2. Results and Discussion
As mentioned in the experimental section, the thermal treatment for the synthesis of the samples belonging to the BiCuSe
1−xS
xO solid solution needed to be optimized. Indeed, the decomposition temperature of the ZrCuSiAs phase depends on the actual composition of the samples, as it can be observed in
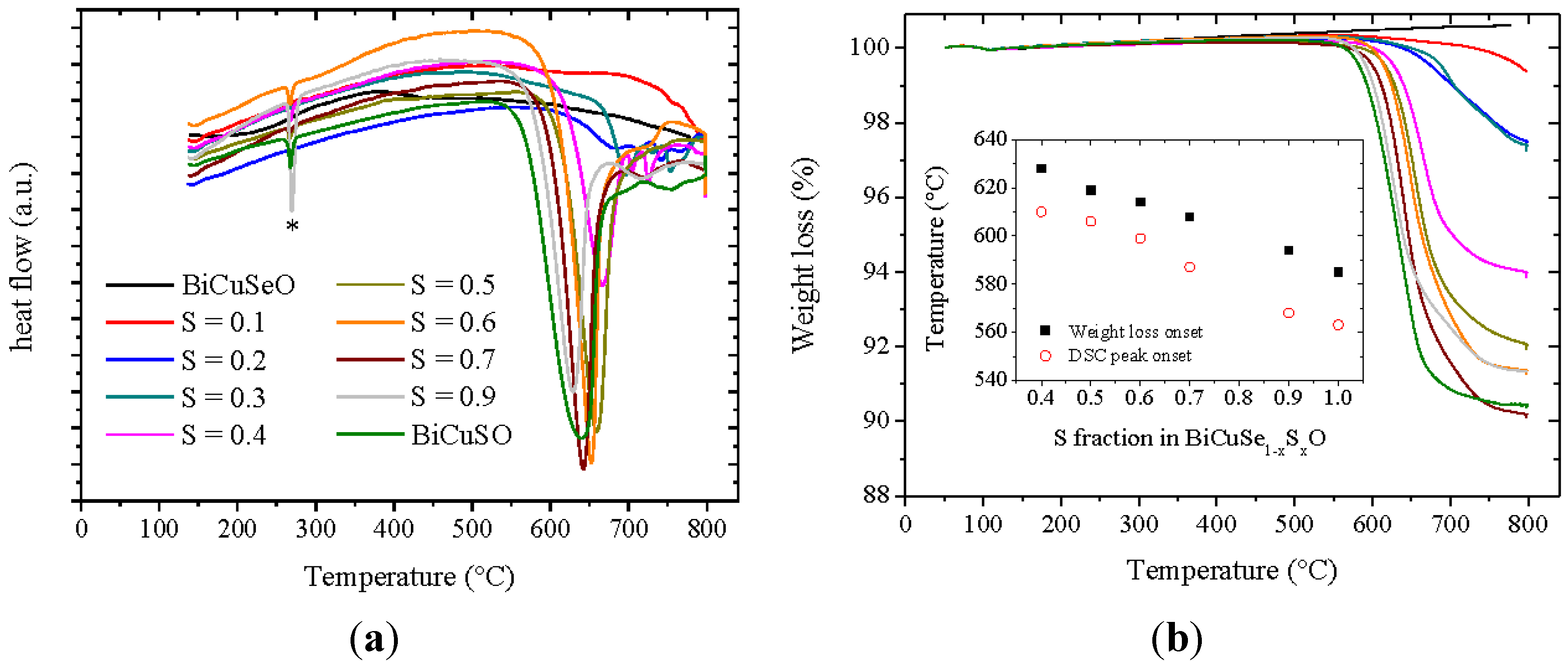
Figure 1, which shows the differential scanning calorimetry and thermogravimetric analysis curves of BiCuSe
1−xS
xO powders under flowing argon. No DSC peak can be observed for BiCuSeO up to 790 °C, which confirms the stability of this compound up to about 800 °C. When increasing the sulfur content to x = 0.3, a clear change of the slope of the curve is present below about 700 °C, although no clear peak can be seen. Above x = 0.4, a well-resolved peak is clearly observed, and the onset temperature of this peak decreases when the sulfur content increases, down to 560 °C for BiCuSO. It shows that the stability of the phase is strongly reduced when substituting selenium by sulfur. Besides, a small peak is observed around 270 °C when the fraction of sulfur exceeds 0.4, which can be explained by the presence of a faint amount of Bi in the samples, see later. This decrease of the decomposition temperature of BiCuSe
1−xS
xO when increasing the sulfur fraction is correlated to volatilization in sulfur-rich samples. No weight loss is observed in the TGA curve of BiCuSeO up to 790 °C, which is consistent with our previous study reporting a good stability of this compound
versus volatilization under argon [
25]. However, this is not the case when the fraction of sulfur increases, which leads to an increase of the weight loss and to a decrease of the temperature triggering the onset of weight loss, as it can be seen in the inset of
Figure 1. Therefore, as the volatilization could probably be slightly reduced during the synthesis in sealed silica tubes as compared to flowing argon, a synthesis temperature as large as 600 °C could be used, whereas the SPS temperature had to be limited to 500 °C for the sulfur-containing samples, which contrasts with BiCuSeO that can be synthesized at higher temperature [
9]. Although the volatilization is unambiguously linked to the presence of sulfur, we have not been able to determine precisely the nature and amount of the elements lost. Indeed, XRD patterns recorded after the thermogravimetric analysis of BiCuSO have shown that the main residual phase is Bi, with a minor presence of poorly crystallized Cu
2-xS and possibly a bismuth oxide and/or a mixed bismuth-selenium oxide (see
Figure S1). Moreover, the total weight loss observed during the thermogravimetric analysis does not correspond to the total mass fraction of sulfur in the compounds. Therefore, it is not possible to determine precisely which element(s) is lost besides sulfur and with which proportion, which could have been used to partly compensate the volatilization during the synthesis process, although the limited amount of oxygen-containing phases after DSC (
Figure S1) could be an indication of S
xO
y losses. Moreover, it is not possible to determine whether the decomposition of sulfur-rich samples is due to the volatilization of some elements, or the decomposition leads to degradation-products that volatilize. Besides giving clues for the synthesis of the BiCuSe
1−xS
xO compounds, these results show that sulfur-substituted samples are much less stable than sulfur-free BiCuSeO, even under argon and with S fraction as low as 20%. Therefore, although the use of sulfur to substitute selenium could lead to a significant decrease of the raw material price of the thermoelectric oxychalcogenides, this lack of stability could preclude its industrial use without appropriate coatings to limit the sulfur volatilization and the materials degradation.
Figure 1.
Differential scanning calorimetry curves of BiCuSe1−xSxO powders under argon (a) and thermogravimetric analysis of BiCuSe1−xSxO powders under argon (b). The peak marked with a * in the DSC curves corresponds to the melting of Bi. The inset shows the onset of the DSC peak corresponding to the decomposition of BiCuSe1−xSxO and the onset of weight loss.
Figure 1.
Differential scanning calorimetry curves of BiCuSe1−xSxO powders under argon (a) and thermogravimetric analysis of BiCuSe1−xSxO powders under argon (b). The peak marked with a * in the DSC curves corresponds to the melting of Bi. The inset shows the onset of the DSC peak corresponding to the decomposition of BiCuSe1−xSxO and the onset of weight loss.
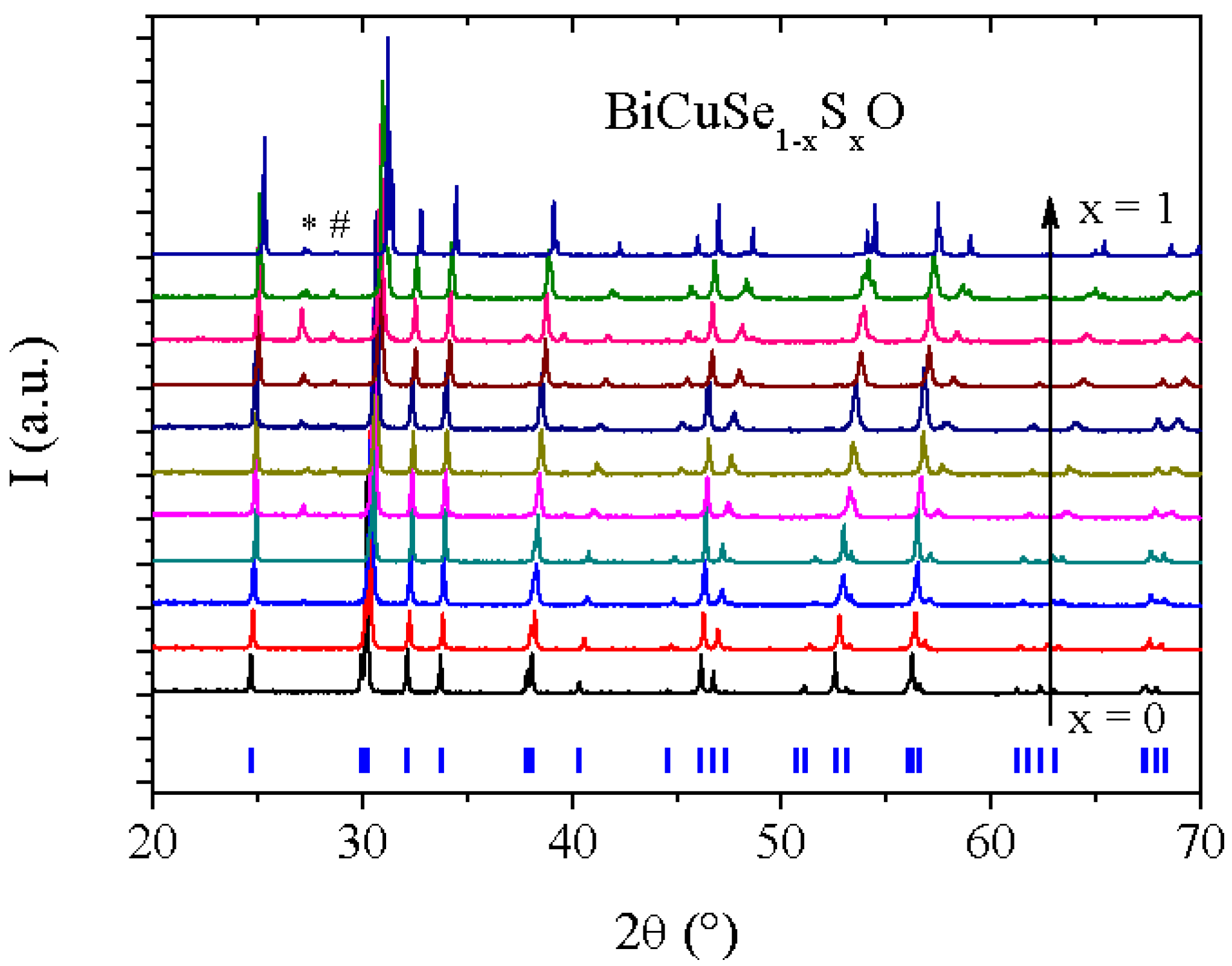
Figure 2 shows the XRD patterns of the samples belonging to the BiCuSe
1−xS
xO solid solution. Both BiCuSeO and BiCuSO crystallize in the ZrCuSiAs structure type with P4/nmm space group, and consist in [Bi
2O
2] and [Cu
2Ch
2] layers stacked along the c axis of the tetragonal unit cell. Between x = 0 and x = 0.3, the samples are well-crystallized and are single phase, within the detection limit of the XRD. When x exceeds 0.4, two supplementary peaks can be observed in the diffraction patterns, one unattributed and the other one corresponding to Bi, which is consistent with the small endothermic peak observed in the DSC curves at ~270 °C. However, all samples remain well-crystallized, and a monotonous shift of the Bragg peaks can unambiguously be observed, which shows that a complete solid solution exists between the end members BiCuSeO and BiCuSO after sufficient heating time. As the presence of the secondary phases for x > 0.4 could have originated from insufficient annealing time due to lower reaction temperature as compared to BiCuSeO, we have tried to use longer heating times or successive annealing with intermediate grinding, but no improvement of the quality of the samples was observed. Worst, the amount of secondary phases tended to increase with successive grinding and annealing, which rather suggests that the presence of these phases could be linked to the volatilization occurring in sulfur-rich samples during the thermal treatment. All attempts to obtain single phase samples for sulfur-rich compositions have been unsuccessful, with always a few percent of secondary phases in the samples (<3% of Bi except for x = 0.8 where it reaches about 8%).
Figure 2.
XRD patterns of BiCuSe1−xSxO samples. The ticks below the patterns indicate the Bragg positions of BiCuSeO. The peak marked with a * corresponds to Bi, and we have not been able to attribute the one marked with a #.
Figure 2.
XRD patterns of BiCuSe1−xSxO samples. The ticks below the patterns indicate the Bragg positions of BiCuSeO. The peak marked with a * corresponds to Bi, and we have not been able to attribute the one marked with a #.
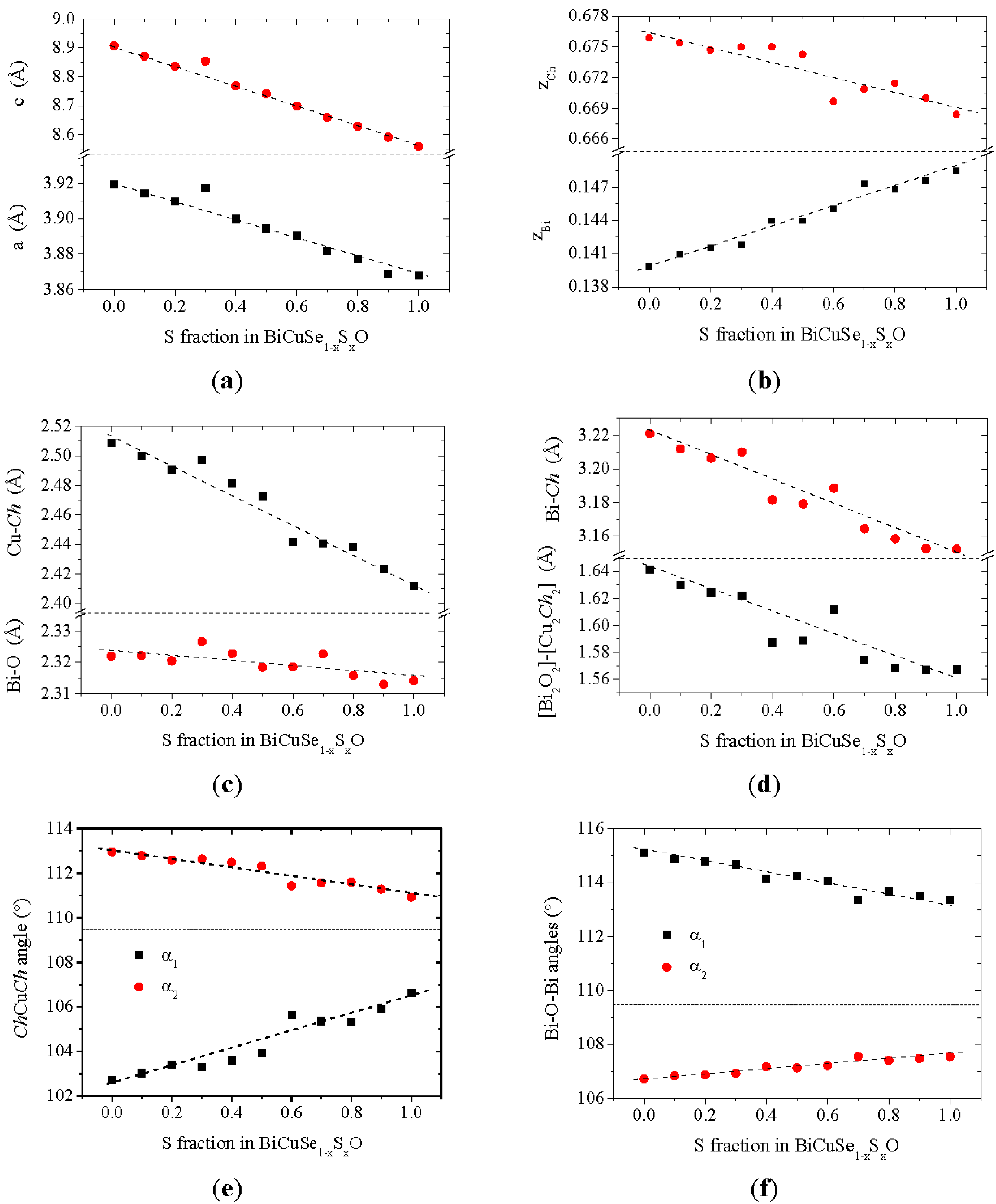
The lattice parameters obtained using Rietveld refinement of the XRD patterns are plotted in
Figure 3a. The lattice parameters of the end members of the solid solution are a = 3.919 Å, a = 3.868 Å and c = 8.907 Å, c = 8.559 Å for BiCuSeO and BiCuSO respectively. They are consistent with the values previously reported in the literature [
8]. Regarding the compounds belonging to the BiCuSe
1−xS
xO solid solution, the dependence of their lattice parameters on the sulfur fraction can be well described using Vegard’s law. The strong decrease of both
a and
c is consistent with the atomic radius of Se being much larger than that of S. However, the evolution of the lattice parameters is strongly anisotropic, as the relative decrease of
c (3.9% between BiCuSeO and BiCuSO) is much larger than the relative decrease of
a (1.3% between BiCuSeO and BiCuSO). This anisotropic evolution of the lattice parameters in the solid solution can be related to the layered structure of the ZrCuSiAs phase. Indeed, as the substitution of selenium by sulfur is an isovalent substitution, which takes place only in the [Cu
2Ch2] layer of the structure, it should not significantly influence the local structure of the [Bi
2O
2] layer, as the Bi-O ionic bond should not be significantly affected. As it can be seen in
Figure 3c, the Bi-O distance is almost unchanged (the decrease of this distance is about 0.3% between BiCuSeO and BiCuSO), which limits the decrease of
a as compared to
c. The almost unchanged Bi-O distance is compensated by a large change in the position of the Bi atoms in the unit cell (z
Bi strongly increases between BiCuSeO and BiCuSO as it can be observed in
Figure 3b), which is due to a slight change in the OBi
4 tetrahedra angles whose distortion decreases (
Figure 3f). The large decrease of
c coupled with the slight evolution of the geometry of the OBi
4 tetrahedra leads to a strong decrease of the distance between the Bi plane and the
Ch plane and to a decrease of the Bi-
Ch distance, by 4.5% and 2.1% respectively between BiCuSeO and BiCuSO. Although this decrease of the Bi-
Ch distance is significant, it is rather limited as compared to the one that could have been expected by the complete substitution of selenium by sulfur, which can be roughly estimated as 6% by using the sum of Bi
3+ ionic radius and
Ch covalent radii (the order of magnitude of this estimation would remain the same if using other radii). These ones were chosen because previous electronic structure calculations showed that the formal charge of bismuth in BiCuSeO is large, which can be explained by the ionic nature of the Bi-O bond in the [Bi
2O
2] layer, whereas that of selenium is moderate, which can be explained by the covalent nature of the Cu-Se bond in the [Cu
2Se
2] layer [
9]. This limited decrease means that the overlap between the
Ch-np and the Bi-6p orbitals should decrease when increasing the sulfur fraction, possibly leading to an increase of the 2D characters of the electrical transport in this material. This evolution is opposite to the one reported previously for the BiCuSe
1−xTe
xO solid solution [
23].
Figure 3.
Structural parameters of the BiCuSe1−xSxO samples: lattice parameters (a); Bi and Ch atomic positions (b); Bi-O and Cu-Ch distances (c); Bi-Ch distance and distance between [Bi2O2]-[Cu2Ch2] planes (d); CuCh4 tetrahedra angles (e) and OBi4 tetrahedra angles (f).
Figure 3.
Structural parameters of the BiCuSe1−xSxO samples: lattice parameters (a); Bi and Ch atomic positions (b); Bi-O and Cu-Ch distances (c); Bi-Ch distance and distance between [Bi2O2]-[Cu2Ch2] planes (d); CuCh4 tetrahedra angles (e) and OBi4 tetrahedra angles (f).
As the radius of sulfur is smaller than that of selenium, the Cu-
Ch distance decreases when Se is substituted for S by 3.9% between BiCuSeO and BiCuSO (
Figure 3c). This decrease of the Cu–
Ch distance is coupled to an evolution of the geometry of the Cu
Ch4 tetrahedra. Indeed, as the relative decrease of
c is larger than the relative decrease of
a (
Figure 3a), the Cu
Ch4 tetrahedra are “compressed” along the
c axis, which leads to a significant decrease of their distortion (
Figure 3e), although the chemical pressure applied when completely substituting selenium by sulfur is not sufficient to obtain regular tetrahedra. It has been previously shown using band structure calculations that the electrical properties of BiCuSeO are mostly driven by the [Cu
2Se
2] layer, with an almost equal contribution of the Cu-3d and Se-4p orbitals to the density of states close to the Fermi level [
8,
23]. Therefore, the structural evolution observed here when substituting Se with S could lead to changes in the band structure, and therefore the transport properties. On the one side, the change in the geometry of the Cu
Ch4 tetrahedra should influence the overlap between the Cu-3d and Se-4p orbitals, as both d and p orbitals are directional, and the nature of the bands close to the Fermi level could be affected. On the other side, the decrease of the Cu-
Ch distance (3.9% between the two end members of the solid solution) is smaller than the one that could be expected, which can be roughly estimated as 5.5% by using the sum of the Cu and
Ch covalent radii. It should result in a decrease of the Cu 3d-
Ch np overlap, leading to a narrowing of the Cu-
Ch antibonding levels and less dispersive bands, with a slightly lower covalent character of the Cu-
Ch bonds.
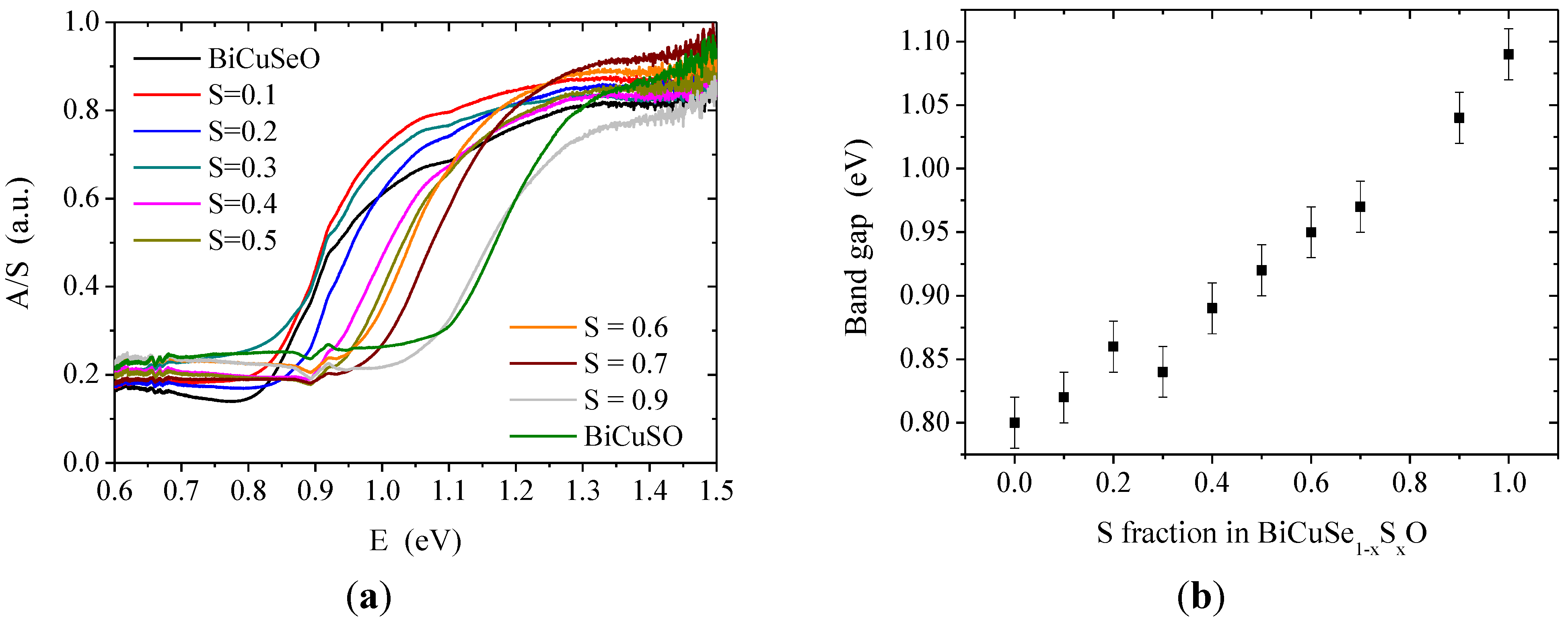
As expected, there is an evolution with the substitution of selenium with sulfur of the band gap determined by UV-Vis spectroscopy. The diffused reflectance spectra observed at room temperature where converted using Kubelka-Munk relation:
with R the absolute reflectance of the sample; A the molar absorption coefficient; and S the scattering coefficient. As the Kubelka-Munk relation assumes a thick infinitely diluted sample in a non-absorbing matrix with a diffuse-diffuse geometry, which was not the case here, the absolute values obtained for A/S do not have a physical meaning. However, the optical band gap can be estimated confidently through the band-edge structure. A clear band-edge structure can be observed in
Figure 4, which indicates that the band gap is in the near-infrared region for all samples. The estimated band gap is 0.8 eV for BiCuSeO and 1.1 eV for BiCuSO, which is consistent with the values reported in the literature for these two compounds [
8]. Within the solid solution, the band gap monotonously increases when increasing the sulfur fraction, with an almost linear trend.
The structural parameters of BiCuSeO and BiCuSO obtained using the
ab initio calculations are summarized in
Table 1. The lattice parameters agree reasonably well with the experimental data, although they are slightly larger, 2.2% and 4.3% respectively for
a and
c in the case of BiCuSeO, and 1.2% and 3.4% in the case of BiCuSO. However, the experimental trends, namely a decrease of the Cu-
Ch and of the Bi-
Ch distances slightly smaller than the ones expected from the evolution of the
Ch radii and a decrease of the distortion of the Cu
Ch4 tetrahedra, are still observed with the structural parameters obtained by the calculations. Another calculation of the electronic band structure has been performed by setting the structural parameters to the experimental values, but no significant difference was observed between the results (except slightly larger Mulliken overlap populations due to reduced distances).
Figure 4.
UV-Vis spectroscopy (
a), for the definition of the
y-axis, see text and evolution of the band-gap in the BiCuSe
1−xS
xO solid solution (
b). The spectrum corresponding to BiCuSe
0.2S
0.8O was not recorded due to the two large amounts of secondary phases in this compound. The small feature in the spectra around 0.9 eV is due to a small glitch in the baseline, see
Figure S2 in the supplementary information.
Figure 4.
UV-Vis spectroscopy (
a), for the definition of the
y-axis, see text and evolution of the band-gap in the BiCuSe
1−xS
xO solid solution (
b). The spectrum corresponding to BiCuSe
0.2S
0.8O was not recorded due to the two large amounts of secondary phases in this compound. The small feature in the spectra around 0.9 eV is due to a small glitch in the baseline, see
Figure S2 in the supplementary information.
The atomic valence charges resulting from a Mulliken population analysis are shown in
Table 1, and the Mulliken population overlap for BiCuSeO and BiCuSO are summarized in
Table 2. As already mentioned [
9], the charges are far from the ones expected from a simple ionic model, namely Bi
2+, Cu
+,
Ch− and O
2−, which proves a mostly covalent bonding between copper and selenium/sulfur, as compared to the Bi–O bonding, which is partly ionic, and a charge transfer between the [Bi
2O
2] and the [Cu
2Ch2] layers. The covalent behavior of the Cu-
Ch bonding is confirmed by the large calculated Cu-
Ch Mulliken overlap population. As expected from the evolution of the Cu-
Ch distances when substituting Se with S (see above), there is a decrease of the covalent character of the Cu-
Ch bond when going from Se to S, which is evidenced by the decrease of the overlap population from 0.116 for Cu-Se to 0.081 for Cu-S. It results in an increase of the absolute value of the charges of Cu and
Ch. As mentioned previously [
9], the interlayer bonding in BiCuSeO is partly covalent, which is evidenced by the moderate Bi-Se overlap population. As expected from the evolution of the Bi-
Ch distance with the substitution (see above), this overlap is strongly reduced in BiCuSO as compared to BiCuSeO, corresponding to an increase of the ionic character of the interlayer bonding.
Table 1.
Structural parameters and atomic charges obtained by the ab initio calculations.
Table 1.
Structural parameters and atomic charges obtained by the ab initio calculations.
| Element | x | y | z | Charge | Element | x | y | z | Charge |
|---|
| Bi | 0.2500 | 0.2500 | 0.1316 | +1.50 | Bi | 0.2500 | 0.2500 | 0.1423 | +1.33 |
| Cu | 0.7500 | 0.2500 | 0.5000 | +0.23 | Cu | 0.7500 | 0.2500 | 0.5000 | +0.32 |
| Se | 0.2500 | 0.2500 | 0.6789 | −0.63 | S | 0.2500 | 0.2500 | 0.6697 | −0.73 |
| O | 0.7500 | 0.2500 | 0.0000 | −1.10 | O | 0.7500 | 0.2500 | 0.0000 | −0.92 |
| Lattice parameters | a = 4.004 Å | Lattice parameters | a = 3.9149 Å |
| c = 9.2873 Å | c = 8.8536 Å |
Table 2.
Mulliken population analysis.
Table 2.
Mulliken population analysis.
| Element 1 | Element 2 | Calc Distance (Å) | Mulliken Overlap Population | Element 1 | Element 2 | Calc Distance (Å) | Mulliken Overlap Population |
|---|
| Bi | O | 2.346 | 0.002 | Bi | O | 2.328 | 0.004 |
| Se | 3.333 | 0.036 | S | 3.230 | 0.017 |
| Cu | Se | 2.601 | 0.116 | Cu | S | 2.467 | 0.081 |
| Cu | 2.831 | 0.045 | Cu | 2.768 | 0.031 |
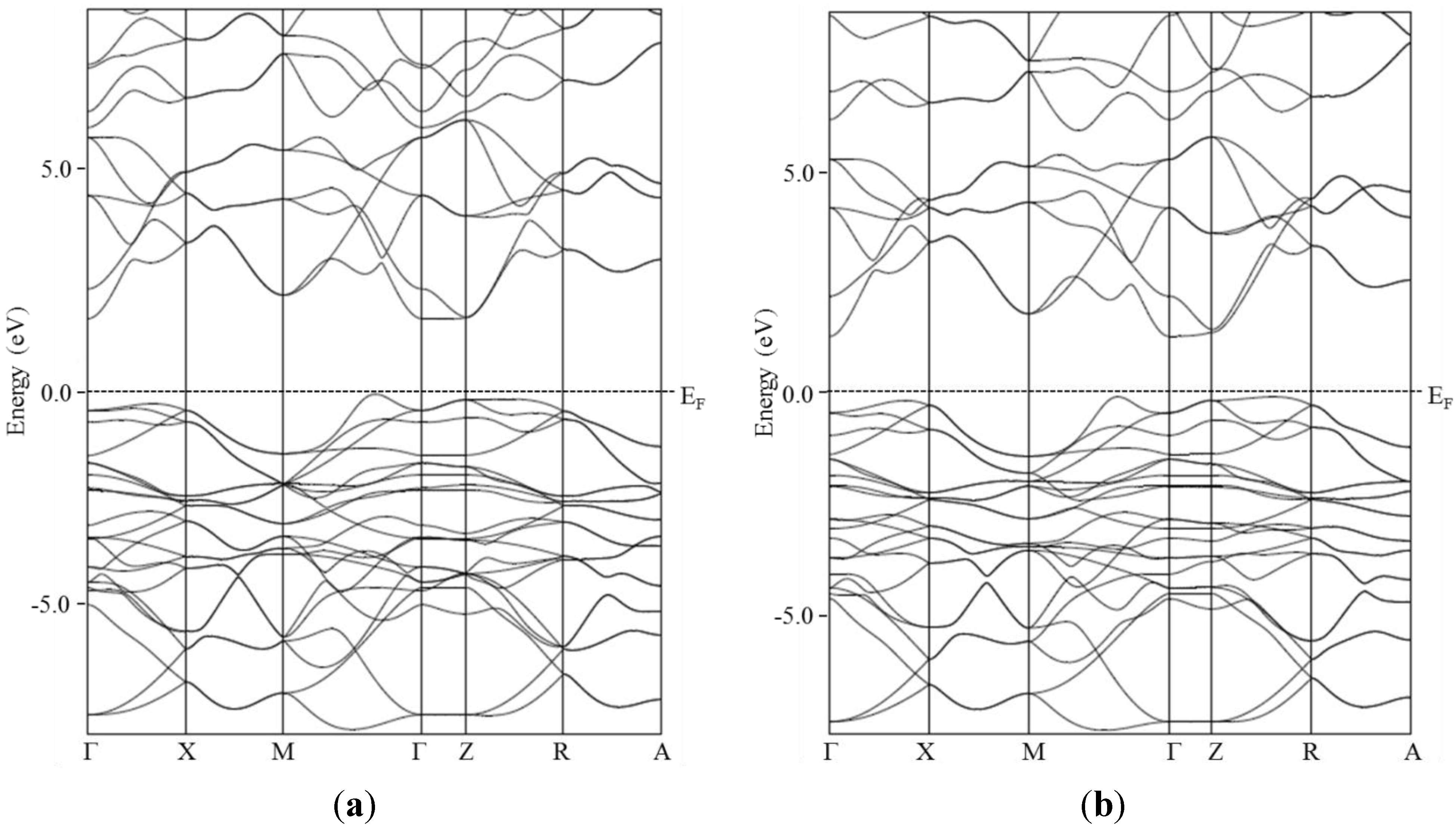
The electronic band structures close to the Fermi level of BiCuSeO and BiCuSO are plotted in
Figure 5. Both of them are consistent with the ones previously reported in the literature [
8,
9]. The band gap is slightly larger in BiCuSO than in BiCuSeO, which is consistent with the experimental observations. Except for the increase of the band gap when substituting Se with S, both electronic band structures are almost the same, which was somehow unexpected because of the evolution of the structural parameters (decrease of the distortion of the CuCh4 tetrahedra and evolution of the distances between Cu and
Ch and Bi and
Ch). Therefore, electrical transport properties should not be affected very much by the substitution.
Figure 5.
Electronic band structure close to the Fermi level of BiCuSO (a) and BiCuSeO (b).
Figure 5.
Electronic band structure close to the Fermi level of BiCuSO (a) and BiCuSeO (b).
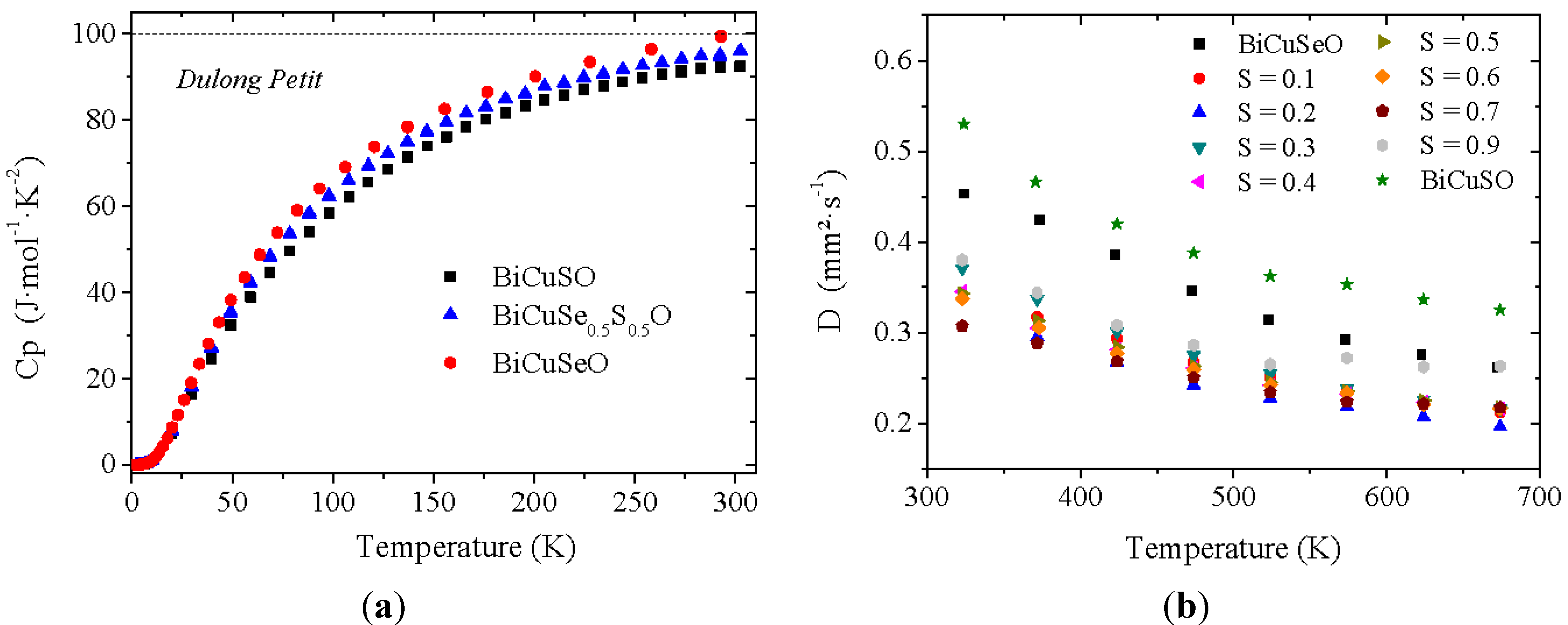
Figure 6a shows the specific heat of the samples belonging to the BiCuSe
1−xS
xO solid solution (for the clarity of the figure, only 3 curves were plotted). As expected, for unintentionally doped semiconductors, the Sommerfeld coefficient γ, which is linked to the density of states at the Fermi level by the relation:
where λ
e-ph is the electron-phonon coupling constant, is very close to zero. A slight difference can be noticed between the different samples: the specific heat of BiCuSeO reaches the Dulong-Petit limit close to room temperature, whereas it is not the case for BiCuSO. Moreover, the specific heat of BiCuSO is slightly lower than that of BiCuSeO in the whole temperature range, and the intermediate compound BiCuSe
0.5S
0.5O lies in between. More generally, the room temperature value of C
p decreases when increasing the sulfur fraction. This result indicates an increase of the Debye temperature θ
D when substituting Se for S, which is consistent with the values calculated in the literature for BiCuSeO (243 K) [
12] and BiCuSO (289 K) [
26].
Figure 6.
Temperature dependence of the specific heat (a), for the clarity of the figure, only 3 curves were plotted, and temperature dependence of the thermal diffusivity of BiCuSe1−xSxO pellets (b).
Figure 6.
Temperature dependence of the specific heat (a), for the clarity of the figure, only 3 curves were plotted, and temperature dependence of the thermal diffusivity of BiCuSe1−xSxO pellets (b).
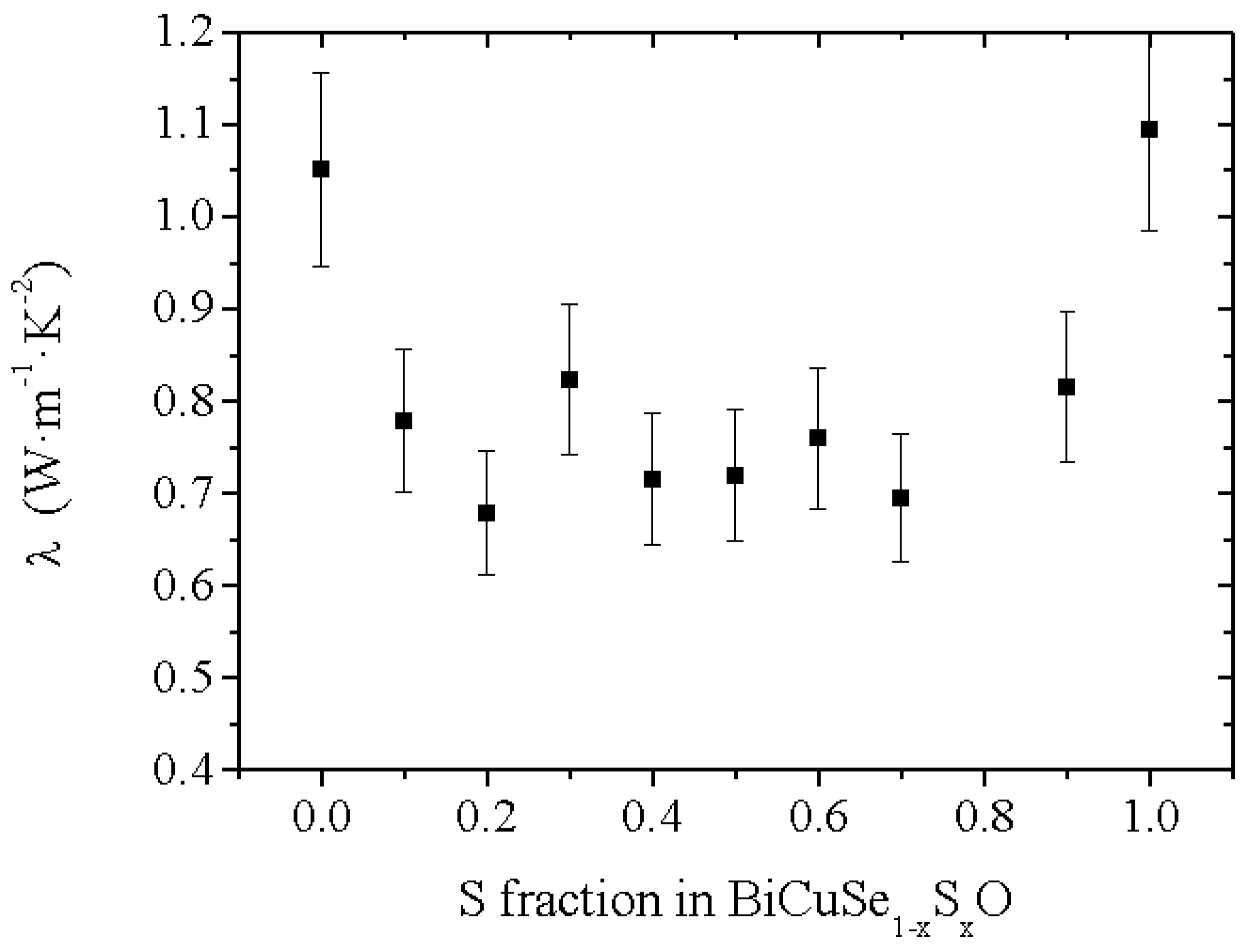
The thermal conductivity in the BiCuSe
1−xS
xO solid solution has been calculated close to room temperature using the interpolated values of the measured specific heat, thermal diffusivity (
Figure 6b) and geometrical density, and the values are plotted in
Figure 7. As the electrical resistivity at room temperature is large (>200 mΩ.cm in the most conductive sample), see later, the thermal conductivity plotted here is almost only constituted by the lattice contribution λ
lat. The thermal conductivity of BiCuSeO is consistent with the value already reported for this compound, with λ~1 W·m
−1·K
−1 [
10]. Interestingly, the room temperature thermal conductivity value of BiCuSO is almost the same within the uncertainty of the measurement, despite a significantly larger thermal diffusivity, because of both lower specific heat and theoretical density. However, the thermal conductivity is strongly reduced for the intermediate compositions. As the microstructure of all samples is micron-size, with no influence of the composition on the grain size or the preferential orientation, the lower values for the intermediate compositions as compared to the end members could not be explained solely by a change in grain-boundary scattering of the phonons. Moreover, as no impurity phase can be detected between S = 0 and S = 0.3, the decrease of λ from BiCuSeO to BiCuSe
0.7S
0.3O cannot originate from impurity scattering of the phonons. However, this decrease can be explained by the diffusion of the phonons by point defect scattering [
27] due to the mass and volume difference between Se and S, and it shows that the partial substitution of Se by S to form the BiCuSe
1−xS
xO solid solution is a good strategy to decrease the thermal conductivity of the thermoelectric oxychalcogenides.
Figure 7.
Evolution of the room temperature value of the thermal conductivity in the BiCuSe1−xSxO solid solution.
Figure 7.
Evolution of the room temperature value of the thermal conductivity in the BiCuSe1−xSxO solid solution.
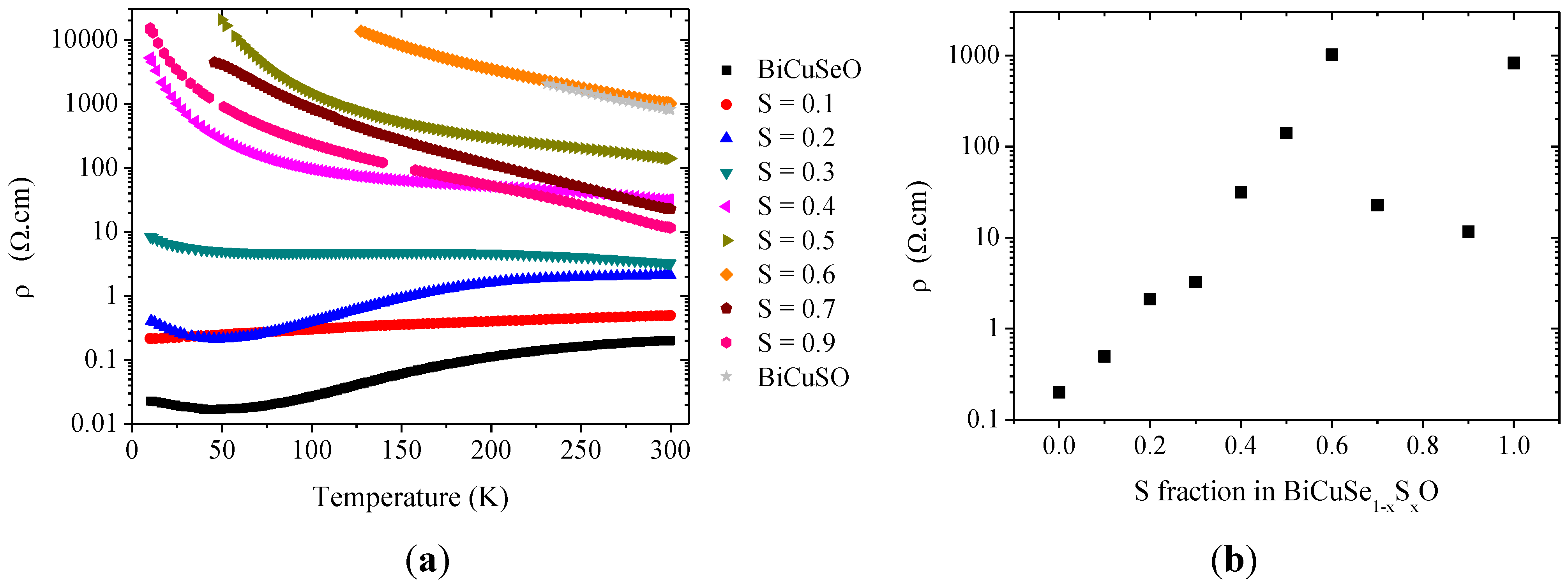
As the electronic band structures of BiCuSeO and BiCuSO are very similar, with the most significant difference being the slight increase of the band gap, no major evolution of the electrical resistivity was expected within the solid solution. However, it is clearly not the case, as it can be observed in
Figure 8, which shows the temperature dependence and the room temperature values of the electrical resistivity in the solid solution, and as it had already been reported by Hiramatsu
et al. [
8]. The electrical resistivity of BiCuSeO exhibits a “bad metal” behavior, with room temperature ρ
300K~200 mΩ.cm and a RRR = 12, as already reported [
8,
9]. It has been shown that this behavior, rather unexpected for a semiconductor with a band-gap of 0.8 eV, can be well explained by an unintentional doping due to the presence of faint amount of copper vacancies [
8,
9], which has been confirmed both experimentally [
19] and theoretically [
28], although it could also be linked to a more complex defect chemistry with complex Cu and Se defects [
29]. When Se is partly substituted by S, the rough tendency is an increase of the electrical resistivity coupled to a change to a semiconductor behavior, consistent with the band-gap of 1.1 eV observed in BiCuSO (the relatively large dispersion of the data for S > 0.6 originates from the presence of Bi secondary phase with low resistivity in the samples for large sulfur fractions, most especially for x = 0.7 and x = 0.9, as it has been observed in the XRD patterns). The increase of the room temperature value is very large, about 4 order of magnitude between x = 0 and x = 0.6. We have tried to evaluate the evolution of the holes concentration in the solid solution using Hall effect measurements, but the samples revealed themselves to be too resistive for an accurate determination of the Hall coefficient. However, we can reasonably hypothesize that the large increase of the electrical resistivity when the sulfur fraction increases is caused by a large decrease of the holes’ concentrations in the samples. This decrease could originate from a decrease of the copper vacancies’ concentrations with sulfur substitution, due to an evolution of the energy of formation of this acceptor defect. Indeed, a large evolution of the energy of formation of Cu and Bi vacancies has been recently suggested in Bi
1−xPb
xCuTeO by DFT calculations, for Pb concentrations as low as 0.1 [
22]. Therefore, a similar evolution could be observed in BiCuSe
1−xS
xO.
Figure 8.
Temperature dependence (a) and evolution of the room temperature value of the electrical resistivity (b) in the BiCuSe1−xSxO solid solution.
Figure 8.
Temperature dependence (a) and evolution of the room temperature value of the electrical resistivity (b) in the BiCuSe1−xSxO solid solution.
As the electrical resistivity strongly increases with the substitution of Se by S, the thermoelectric performances of the materials are obviously largely degraded. However, it should be reminded that this substitution is isovalent and that all samples are far from the optimum carriers concentration, which is close to 10
21 cm
−3 in BiCuSeO [
9,
11]. Therefore, the electrical properties would obviously be improved by aliovalent doping, for example using a 2+ cation to substitute Bi. Coupled to ~25% reduction of the thermal conductivity in intermediate compositions of the BiCuSe
1−xS
xO solid solution, it could lead to an increase of the thermoelectric figures of merit ZT as compared to sulfur-free compounds.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



