3.2.1. Glass Transition Temperature Determined by Differential Scanning Calorimetry
During the glass transition process, one of the properties that exhibit abrupt change is the heat capacity. The DSC technique serves to investigate this, and determine the glass transition temperature (
Tg). The
Tg measured in this way is sometimes referred to as “calorimetric”. Due to the fact that the sample is not exposed to external deformations, it is also called “static”
Tg.
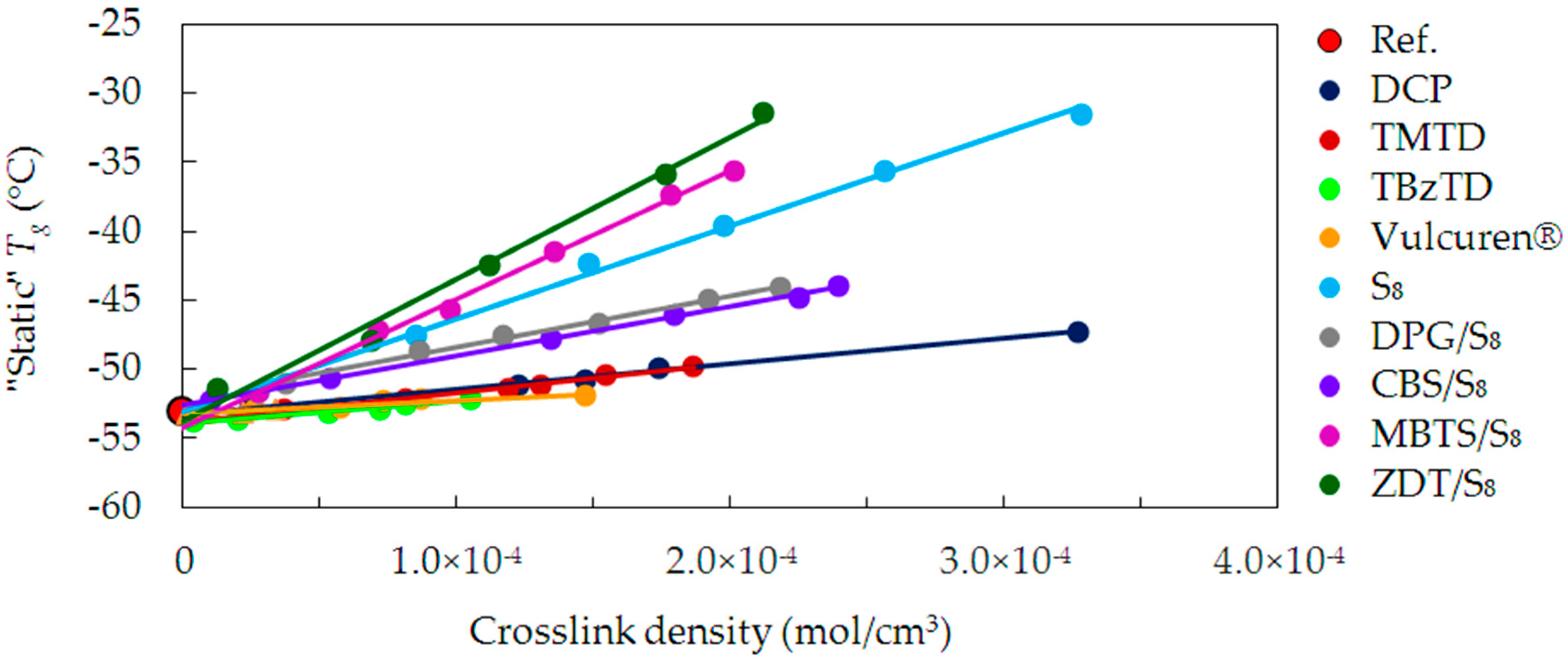
Figure 4 shows changes in
Tg as a function of crosslink density, for the uncrosslinked reference and the series of samples cured with various curatives.
For the uncrosslinked reference (Ref.), the “static” Tg is approx. −53 °C. It has no chemical crosslinks and the chains interact with each other via weak van der Waals forces. Hence, the value of Tg comes from the microstructure of SBR—its building blocks, the relative participation of styrene and butadiene, and molecular weight.
Compared to the reference, for all cured samples the Tg values linearly increase with a rise in crosslink density. As a result of the crosslinking, polymer chains were united with short, strong covalent bonds. This made the cured material more compact and stiffer. Crosslinks are topological constraints, which hinder segmental mobility of the chains. The denser the network is, the shorter the molecular segments between crosslinks are. The increase in the crosslink density is, therefore, followed by a rise in Tg. This is caused by higher thermal energy required to enable the molecular mobility of the polymer segments.
Actually, it is a well-known phenomenon [
15,
16] that the presence of crosslinks causes an increase in
Tg. Fox and Loshaek [
17] derived equations, predicting linear dependency of
Tg on the crosslink density. This prediction concerned polymers with a fixed composition and having a sufficiently low crosslink density. It assumed failure of linearity at higher crosslink densities. In the present research, good linearity is evident in the whole range of crosslink density for all series cured with various curatives. However, trends for particular series of samples show significant differences. They come from other structural factors than the total crosslink density.
Figure 4 shows that the increase in
Tg is the lowest for the series cured with single-component curatives, such as DCP, TMTD, TBzTD, and Vulcuren
®. The increase in
Tg is higher in case of CBS/S
8, DPG/S
8 and pure sulfur S
8 series. The largest change is for MTBS/S
8 and ZDT/S
8. To explain differences between the series, their structural features have to be discussed in detail.
As a result of curing with an organic peroxide DCP, C-C crosslinks were formed. They are short and rigid and, hence, provide good elasticity and high stiffness to the material. It would be logical to assume that due to these characteristics they will cause a large increase in Tg. Actually, the situation is the opposite: an increase in the crosslink density caused little changes in Tg. Additionally, the DCP series is very much in line with the dependency for the TMTD series. Curing with TMTD also produced C–C and monosulfidic crosslinks. On the basis of these identical trends, a few remarks can be made. Firstly, DCP and TMTD led to formation of very short and undiversified crosslinks. Secondly, DCP and TMTD are curatives of low molecular weights and uncomplicated structures. Therefore, complex rearrangement reactions of these curatives were not possible. This made their reactivity at elevated temperature uncomplicated. Due to the low molecular weight, a part of the decomposition products possibly volatized and liberated from the system. Further, in addition to the formation of intermolecular crosslinks, the DCP and TMTD curatives modified the polymer chains by grafting on them. However, due to their small size, the DCP and TMTD did not stiffen the polymer chains in this way. As a result, the chains remained flexible and mobile. Due to all these factors, an increase in crosslink density caused very low rise in Tg.
For TBzTD and Vulcuren® series, the change of Tg with an increase in the crosslink density is also small. This might be caused by the presence of benzyl groups in the structure of those curatives. These groups are also present in the SBR structure as styrene units, which makes the TBzTD and Vulcuren® molecules alike to the polymer matrix. In spite of their large amount and bulkiness, they led to a minor change in heat capacity. Therefore, the increase in Tg was small. Additionally, despite the difference in the crosslink structure of TBzTD and Vulcuren® samples, their Tg changes are similar. This suggest that the crosslink structure is of minor importance.
Curing with rhombic sulfur S
8 led to a large increase in
Tg as a function of the crosslink density. This increase is evidently higher than for the above discussed single-component curatives. Curing with pure sulfur is inefficient—it has a slow curing rate. Moreover, it produces a large number of polysulfidic and intramolecular cyclic structures [
18,
19]. These numerous modifications restricted the free rotation possibilities of a single polymer chain. In this way, they hindered the mobility of the chains and considerably stiffened them. As a result, the
Tg significantly increased.
In case of accelerated sulfur curing, the increase in
Tg as a function of crosslink density is strongly dependent on the type of accelerator used. The increase for the DPG/S
8 and CBS/S
8 series is lower, when compared to the sulfur cured samples (S
8). This can arise from two facts. Firstly, when the accelerator is combined with sulfur, the formation of intermolecular crosslinks is more efficient. It leads to lower quantity of intramolecular modifications of the polymer chains. Secondly, the DPG and CBS molecules have a low molecular weight (211.26 g/mol and 264.41 g/mol, respectively) and are small in size. Hence, the pendant groups of the accelerator fragments are not bulky. Therefore, they do not hinder the free rotation of the macromolecular segments to such a high degree like the cyclic sulfur structures do. These two effects can contribute to the observed trend lines as shown in
Figure 4. Additionally, DPG/S
8 and CBS/S
8 series exhibit very alike dependencies, because the accelerators are similar in molecular weight and size. As mentioned before, the ratio of accelerator to sulfur, expressed in “phr” unit, was 1. Considering the molecular weight of the curatives, and calculating their amount in moles, this ratio is slightly different. For DPG/S
8 it equals 1.18, whereas for CBS/S
8 it is 0.92 (
Table 3).
For the MBTS/S8 series, the increase in Tg is higher than for the series cured with pure sulfur. This is due to the structure of the MBTS molecule. It has a high molecular weight (332.49 g/mol) and aromatic heterocyclic moieties, which exhibit high stiffness. Fragments of the accelerator, together with sulfur, grafted onto the polymer chains during the curing process. Due to the bulkiness and inflexibility of these structures, this modification caused considerable stiffening of the polymer chains. Consequently, it was followed by a large increase in Tg. The ratio between the number of moles of MBTS and S8 equals 0.73. It is lower than for the DPG/S8 and CBS/S8 series, in which the accelerator molecules are smaller. Furthermore, the MBTS/S8 samples have a similar crosslink structure compared to the DPG/S8 and CBS/S8 samples. However, the Tg values differ between those series. This proves that not the crosslink structure, but the accelerator type influences the behavior of the polymer chains.
In case of the samples cured with ZDT/S8, the increase in Tg with rise in crosslink density is very large. It is much higher than for the S8 series, and the greatest from all the studied curing systems. This effect is due to the ZDT molecular weight of 772.47 g/mol, which is the largest of all used accelerators. The ZDT structure is complex and has the bulky alkyl moieties. Similarly to the above discussed systems, modifications of the chains were formed by the curatives during the curing process. Due to their bulkiness, the mobility of the macromolecular segments was limited to a high extent. This caused a significant increase in the Tg. Furthermore, ZDT is a polar molecule, which also possibly contributed to this rise. The ZDT fragments bound to the chains could interact with other similar fragments via intermolecular specific interactions. This additionally restricted the mobility of the polymer chains. As a result of curing, the crosslink structure developed in ZDT/S8 and sulfur S8 samples was similar. However, the Tg values of these two series differ significantly. The comparable crosslink structure does not result in the same behavior of the polymer chains. It is the various types of intramolecular modifications, which explain the differences. In case of ZDT/S8, the bulky fragments of accelerator, together with sulfur, grafted of the polymer chains. In case of S8 series, the modifications were in a form of cyclic structures and pendant groups composed of sulfur. The combined ZDT/S8 curatives stiffened the polymer chains to a greater extent than solely sulfur S8.
As shown in
Table 3, one-to-one ratio between ZDT and S
8 is as low as 0.17, when their number of moles is considered. This means that the number of ZDT molecules was in a very small amount in relation to the molecules of rhombic sulfur. In fact, “phr” is a simplified unit, used to easily calculate the amount of various additives in a rubber mix formulation on a weight basis. Due to its universality, it was widely accepted by rubber technologists and research scientists. However, as the above described examples show, the “phr” unit used to calculate the amount of curatives can be misleading. Curatives can exhibit diversified structure and molecular weight and this unit does not consider these important variables. Therefore, in case of substances, such as curatives, it is non-informational and indirect. To overcome this problem, the amount of curatives should be expressed in “mol” unit. It considers the molecular weight, so that the number of reacting molecules is clearly expressed.
3.2.2. Glass Transition Process Monitored by Dynamic Mechanical Analysis
DMA tests were performed to study microscale changes in molecular mobility, as a response to externally applied mechanical deformations on the samples. In this way the “dynamic”
Tg values were determined by the maximum of the tan delta peak.
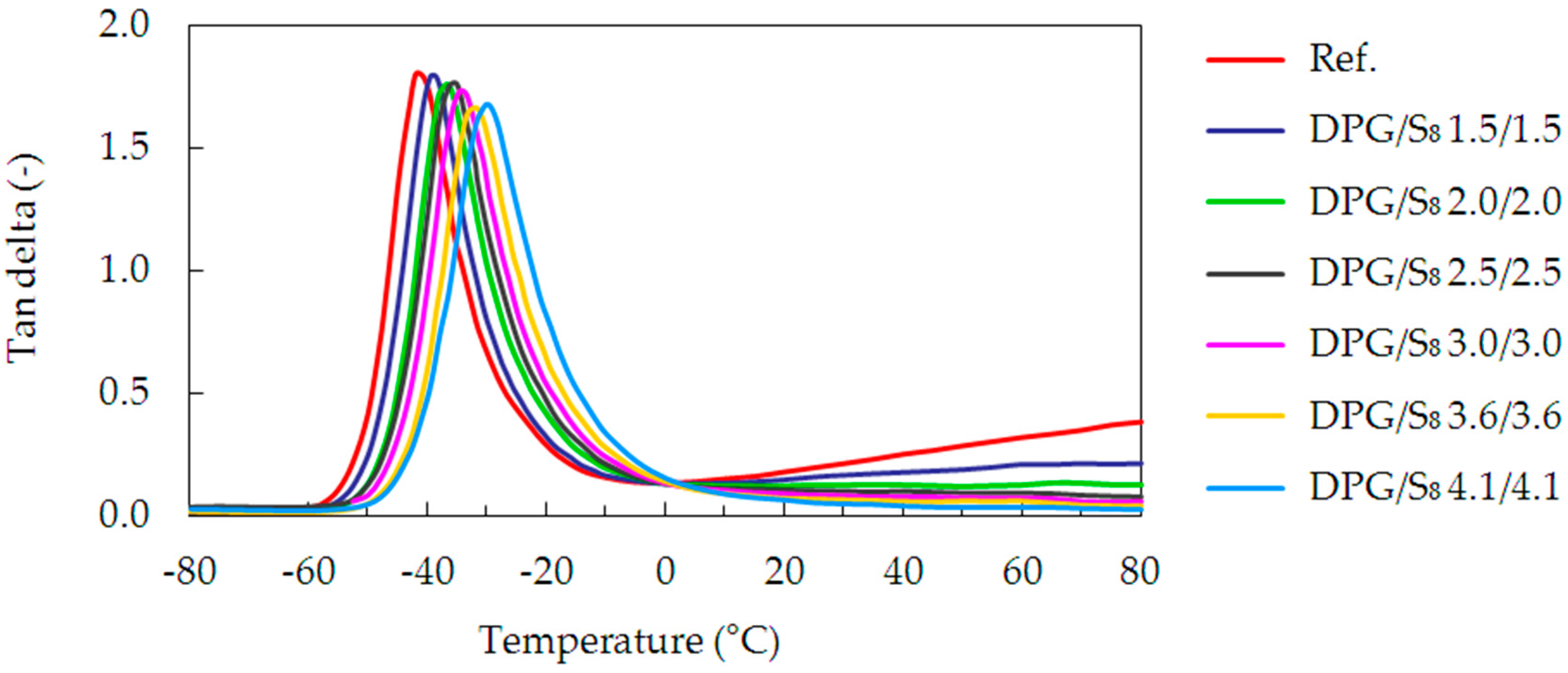
Figure 5 presents exemplary data from the DMA measurements, represented by the DPG/S
8 series. They show the tan delta plotted versus the temperature for different crosslink densities.
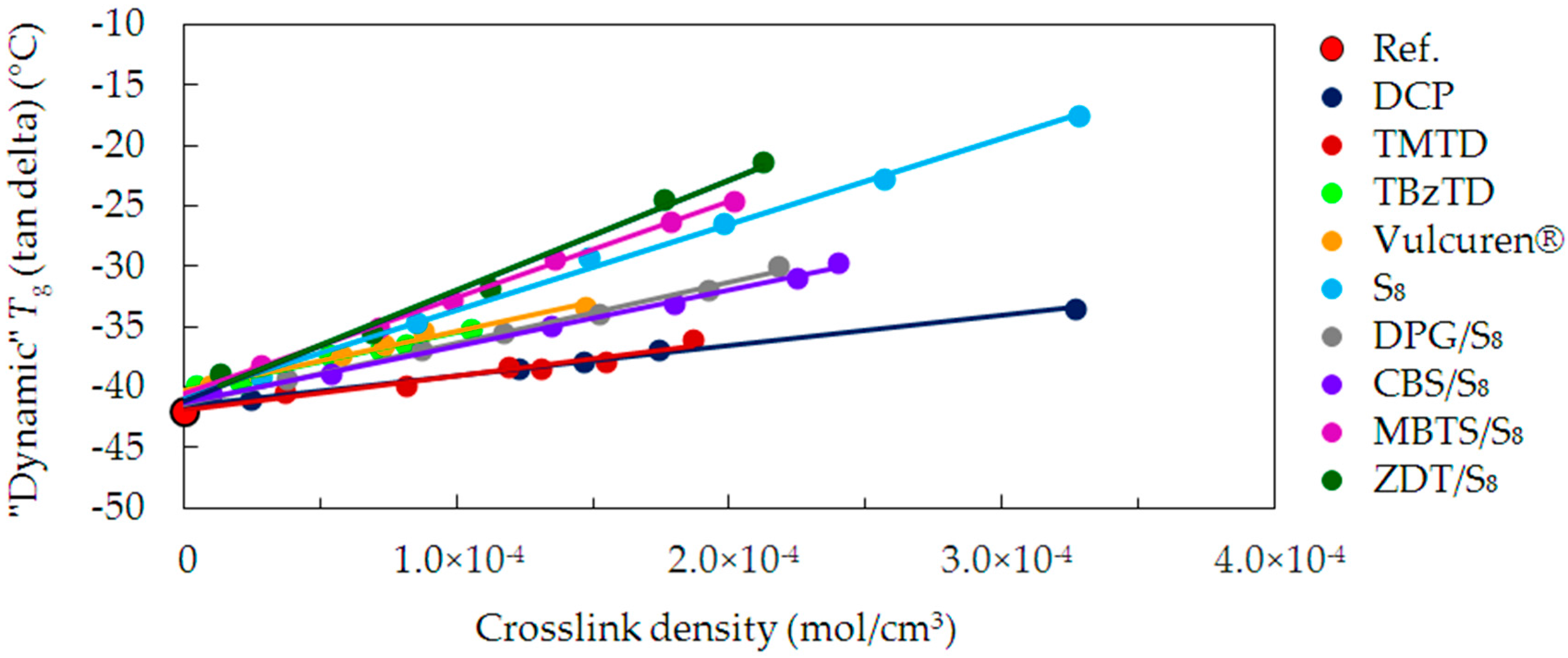
Figure 6 depictures the “dynamic”
Tg values as a function of the crosslink density, for all of the investigated samples. The “dynamic“
Tg shifts to higher values with increasing crosslink density when compared to the uncrosslinked reference. These changes of
Tg are related to the chains’ relaxation. The “dynamic”
Tg values show significant differences between the series cured with various curatives, as already observed in case of the “static”
Tg.
For the uncrosslinked reference, the “dynamic” Tg is approx. −42 °C. It is significantly larger than the “static” Tg (−53 °C). Such a difference is caused by frequency of 10 Hz in DMA measurement. Due to this, the “dynamic” Tg shifted to a higher temperature.
In case of the cured samples, the lowest increase in the “dynamic” Tg is for the DCP and TMTD series. This corresponds to the dependencies in the “static” Tg. However, for the TBzTD and Vulcuren® samples, the increase in “dynamic” Tg is significantly larger than in case of the “static” Tg. This effect arises from differences in the measurement techniques of DSC and DMA. Both in DMA and DSC measurements, molecular motions of the polymer segments begin at a temperature associated with the glass transition. The polymer segments are restricted by the crosslinks and modifications. In DSC, the sample is static, whereas in DMA it is deformed. Thus, in DMA, the mobility of the chains is a response to the deformations of a particular frequency. As a result, segments of the chains undergo conformational rearrangements. It minimizes localized stress, as it is possible on an experimental time scale. Curing with TBzTD and Vulcuren® led to the formation of stiff and bulky structures. They require high thermal energy to enable molecular mobility under dynamic conditions. The trend lines for the TBzTD and Vulcuren® series are similar to those of DPG/S8 and CBS/S8. The “dynamic” Tg of the sulfur cured samples (S8), show a further increase in Tg. In case of MBTS/S8 cured samples, a higher increase in Tg is observed. This is due to the inflexible and bulky moieties of MBTS, combined with sulfur, causing a significant stiffening of the polymer chains. Finally, the most evident increase in Tg was observed for ZDT/S8. In this series the accelerator molecules are the largest and cause the highest stiffening of the polymer chains.
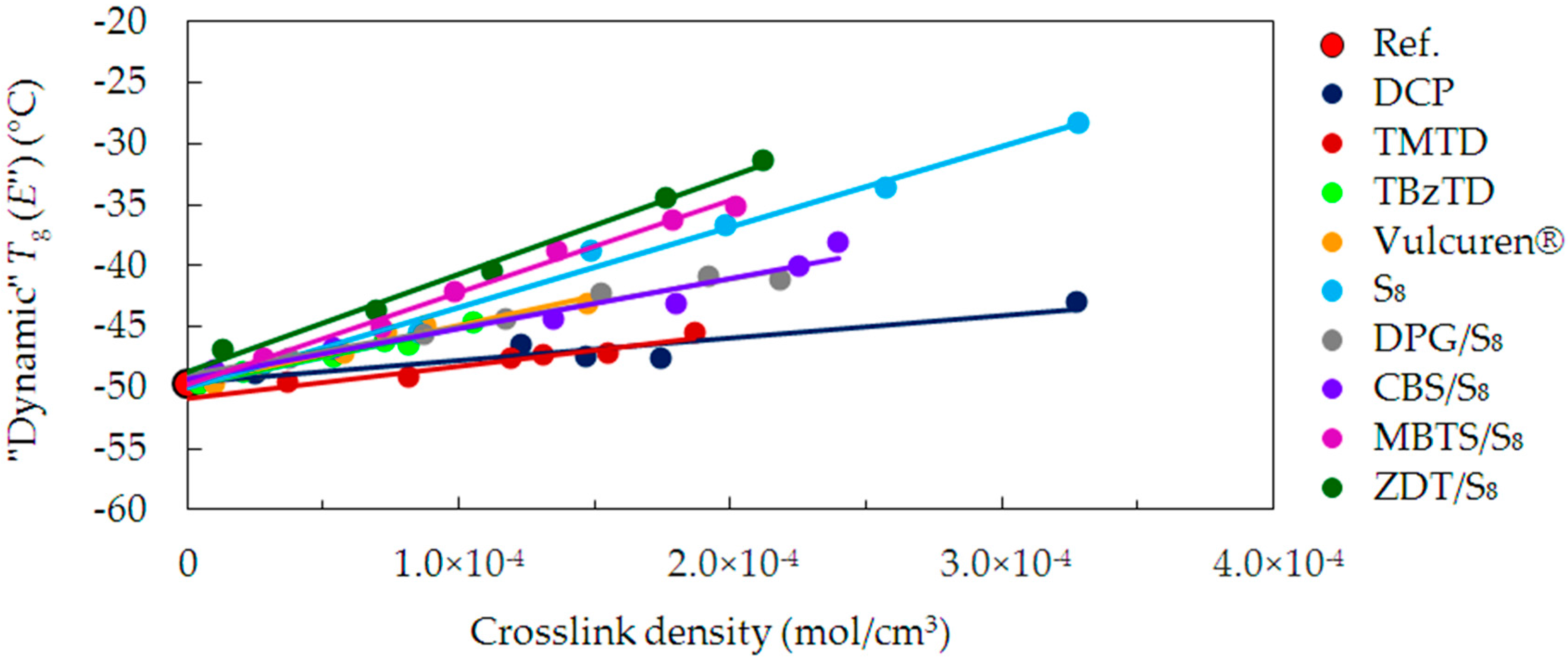
The “dynamic” glass transition process is sometimes also ascribed to the maximum of the loss modulus (
E’’) peak.
Figure 7 presents the
Tg values determined from
E’’. The
Tg for the uncrosslinked reference is approx. −50 °C. All cured series of samples show a tendency corresponding to the tan delta “dynamic”
Tg. Differences between the maxima of the tan delta and
E’’ peaks are within the range of 8–11 °C. The values of
E’’ “dynamic”
Tg are close to values of “static”
Tg.
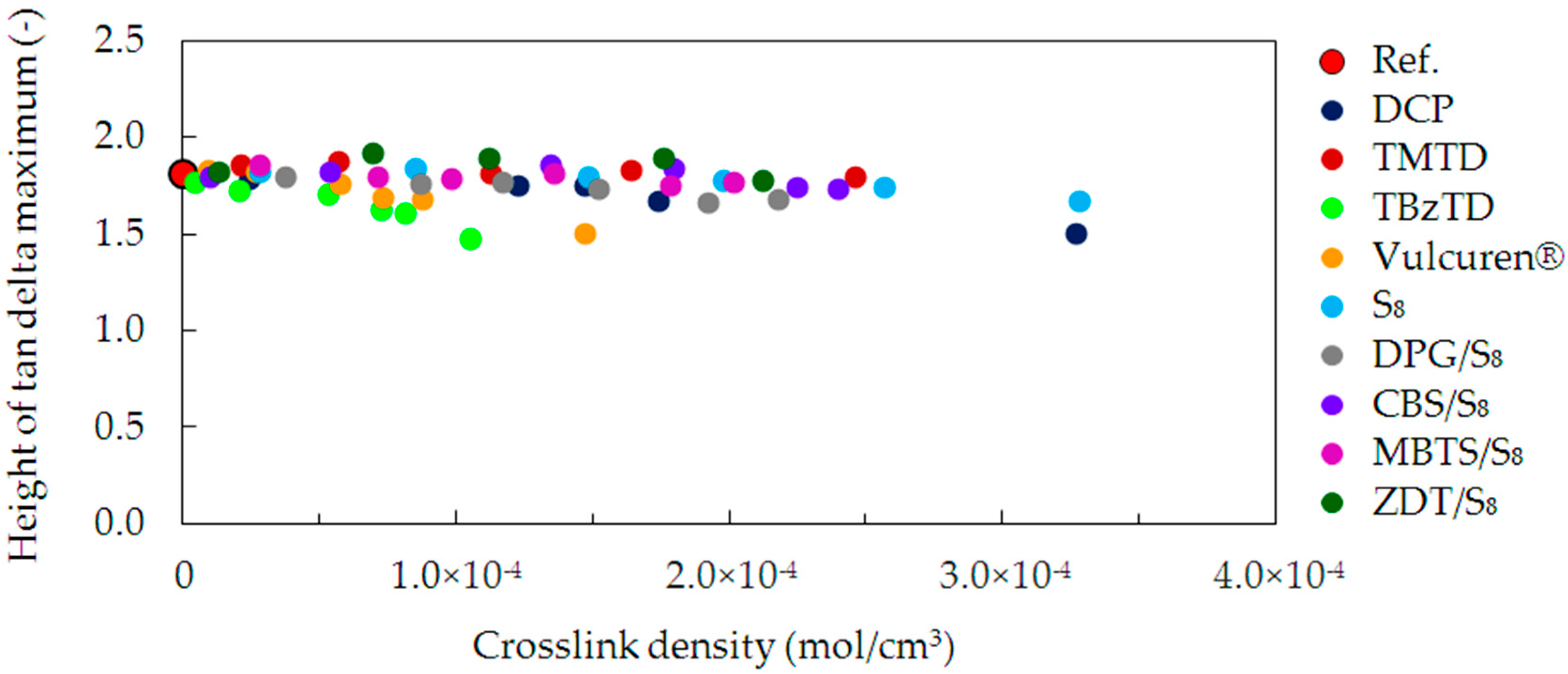
To evaluate the elastic properties of the studied samples, the height of the tan delta peaks was correlated with the crosslink density (
Figure 8). The largest value was observed for the uncrosslinked reference, due to its high plasticity. In case of the cured samples, the increase in the crosslink density was followed by a decrease in the tan delta height. The elastic properties were, therefore, improved. This effect is caused by the higher number of crosslinks connecting the polymer chains. The crosslinks serve to efficiently recover to the unstrained spatial network. The trend is the strongest for the TBzTD and Vulcuren
® cured samples.
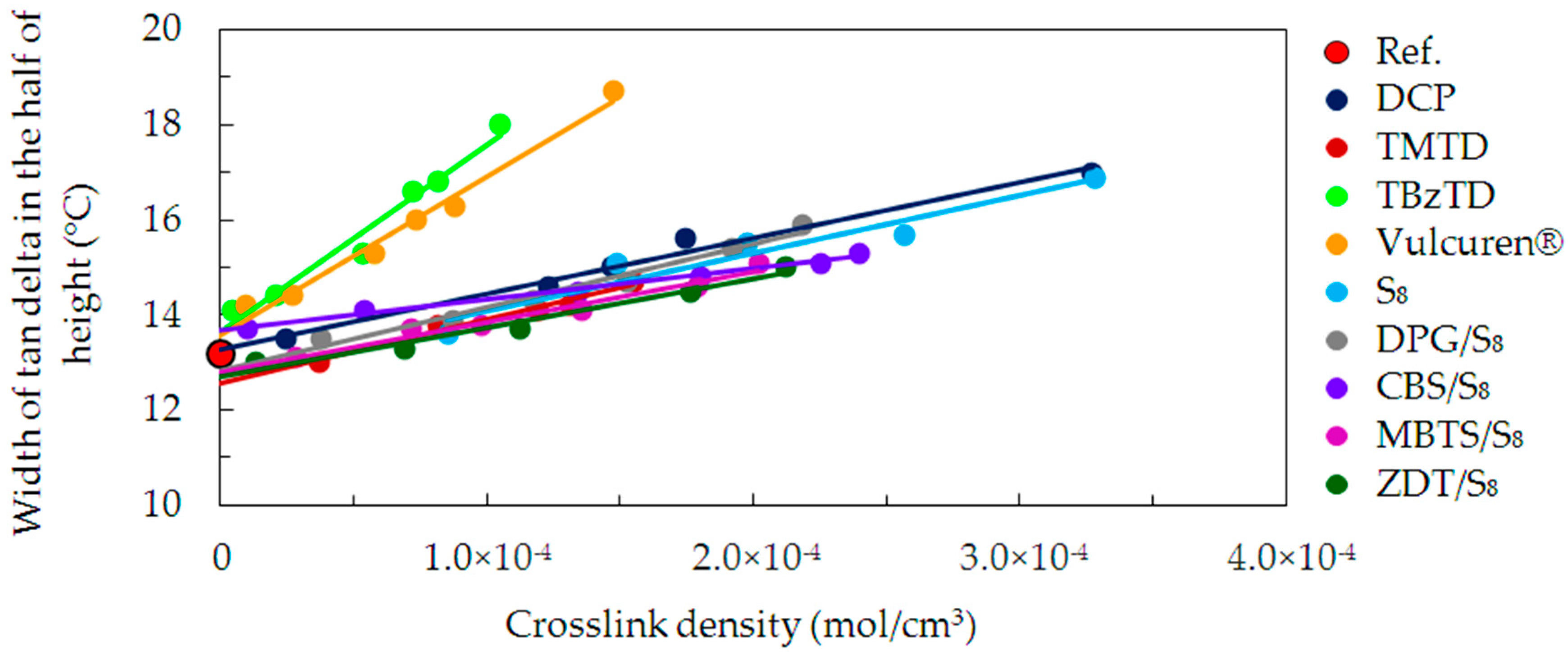
The range of temperatures, in which the molecular mobility begins, was determined. For this, the width of the tan delta peak in the half of its height was calculated.
Figure 9 shows values of this parameter, correlated with the crosslink density. An increase in the crosslink density resulted in a broadening of the tan delta peak. This is probably caused by more densely located structures on the chains formed by curatives. Additionally, the number and size of heterogenic microregions increased. These structures relaxed over a broader range of temperature. Changes in width of the tan delta peak are similar for most of the investigated series of samples, such as DCP, TMTD, S
8, DPG/S
8, CBS/S
8, MBTS/S
8, and ZDT/S
8. However, in case of the TBzTD or Vulcuren
® samples, this increase is much higher. This can be caused by the large amounts of TBzTD and Vulcuren
® present in the samples. Consequently, the molecular movements during the glass transition process occurred over a broad range of temperature.
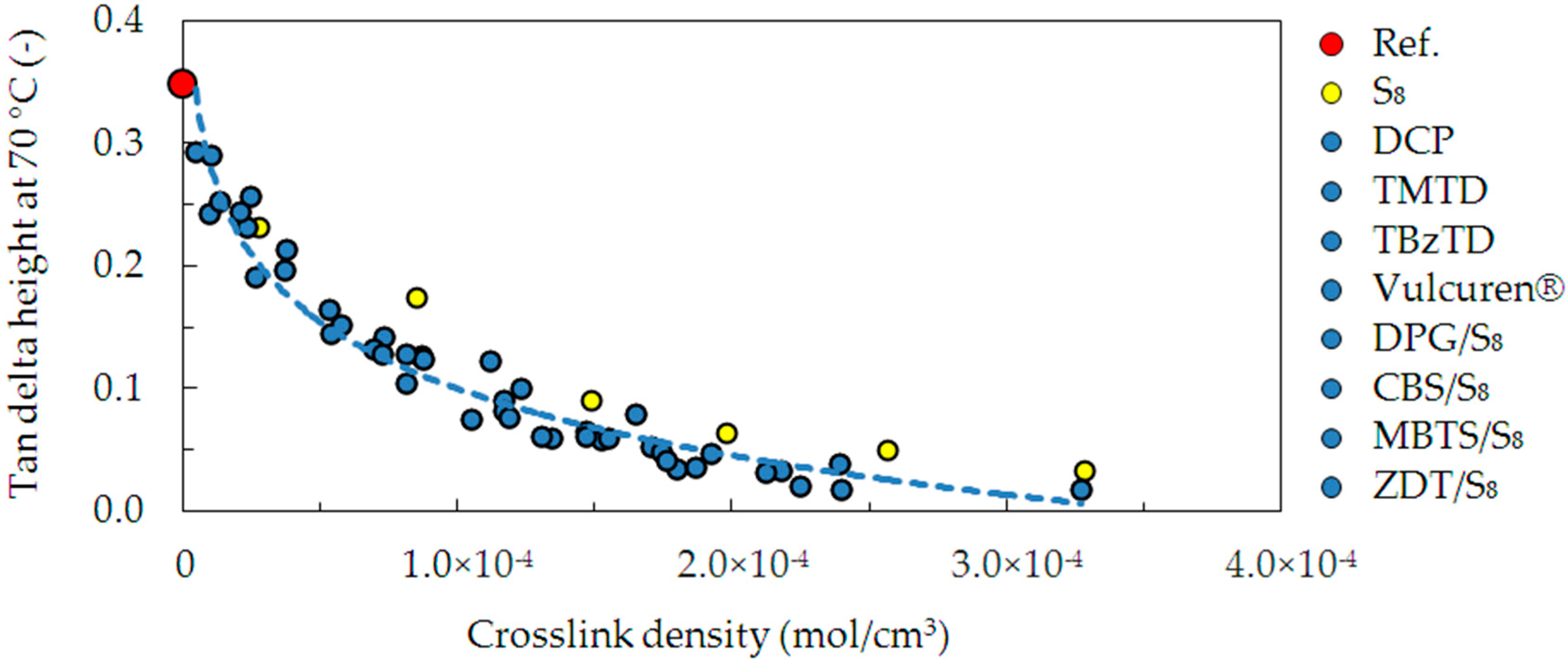
The damping properties at elevated temperature, i.e., 70 °C, were evaluated by determination of the tan delta.
Figure 10 presents these values, correlated with the crosslink density. The dependence shows that an increase in the crosslink density resulted in a large decrease in damping. This is due to an increase in the number of crosslinks, which provide elastic response and lower dissipation of energy. This dependence is similar in most of the investigated series of samples: DCP, TMTD, TBzTD, Vulcuren
®, DPG/S
8, CBS/S
8, MBTS/S
8, and ZDT/S
8. However, in the case of samples cured with pure sulfur S
8, the investigated ability to dissipate energy is slightly larger. This can be attributed to the fact that in addition to crosslinking, pure sulfur produced numerous modifications of the polymer chains. They are not elastically effective, hence the slightly larger dissipation of the energy. Additionally, the S
8 series was cured at 180 °C, whereas all the other series at 160 °C. The higher temperature could result in increased chain scission in the sulfur-cured samples. This could contribute to different behavior of the S
8 cured samples.
As discussed above, the presence of crosslinks caused an increase in the
Tg. This effect considerably differs between the samples cured with the various curatives. Therefore, other structural parameters than the total crosslink density contribute greatly to the changes in
Tg. The obtained results show that the crosslink structure does not explain them. It can be concluded that this parameter is not of a crucial importance. The differences between the
Tg values of particular samples are ascribed to the presence of the curatives. They chemically attach to the polymer chains and form intramolecular modifications. If the curatives substances are small in size, i.e., have a low molecular weight, they do not affect the flexibility of the polymer chains. However, curatives having a high molecular weight, and bulky inflexible moieties, stiffen the polymer chains. It causes significant decrease of mobility of the chains, especially in vinyl polymers [
20], such as SBR. This rotational restriction of these macromolecules influences the glass transition process and increases the
Tg values considerably. Furthermore, the chemical structures of the curatives are usually very different from the structure of the basic polymers. It can, therefore, be considered that as a result of curing, a copolymer is formed, as already proposed by Fox and Loshaek [
17]. In this way, curatives grafted on the chains can be considered as functional groups, which modify the polymer chains. The
Tg of the cured system is not only dependent on the crosslinks themselves. Additionally, the average composition of the system plays a role.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








