RAD-Seq Data Point to a Distinct Split in Liriodendron (Magnoliaceae) and Obvious East–West Genetic Divergence in L. chinense


Abstract
:1. Introduction
2. Methods and Materials
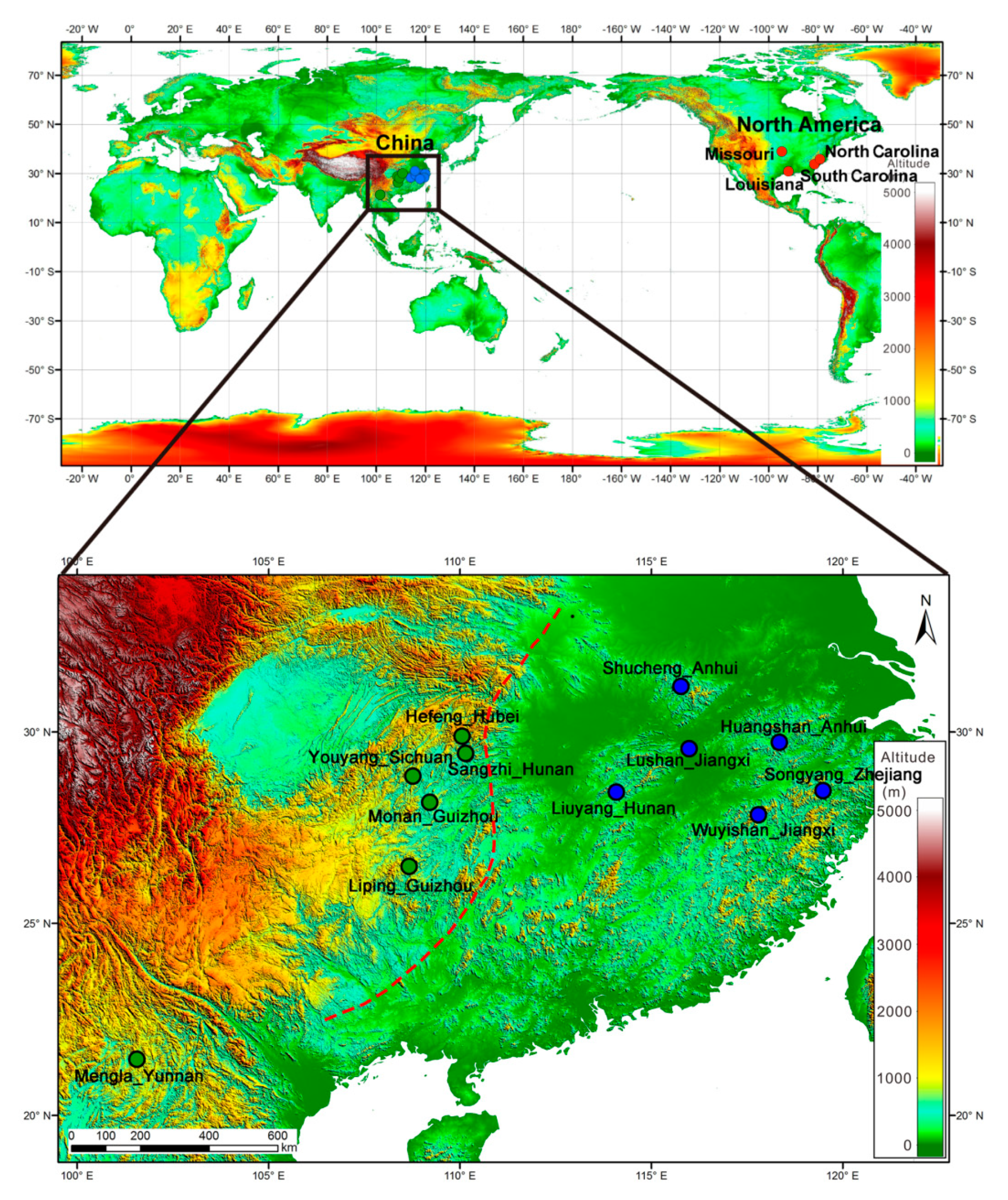
2.1. Sample Collection
2.2. RAD Sequencing, Reads Clustering, and SNP Calling
2.3. Phylogeny Construction
2.4. Genetic Structure Analysis
3. Results
3.1. Sequencing Data Quality and Processing
3.2. Phylogenetic Analysis
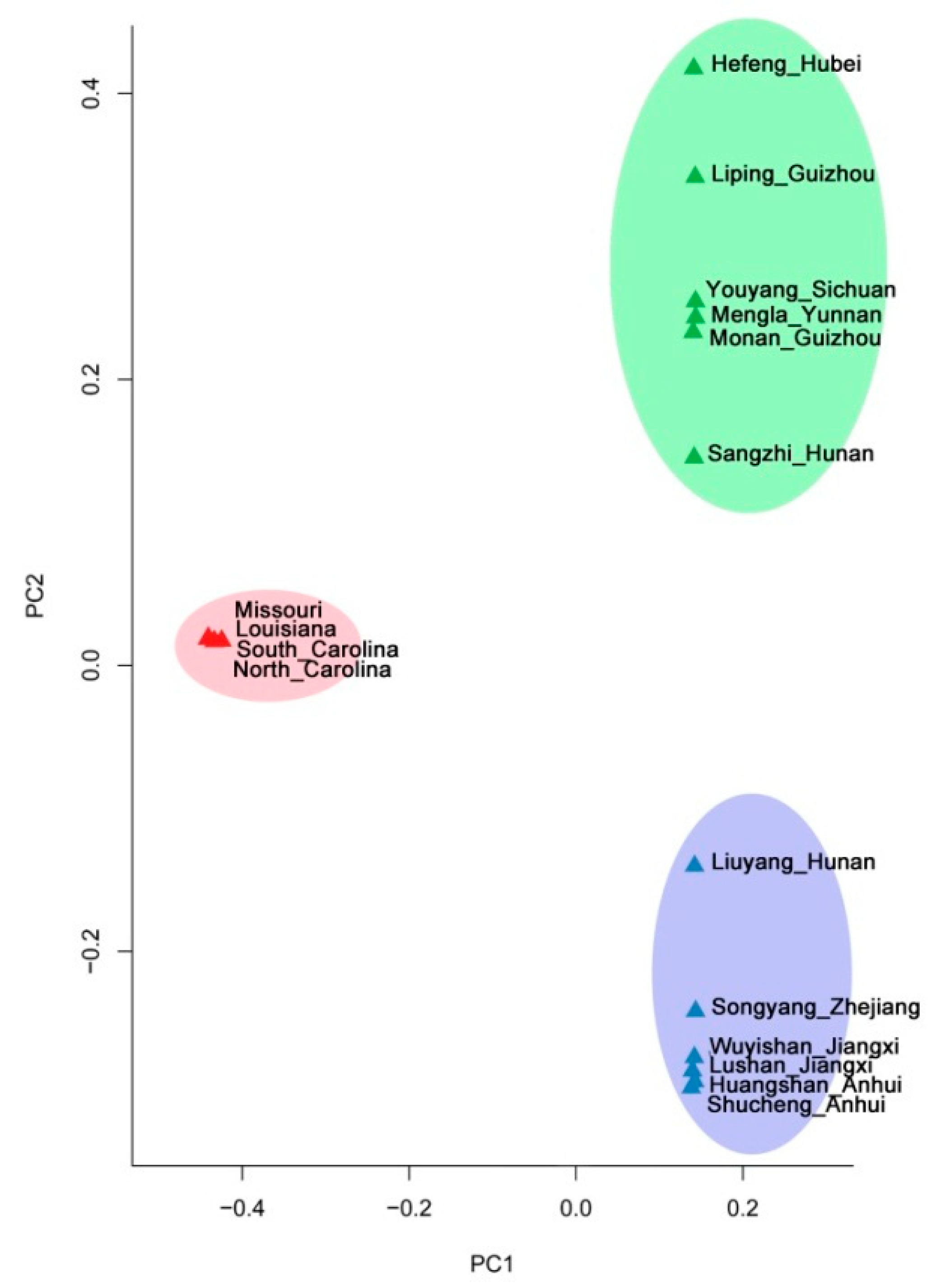
3.3. Population Structure Analyses
4. Discussion
4.1. The Extensive Genetic Divergence in Liriodendron
4.2. Demarcations between Eastern and Western China for L. chinense
4.3. Genetic Relationship within Liriodendron
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gray, A. Analogy between the flora of Japan and that of the United States. Am. J. Sci. Arts 1846, 2, 135–136. [Google Scholar]
- Wen, J. Evolution of Eastern Asian and Eastern North American disjunct distributions in flowering plants. Annu. Rev. Ecol. Syst. 1999, 30, 421–455. [Google Scholar] [CrossRef]
- Zhao, Y.; Qi, Z.; Ma, W.; Dai, Q.; Li, P.; Cameron, K.M.; Lee, J.; Xiang, Q.Y.; Fu, C. Comparative phylogeography of the Smilax hispida group (Smilacaceae) in eastern Asia and North America—Implications for allopatric speciation, causes of diversity disparity, and origins of temperate elements in Mexico. Mol. Phylogenet. Evol. 2013, 68, 300–311. [Google Scholar] [CrossRef]
- Zhu, W.-D.; Nie, Z.-L.; Wen, J.; Sun, H. Molecular phylogeny and biogeography of Astilbe (Saxifragaceae) in Asia and eastern North America. Bot. J. Linn. Soc. 2013, 171, 377–394. [Google Scholar] [CrossRef]
- Qian, H.; Jin, Y.; Ricklefs, R.E. Phylogenetic diversity anomaly in angiosperms between eastern Asia and eastern North America. Proc. Natl. Acad. Sci. USA 2017, 114, 11452–11457. [Google Scholar] [CrossRef] [Green Version]
- Parks, C.; Miler, N.; Wendel, J.; Mcdouglas, K. Genetic divergence within the genus Liriodendron (Magnoliaceae). Ann. Mo. Bot. Gard. 1983, 70, 658–666. [Google Scholar] [CrossRef]
- Nie, Z.-L.; Wen, J.; Azuma, H.; Qiu, Y.-L.; Sun, H.; Meng, Y.; Sun, W.-B.; Zimmer, E.A. Phylogenetic and biogeographic complexity of Magnoliaceae in the Northern Hemisphere inferred from three nuclear data sets. Mol. Phylogenet. Evol. 2008, 48, 1027–1040. [Google Scholar] [CrossRef] [Green Version]
- Parks, C.R.; Wendel, J.F. Molecular divergence between Asian and North American species of Liriodendron (Magnoliaceae) with implications for interpretation of fossil floras. Am. J. Bot. 1990, 77, 1243–1256. [Google Scholar] [CrossRef]
- Zhang, F.; Zhao, Y. Advance of study on Liriodendron. J. Yunnan Agric. Univ. 2005, 20, 697–701. [Google Scholar]
- Hao, R.-M.; He, S.-A.; Tang, S.-J.; Wu, S.-P. Geographical distribution of Liriodendron chinense in China and its significance. J. Plant Resour. Environ. 1995, 4, 1–6. [Google Scholar]
- Zhang, X.; Carlson, A.; Tian, Z.; Staton, M.; Schlarbaum, S.E.; Carlson, J.E.; Liang, H. Genetic characterization of Liriodendron seed orchards with EST-SSR markers. J. Plant Sci. Mol. Breed. 2015, 4, 1. [Google Scholar] [CrossRef]
- Sewell, M.; Parks, C.; Chasem, M. Intraspecific chloroplast DNA variation and biogeography of north American Liriodendron L. (Magnoliaceae). Evolution 1996, 50, 1147–1154. [Google Scholar] [CrossRef]
- Merkle, S.A.; Hoey, M.T.; Watson-Pauley, B.A.; Schlarbaum, S.E. Propagation of Liriodendron hybrids via somatic embryogenesis. Plant Cell Tissue Organ Cult. 1993, 34, 191–198. [Google Scholar] [CrossRef]
- Kim, S.; Koh, J.; Yoo, M.J.; Kong, H.; Hu, Y.; Ma, H.; Soltis, P.S.; Soltis, D.E. Expression of floral MADS-box genes in basal angiosperms: Implications for the evolution of floral regulators. Plant J. 2005, 43, 724–744. [Google Scholar] [CrossRef]
- Liang, H.; Carlson, J.E.; Leebens-Mack, J.H.; Wall, P.K.; Mueller, L.A.; Buzgo, M.; Landherr, L.L.; Hu, Y.; DiLoreto, D.S.; Ilut, D.C.; et al. An EST database for Liriodendron tulipifera L. floral buds: The first EST resource for functional and comparative genomics in Liriodendron. Tree Genet. Genomes 2008, 4, 419–433. [Google Scholar] [CrossRef]
- Fetter, K.C. Migration, adaptation, and speciation—A post-glacial history of the population structure, phylogeography, and biodiversity of Liriodendron tulipifera L. (Magnoliaceae). Diss. Theses Gradworks 2014, 62, 304–309. [Google Scholar]
- Parks, C.R.; Wendel, J.F.; Sewell, M.M.; Qiu, Y.-L. The significance of allozyme variation and introgression in the Liriodendron tulipifera complex (Magnoliaceae). Am. J. Bot. 1994, 81, 878–889. [Google Scholar] [CrossRef]
- Li, K.; Chen, L.; Feng, Y.; Yao, J.; Li, B.; Xu, M.; Li, H. High genetic diversity but limited gene flow among remnant and fragmented natureal populations of Liriodendron chinense Sarg. Biochem. Syst. Ecol. 2014, 54, 230–236. [Google Scholar] [CrossRef]
- Yang, A.; Dick, C.W.; Yao, X.; Huang, H. Impacts of biogeographic history and marginal population genetics on species range limits: A case study of Liriodendron chinense. Sci. Rep. 2016, 6, 25632. [Google Scholar] [CrossRef]
- Zhong, Y.D.; Zhang, X.; Li, Y.Q.; Liu, L.P.; Yu, F.X. Genome survey of Liriodendron chinense (Hemsl.) Sarg. Mol. Plant Breed. 2017, 15, 507–512. [Google Scholar]
- Xu, P.; Xu, S.; Wu, X.; Tao, Y.; Wang, B.; Wang, S.; Qin, D.; Lu, Z.; Li, G. Population genomic analyses from low-coverage RAD-Seq data: A case study on the non-model cucurbit bottle gourd. Plant J. Cell Mol. Biol. 2014, 77, 430–442. [Google Scholar] [CrossRef]
- You, F.M.; Huo, N.; Deal, K.R.; Gu, Y.Q.; Luo, M.-C.; McGuire, P.E.; Dvorak, J.; Anderson, O.D. Annotation-based genome-wide SNP discovery in the large and complex Aegilops tauschii genome using next-generation sequencing without a reference genome sequence. BMC Genomics 2011, 12, 59. [Google Scholar] [CrossRef]
- Novaes, E.; Drost, D.R.; Farmerie, W.G.; Pappas, G.J.; Grattapaglia, D.; Sederoff, R.R.; Kirst, M. High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genomics 2008, 9, 312. [Google Scholar] [CrossRef]
- Lai, J.; Li, R.; Xu, X.; Jin, W.; Xu, M.; Zhao, H.; Xiang, Z.; Song, W.; Ying, K.; Zhang, M.; et al. Genome-wide patterns of genetic variation among elite maize inbred lines. Nat. Genet. 2010, 42, 1027–1030. [Google Scholar] [CrossRef]
- Xu, X.; Liu, X.; Ge, S.; Jensen, J.D.; Hu, F.; Li, X.; Dong, Y.; Gutenkunst, R.N.; Fang, L.; Huang, L.; et al. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat. Biotechnol. 2012, 30, 105–111. [Google Scholar] [CrossRef]
- Hou, Y.; Nowak, M.D.; Mirre, V.; Bjora, C.S.; Brochmann, C.; Popp, M. RAD-seq data point to a northern origin of the arctic-alpine genus Cassiope (Ericaceae). Mol. Phylogenet. Evol. 2016, 95, 152–160. [Google Scholar] [CrossRef]
- Bateman, R.M.; Sramko, G.; Paun, O. Integrating restriction site-associated DNA sequencing (RAD-seq) with morphological cladistic analysis clarifies evolutionary relationships among major species groups of bee orchids. Ann. Bot. 2018, 121, 85–105. [Google Scholar] [CrossRef]
- Takahashi, T.; Nagata, N.; Sota, T. Application of RAD-based phylogenetics to complex relationships among variously related taxa in a species flock. Mol. Phylogenet. Evol. 2014, 80, 137–144. [Google Scholar] [CrossRef]
- Herrera, S.; Shank, T.M. RAD sequencing enables unprecedented phylogenetic resolution and objective species delimitation in recalcitrant divergent taxa. Mol. Phylogenet. Evol. 2016, 100, 70–79. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Ji, X.; Obata, S.; Pais, A.; Dong, Y.; Peet, R.; Xiang, Q.J. Resolving relationships and phylogeographic history of the Nyssa sylvatica complex using data from RAD-seq and species distribution modeling. Mol. Phylogenet. Evol. 2018, 126, 1–16. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small amounts of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Catchen, J.M.; Amores, A.; Hohenlohe, P.; William, C.; Postlethwait, J.H. Stacks: Building and Genotyping Loci De Novo from Short-Read Sequences. Genes Genomes Genet. 2011, 1, 171–182. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Zerbino, D.R. Using the Velvetde novoassembler for short-readsequencing technologies. Curr. Protoc. Bioinf. 2010, 31, 11.5.1–11.5.12. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Retief, J.D. Phylogenetic analysis using PHYLIP. Methods Mol. Biol. 2000, 132, 243–258. [Google Scholar]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Patterson, N.; Price, A.L.; Reich, D. Population structure and eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef]
- Tang, H.; Peng, J.; Wang, P.; Risch, N.J. Estimation of Individual Admixture: Analytical and Study Design Considerations. Genet. Epidemiol. 2005, 28, 289–301. [Google Scholar] [CrossRef]
- Manni, F.; Guerard, E.; Heyer, E. Geographic patterns of (genetic, morphologic, linguistic) variation: How barriers can be detected by using Monmonier’s algorithm. Hum. Biol. 2004, 76, 173–190. [Google Scholar] [CrossRef]
- Takahashi, T.; Moreno, E. A RAD-based phylogenetics for Orestias fishes from Lake Titicaca. Mol. Phylogenet. Evol. 2015, 93, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Wei, N.; Fritsch, P.W.; Yao, X. AFLP genome scanning reveals divergent selection in natural populations of Liriodendron chinense (Magnoliaceae) along a latitudinal transect. Front. Plant Sci. 2016, 7, 698. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Zhong, Y.; Liu, S.; Liu, T.; Liu, L.; Li, Y.; Yu, F. New insight into the phylogeographic pattern of Liriodendron chinense (Magnolicaceae) revealed by chloroplast DNA: East–west lineage split and genetic mixture within western subtropical China. Peer J. 2018. under review. [Google Scholar]
- He, S.A.; Santamour, F.S. Isoenzyme Verification of American-Chinese Hybrids of Liquidambar and Liriodendron. Ann. Mo. Bot. Gard. 1983, 70, 748–749. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Eastern China | Western China | North America |
|---|---|---|---|
| Eastern China | 0.130 a | 0.177 c | 0.465 d |
| Western China | 0.161 b | 0.486 e | |
| North America | 0.127 a |
| Group | r | p |
|---|---|---|
| Liriodendron | 0.820 | 0.001 |
| L. tulipifera | 0.311 | 0.290 |
| L. chinense | 0.566 | 0.002 |
| Eastern China | 0.564 | 0.006 |
| Western China | −0.165 | 0.439 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, Y.; Yang, A.; Liu, S.; Liu, L.; Li, Y.; Wu, Z.; Yu, F. RAD-Seq Data Point to a Distinct Split in Liriodendron (Magnoliaceae) and Obvious East–West Genetic Divergence in L. chinense. Forests 2019, 10, 13. https://doi.org/10.3390/f10010013
Zhong Y, Yang A, Liu S, Liu L, Li Y, Wu Z, Yu F. RAD-Seq Data Point to a Distinct Split in Liriodendron (Magnoliaceae) and Obvious East–West Genetic Divergence in L. chinense. Forests. 2019; 10(1):13. https://doi.org/10.3390/f10010013
Chicago/Turabian StyleZhong, Yongda, Aihong Yang, Shujuan Liu, Lipan Liu, Yanqiang Li, Zhaoxiang Wu, and Faxin Yu. 2019. "RAD-Seq Data Point to a Distinct Split in Liriodendron (Magnoliaceae) and Obvious East–West Genetic Divergence in L. chinense" Forests 10, no. 1: 13. https://doi.org/10.3390/f10010013