---|---|---|---|
| Lewis Contribution (Fc) | |||
| Total | −1650.66185 | −1650.661026 | −0.52 |
| Total Lewis (EL) | −1648.30198 | −1647.402542 | −564.41 |
| Total Non-Lewis (ENL) | −2.35987395 | −3.25848317 | 563.89 |
| Lewis Contribution (No Fe) | |||
| Total | −386.897706 | −386.8974192 | −0.18 |
| Total Lewis (EL) | −385.653328 | −385.6162447 | −23.27 |
| Total Non-Lewis (ENL) | 1.244378 | 1.281174 | 23.09 |
| LP(Fe) – BD *(C-C & C-H) | |||
| Energy of Deletion (ED) | −1650.02738 | −1649.298117 | −457.62 |
| LP(Fe) – RY *(C & H) | |||
| Energy of Deletion (ED) | −1650. 02384 | −1650.013585 | −6.43 |
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kealy, T.J.; Pauson, P.L. A new type of organo-Iron compound. Nature 1951, 168, 1039–1040. [Google Scholar] [CrossRef]
- Mohammadi, N.; Ganesan, A.; Chantler, C.T.; Wang, F. Differentiation of ferrocene D5d and D5h conformers using IR spectroscopy. J. Organomet. Chem. 2012, 713, 51–59. [Google Scholar] [CrossRef]
- Islam, S.; Wang, F. The d-electrons of Fe in ferrocene: The excess orbital energy spectrum (EOES). RSC Adv. 2015, 5, 11933–11941. [Google Scholar] [CrossRef]
- Haaland, A.; Nilsson, J.E. The determination of barriers to internal rotation by means of electron diffraction. Ferrocene and ruthenocene. Acta Chem. Scand. 1968, 22, 2653–2670. [Google Scholar] [CrossRef]
- Haaland, A.; Lusztyk, J.; Novak, D.P.; Brunvoll, J.; Starowieyski, K.B. Molecular structures of dicyclopentadienylmagnesium and dicyclopentadienylchromium by gas-phase electron diffraction. J. Chem. Soc. Chem. Commun. 1974, 54. [Google Scholar] [CrossRef]
- Haaland, A. Molecular structure and bonding in the 3d metallocenes. Acc. Chem. Res. 1979, 12, 415–422. [Google Scholar] [CrossRef]
- Lippincott, E.R.; Nelson, R.D. The vibrational spectra and structure of ferrocene and ruthenocene. J. Chem. Phys. 1953, 21, 1307–1308. [Google Scholar] [CrossRef]
- Lippincott, E.R.; Nelson, R.D. The vibrational spectra and structure of ferrocene and ruthenocene. Spectrochim. Acta 1958, 10, 307–329. [Google Scholar] [CrossRef]
- Cotton, F.A.; Wilkinson, G.; Murillo, C.A.; Bochmann, M. Advanced Inorganic Chemistry, 5th ed.; John Wiley & Sons: New York, NY, USA, 1988. [Google Scholar]
- Yamaguchi, Y.; Ding, W.; Sanderson, C.T.; Borden, M.L.; Morgan, M.J.; Kutal, C. Electronic structure, spectroscopy, and photochemistry of group 8 metallocenes. Coord. Chem. Rev. 2007, 251, 515–524. [Google Scholar] [CrossRef]
- Duhović, S.; Diaconescu, P.L. An experimental and computational study of 1,1′-ferrocene diamines. Polyhedron 2013, 52, 377–388. [Google Scholar] [CrossRef]
- Cooper, D.C.; Yennie, C.J.; Morin, J.B.; Delaney, S.; Suggs, J.W. Development of a DNA-damaging ferrocene amino acid. J. Organomet. Chem. 2011, 696, 3058–3061. [Google Scholar] [CrossRef]
- Gryaznova, T.P.; Katsyuba, S.A.; Milyukov, V.A.; Sinyashin, O.G. DFT study of substitution effect on the geometry, IR spectra, spin state and energetic stability of the ferrocenes and their pentaphospholyl analogues. J. Organomet. Chem. 2010, 695, 2586–2595. [Google Scholar] [CrossRef]
- Mehrotra, R.C.; Singh, A. Organometallic Chemistry, 2nd ed.; New Age International (P) Limited: New Delhi, India, 2007. [Google Scholar]
- Reddy, K.V. Symmetry and Spectroscopy of Molecules; New Age International (P) Limited: New Delhi, India, 1998. [Google Scholar]
- Atkins, P.; Overton, T.; Rourke, J.; Weller, M.; Armstrong, F. Shriver and Atkins' Inorganic Chemistry, 5th ed.; OUP Oxford: Oxford, UK, 2010. [Google Scholar]
- Coriani, S.; Haaland, A.; Helgaker, T.; Jørgensen, P. The Equilibrium Structure of Ferrocene. ChemPhysChem 2006, 7, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.R.; Duley, S.; Chattaraj, P.K. Bonding, reactivity and aromaticity in some novel all-metal metallocenes. Proc. Indian Natl. Sci. Acad. Phys. Sci. 2008, 74, 11–18. [Google Scholar]
- Bean, D.E.; Fowler, P.W.; Morris, M.J. Aromaticity and ring currents in ferrocene and two isomeric sandwich complexes. J. Organomet. Chem. 2011, 696, 2093–2100. [Google Scholar] [CrossRef]
- Frunzke, J.; Lein, M.; Frenking, G. Structures, metal-ligand bond strength, and bonding analysis of ferrocene derivatives with group-15 heteroligands Fe(η5-E5)2 and FeCp(η5-E5) (E = N, P, As, Sb). A Theoretical Study. Organometallics 2002, 21, 3351–3359. [Google Scholar] [CrossRef]
- Rayón, V.M.; Frenking, G. Bis(benzene)chromium is a δ-bonded molecule and ferrocene is a π-bonded molecule. Organometallics 2003, 22, 3304–3308. [Google Scholar] [CrossRef]
- Lein, M.; Frunzke, J.; Timoshkin, A.; Frenking, G. Iron bispentazole Fe(η5-N5)2, a theoretically predicted high-energy compound: Structure, bonding analysis, metal–ligand bond strength and a comparison with the isoelectronic ferrocene. Chem. Eur. J. 2001, 7, 4155–4163. [Google Scholar] [CrossRef]
- Frenking, G.; Fröhlich, N. The nature of the bonding in transition-metal compounds. Chem. Rev. 2000, 100, 717–774. [Google Scholar] [CrossRef] [PubMed]
- Frenking, G.; Wichmann, K.; Fröhlich, N.; Loschen, C.; Lein, M.; Frunzke, J.; Rayón, V.C.M. Towards a rigorously defined quantum chemical analysis of the chemical bond in donor-acceptor complexes. Coord. Chem. Rev. 2003, 238–239, 55–82. [Google Scholar] [CrossRef]
- Cortés-Guzmán, F.; Bader, R.F.W. Complementarity of QTAIM and MO theory in the study of bonding in donor-acceptor complexes. Coord. Chem. Rev. 2005, 249, 633–662. [Google Scholar] [CrossRef]
- Xu, Z.-F.; Xie, Y.; Feng, W.-L.; Schaefer, H.F. Systematic investigation of electronic and molecular structures for the first transition metal series metallocenes M(C5H5)2 (M = V, Cr, Mn, Fe, Co, and Ni). J. Phys. Chem. A 2003, 107, 2716–2729. [Google Scholar] [CrossRef]
- Salzner, U. Modeling photoelectron spectra of conjugated oligomers with time-dependent density functional theory. J. Phys. Chem. A 2010, 114, 10997–11007. [Google Scholar] [CrossRef] [PubMed]
- Zlatar, M.; Schläpfer, C.-W.; Daul, C. A New Method to Describe the Multimode Jahn-Teller Effect Using Density Functional Theory. In The Jahn-Teller Effect; Köppel, H., Yarkony, D.R., Barentzen, H., Eds.; Springer: Berlin, Germany, 2009; Volume 97, pp. 131–165. [Google Scholar]
- Wilkinson, G.; Rosenblum, M.; Whiting, M.C.; Woodward, R.B. The structure of iron bis-cyclopentadienyl. J. Am. Chem. Soc. 1952, 74, 2125–2126. [Google Scholar] [CrossRef]
- Bohn, R.K.; Haaland, A. On the molecular structure of ferrocene, Fe(C5H5)2. J. Organomet. Chem. 1966, 5, 470–476. [Google Scholar] [CrossRef]
- Fischer, V.E.O.; Pfab, W. Cyclopentadien-metallkomplexe, ein neuer Typ metallorganischer verbindungen. Z. Naturforschung 1952, 7, 377–379. [Google Scholar] [CrossRef]
- Lippincott, E.R.; Nelson, R.D. The thermodynamic functions of bis-cyclopentadienyl iron, bis-cyclopentadienylnickel and bis-cyclopentadienylruthenium. J. Am. Chem. Soc. 1955, 77, 4990–4993. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. On the calculation of bonding energies by the Hartree Fock Slater method. Theoret. Chim. Acta 1977, 46, 1–10. [Google Scholar] [CrossRef]
- Schenter, G.K.; Glendening, E.D. Natural energy decomposition analysis: The linear response electrical self energy. J. Phys. Chem. 1996, 100, 17152–17156. [Google Scholar] [CrossRef]
- Morokuma, K. Molecular Orbital Studies of hydrogen bonds. III. C=O···H–O hydrogen bond in H2CO···H2O and H2CO···2H2O. J. Chem. Phys. 1971, 55, 1236–1244. [Google Scholar] [CrossRef]
- Gomez-Sandoval, Z.; Peña, E.; Guerra, C.F.; Bickelhaupt, F.M.; Mendez-Rojas, M.A.; Merino, G. A helicoid ferrocene. Inorg. Chem. 2009, 48, 2714–2716. [Google Scholar] [CrossRef] [PubMed]
- Swart, M. Metal-ligand bonding in metallocenes: Differentiation between spin state, electrostatic and covalent bonding. Inorg. Chim. Acta 2007, 360, 179–189. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, H.; Sun, M.; Liu, Y.; Pang, X.; Yu, X.; Liu, B.; Li, Z. Substitution effect on the geometry and electronic structure of the ferrocene. J. Comput. Chem. 2007, 28, 2260–2274. [Google Scholar] [CrossRef] [PubMed]
- Mayor-López, M.J.; Weber, J. DFT calculations of the binding energy of metallocenes. Chem. Phys. Lett. 1997, 281, 226–232. [Google Scholar] [CrossRef]
- Gusarov, S.; van den Hoek, F.E.H.P.; Jacob, C.R.; Jacobsen, H.; Jensen, L.; Kaminski, J.W.; van Kessel, G.; Kootstra, F.; Kovalenko, A.; Krykunov, M.V.; et al. ADF2014; Vrije Universiteit: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Glendening, E.D.; Streitwieser, A. Natural energy decomposition analysis: An energy partitioning procedure for molecular interactions with application to weak hydrogen bonding, strong ionic, and moderate donor—Acceptor interactions. J. Chem. Phys. 1994, 100, 2900–2909. [Google Scholar] [CrossRef]
- Glendening, E.D. Natural energy decomposition analysis: Explicit evaluation of electrostatic and polarization effects with application to aqueous clusters of alkali metal cations and neutrals. J. Am. Chem. Soc. 1996, 118, 2473–2482. [Google Scholar] [CrossRef]
- Glendening, E.D. Natural energy decomposition analysis: Extension to density functional methods and analysis of cooperative effects in water clusters. J. Phys. Chem. A 2005, 109, 11936–11940. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Wolters, L.P.; Bickelhaupt, F.M. The activation strain model and molecular orbital theory. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2015, 5, 324–343. [Google Scholar] [CrossRef]
- Hopffgarten, M.V.; Frenking, G. Energy decomposition analysis. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 43–62. [Google Scholar] [CrossRef]
- Hirao, H. Reactive bond orbitals: A localized resonance-structure approach to charge transfer. Chem. Phys. Lett. 2007, 443, 141–146. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. A theoretical study of the ethylene-metal bond in complexes between copper(1+), silver(1+), gold(1+), platinum(0) or platinum(2+) and ethylene, based on the Hartree-Fock-Slater transition-state method. Inorg. Chem. 1979, 18, 1558–1565. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. Carbon monoxide, carbon monosulfide, molecular nitrogen, phosphorus trifluoride, and methyl isocyanide as sigma. donors and pi. acceptors. A theoretical study by the Hartree-Fock-Slater transition-state method. Inorg. Chem. 1979, 18, 1755–1759. [Google Scholar] [CrossRef]
- Su, P.; Li, H. Energy decomposition analysis of covalent bonds and intermolecular interactions. J. Chem. Phys. 2009, 131. [Google Scholar] [CrossRef] [PubMed]
- Stevens, W.J.; Fink, W.H. Frozen fragment reduced variational space analysis of hydrogen bonding interactions. Application to the water dimer. Chem. Phys. Lett. 1987, 139, 15–22. [Google Scholar] [CrossRef]
- Mo, Y.; Gao, J.; Peyerimhoff, S.D. Energy decomposition analysis of intermolecular interactions using a block-localized wave function approach. J. Chem. Phys. 2000, 112, 5530–5538. [Google Scholar] [CrossRef]
- Mo, Y.; Bao, P.; Gao, J. Energy decomposition analysis based on a block-localized wavefunction and multistate density functional theory. Phys. Chem. Chem. Phys. 2011, 13, 6760–6775. [Google Scholar] [CrossRef] [PubMed]
- Misquitta, A.J.; Podeszwa, R.; Jeziorski, B.; Szalewicz, K. Intermolecular potentials based on symmetry-adapted perturbation theory with dispersion energies from time-dependent density-functional calculations. J. Chem. Phys. 2005, 123. [Google Scholar] [CrossRef] [PubMed]
- Kitaura, K.; Morokuma, K. A new energy decomposition scheme for molecular interactions within the Hartree-Fock approximation. Int. J. Quantum Chem. 1976, 10, 325–340. [Google Scholar] [CrossRef]
- Khaliullin, R.Z.; Cobar, E.A.; Lochan, R.C.; Bell, A.T.; Head-Gordon, M. Unravelling the origin of intermolecular interactions using absolutely localized molecular orbitals. J. Phys. Chem. A 2007, 111, 8753–8765. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation theory approach to intermolecular potential energy surfaces of van der waals complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Blanco, M.A.; Martín Pendás, A.; Francisco, E. Interacting quantum atoms: A correlated energy decomposition scheme based on the quantum theory of atoms in molecules. J. Chem. Theory Comput. 2005, 1, 1096–1109. [Google Scholar] [CrossRef]
- Mitoraj, M.P.; Michalak, A.; Ziegler, T. A combined charge and energy decomposition scheme for bond analysis. J. Chem. Theory Comput. 2009, 5, 962–975. [Google Scholar] [CrossRef]
- Szalewicz, K. Symmetry-adapted perturbation theory of intermolecular forces. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 254–272. [Google Scholar] [CrossRef]
- Ziegler, T.; Tschinke, V.; Becke, A. Theoretical study on the relative strengths of the metal-hydrogen and metal-methyl bonds in complexes of middle to late transition metals. J. Am. Chem. Soc. 1987, 109, 1351–1358. [Google Scholar] [CrossRef]
- Ziegler, T.; Tschinke, V.; Ursenbach, C. Thermal stability and kinetic lability of the metal carbonyl bond. A theoretical study on M(CO)6 (M = chromium, molybdenum, tungsten), M(CO)5 (M = iron, ruthenium, osmium), and M(CO)4 (M = nickel, palladium, platinum). J. Am. Chem. Soc. 1987, 109, 4825–4837. [Google Scholar] [CrossRef]
- Li, J.; Schreckenbach, G.; Ziegler, T. A reassessment of the first metal-carbonyl dissociation energy in M(CO)4 (M = Ni, Pd, Pt), M(CO)5 (M = Fe, Ru, Os), and M(CO)6 (M = Cr, Mo, W) by a quasirelativistic density functional method. J. Am. Chem. Soc. 1995, 117, 486–494. [Google Scholar] [CrossRef]
- Ehlers, A.W.; Baerends, E.J.; Bickelhaupt, F.M.; Radius, U. Alternatives to the CO ligand: Coordination of the isolobal analogues BF, BNH2, BN(CH3)2, and BO- in mono- and binuclear first-row transition metal complexes. Chem. Eur. J. 1998, 4, 210–221. [Google Scholar] [CrossRef]
- Foster, J.P.; Weinhold, F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Mitin, A.V.; Baker, J.; Pulay, P. An improved 6-31G* basis set for first-row transition metals. J. Chem. Phys. 2003, 118, 7775–7782. [Google Scholar] [CrossRef]
- Chong, D.P.; Van Lenthe, E.; Van Gisbergen, S.; Baerends, E.J. Even-tempered slater-type orbitals revisited: From hydrogen to krypton. J. Comput. Chem. 2004, 25, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO 6.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2013. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Koch, H.; Jorgensen, P.; Helgaker, T. The molecular structure of ferrocene. J. Chem. Phys. 1996, 104, 9528–9530. [Google Scholar] [CrossRef]
- Hohm, U.; Goebel, D.; Grimme, S. Experimental and theoretical study of the dipole polarizability of ferrocene Fe(C5H5)2. Chem. Phys. Lett. 1997, 272, 328–334. [Google Scholar] [CrossRef]
- Pierloot, K.; Persson, B.J.; Roos, B.O. Theoretical study of the chemical bonding in [Ni(C2H4)] and ferrocene. J. Phys. Chem. 1995, 99, 3465–3472. [Google Scholar] [CrossRef]
- Vyboishchikov, S.F.; Krapp, A.; Frenking, G. Two complementary molecular energy decomposition schemes: The Mayer and Ziegler–Rauk methods in comparison. J. Chem. Phys. 2008, 129. [Google Scholar] [CrossRef] [PubMed]
- Bickelhaupt, F.M.; Baerends, E.J. The case for steric repulsion causing the staggered conformation of ethane. Angew. Chem. Int. Ed. 2003, 42, 4183–4188. [Google Scholar] [CrossRef] [PubMed]
- Phung, Q.M.; Vancoillie, S.; Pierloot, K. A Multiconfigurational perturbation theory and density functional theory study on the heterolytic dissociation enthalpy of first-row metallocenes. J. Chem. Theory Comput. 2012, 8, 883–892. [Google Scholar] [CrossRef]
- Wang, F. Assessment of quantum mechanical models based on resolved orbital momentum distributions of n-butane in the outer valence shell. J. Phys. Chem. A 2003, 107, 10199–10207. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Islam, S.; Vasilyev, V. Ferrocene Orientation Determined Intramolecular Interactions Using Energy Decomposition Analysis. Materials 2015, 8, 7723-7737. https://doi.org/10.3390/ma8115419
Wang F, Islam S, Vasilyev V. Ferrocene Orientation Determined Intramolecular Interactions Using Energy Decomposition Analysis. Materials. 2015; 8(11):7723-7737. https://doi.org/10.3390/ma8115419
Chicago/Turabian StyleWang, Feng, Shawkat Islam, and Vladislav Vasilyev. 2015. "Ferrocene Orientation Determined Intramolecular Interactions Using Energy Decomposition Analysis" Materials 8, no. 11: 7723-7737. https://doi.org/10.3390/ma8115419