Use of Nuclear Microsatellite Loci for Evaluating Genetic Diversity of Selected Populations of Picea abies (L.) Karsten in the Czech Republic

Abstract
:1. Introduction
2. Materials and Methods
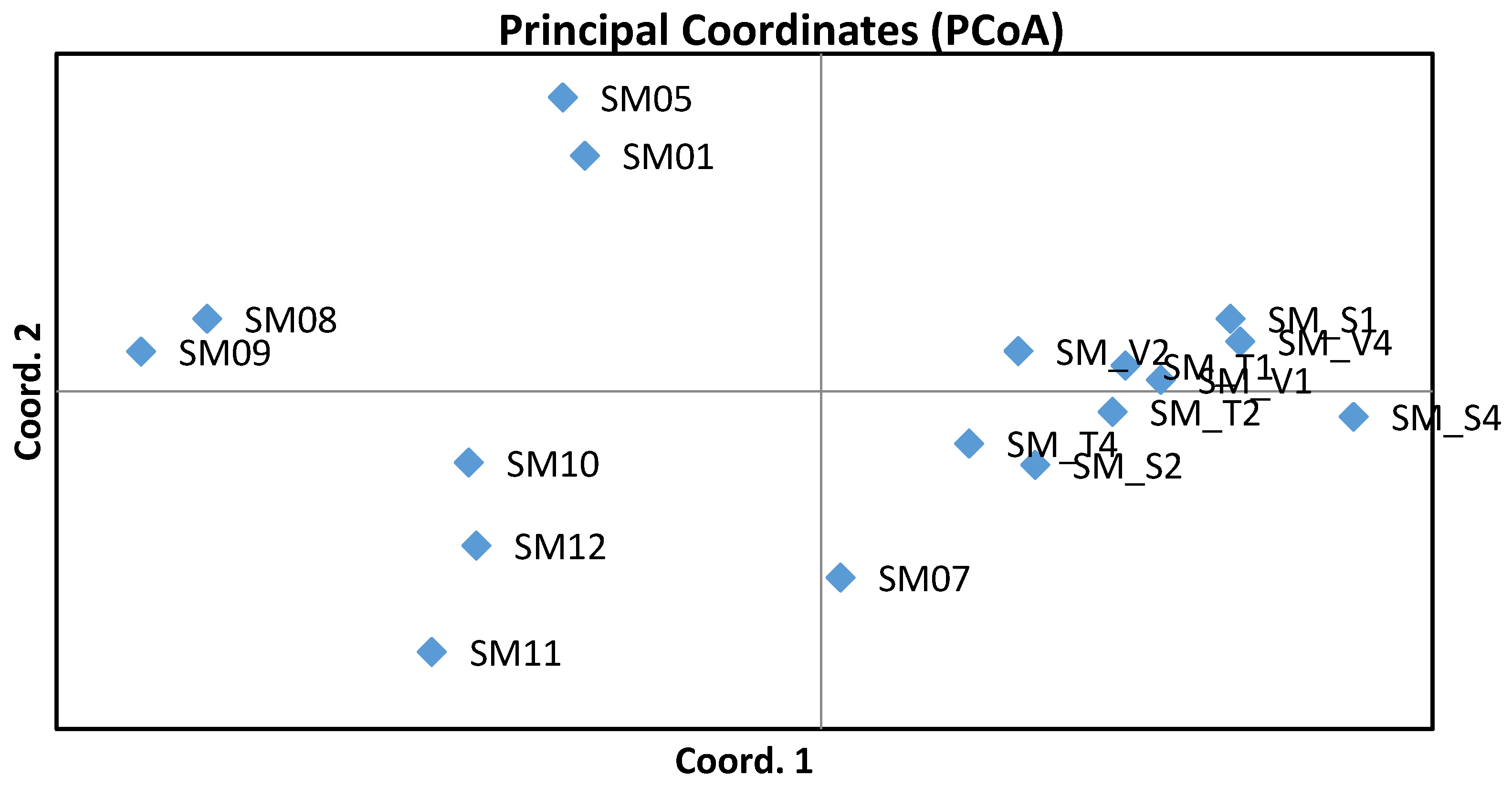
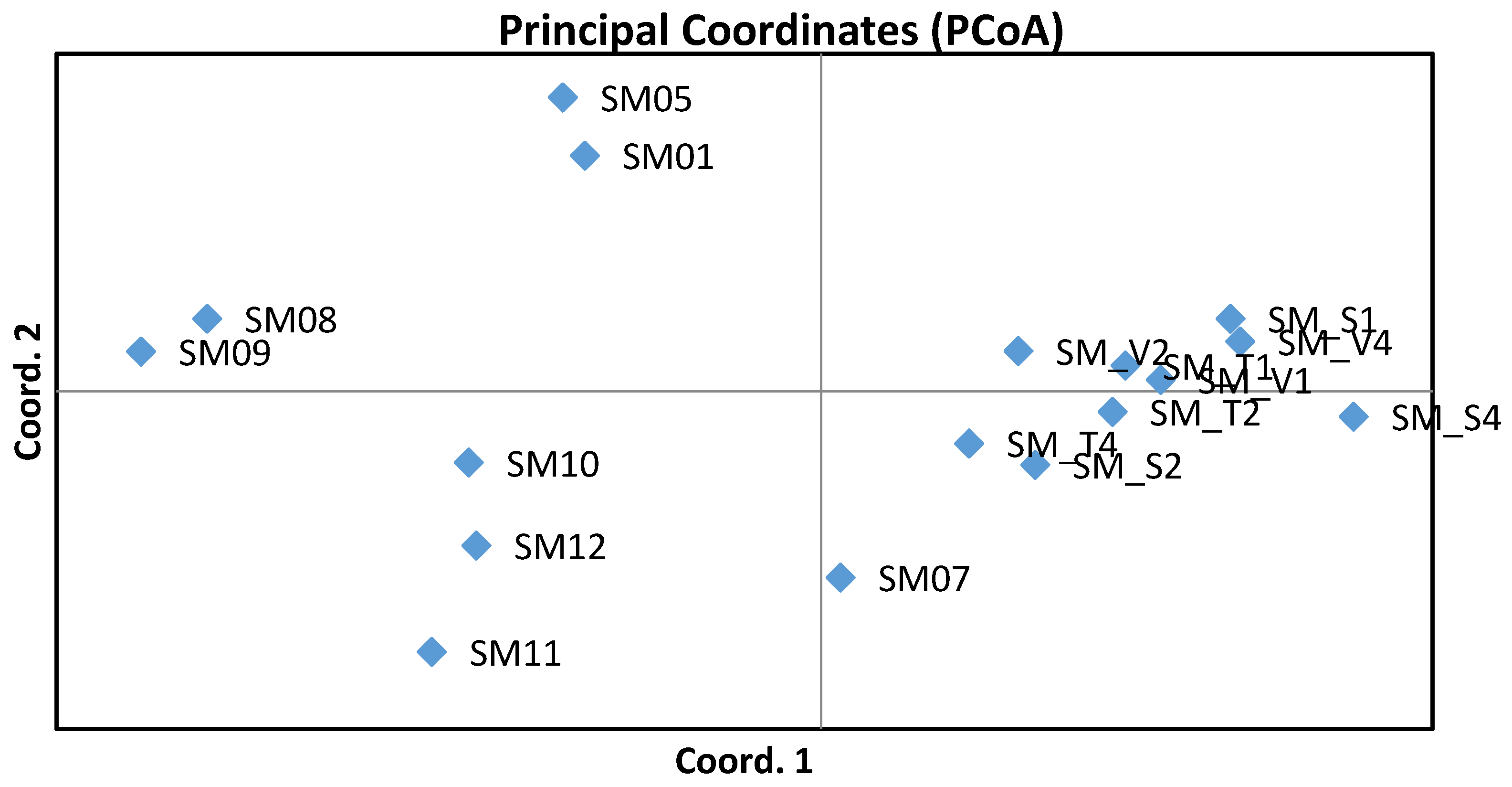
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Musil, I.; Hamerník, J.; Leugnerová, G. Lesnická Dendrologie 1. Jehličnaté Dřeviny. [Forest Dendrology 1. Coniferous Trees]; The Czech Univerzity of Agriculture Prague: Praha, Czech Republic, 2003. [Google Scholar]
- Směrnice pro uznávání a zabezpečení zdrojů reprodukčního materiálu lesních dřevin a pro jeho přenos. [Directives for the Recognition and Security of Forest Resources and the Transfer of Forest Resources]; Ministry of Forestry and Water Management and Woodworking Industry of the Czechoslovak Republic: Praha, Czech Republic, 1988.
- Úradníček, L.; Maděra, P.; Tichá, S.; Koblížek, J. Dřeviny České Republiky [Woody Species of the Czech Republic]; Nakladatelství a vydavatelství Lesnická práce, s.r.o.: Kostelec nad Černými lesy, Czech Republic, 2009. [Google Scholar]
- Ministry of Agriculture of the Czech Republic. Information on Forests and Forestry in the Czech Republic by 2014. Available online: http://eagri.cz/public/web/file/433136/ZZ2014AJ_16112015.pdf (accessed on 27 June 2016).
- Šrámek, V.; Neudertová Hellebrandová, K. Mapy ohrožení smrkových porostů suchem jako nástroj identifikace rizikových oblastí [Maps of drought risk for Norway spruce stands as a decision tool indicating threatened regions in the Czech Republic: Short communication]. Rep. For. Res. 2016, 61, 305–309. [Google Scholar]
- Maghuly, F.; Pinsker, W.; Praznik, W.; Fluch, S. Genetic diversity in managed subpopulations of Norway spruce [Picea abies (L). Karst.]. For. Ecol. Manag. 2006, 222, 266–271. [Google Scholar]
- Hampe, A.; Petit, R.J. Conserving biodiversity under climate change: The rear edge matters. Ecol. Lett. 2005, 8, 461–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neale, D.B.; Kremer, A. Forest tree genomics: Growing resources and applications. Nat. Rev. Genet. 2011, 12, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Whitham, T.G.; Bailey, J.K.; Schweitzer, J.A.; Shuster, S.M.; Bangert, R.K.; LeRoy, C.J.; Lonsdorf, E.V.; Allan, G.J.; DiFazio, S.P.; Potts, B.M.; et al. A framework for community and ecosystem genetics from genes to ecosystems. Nat. Rev. Genet. 2006, 7, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Geburek, T. Genetic variation of Norway spruce (Picea abies [L.] Karst.) populations in Austria. III. Macrospatial allozyme patterns of high elevation populations. For. Genet. 1999, 6, 201–211. [Google Scholar]
- Konnert, M. Genetic variation of Picea abies in southern Germany as determined using isozyme and STS markers. Dendrobiology 2009, 61, 131–136. [Google Scholar]
- Schubert, R.; Mueller-Starck, G.; Riegel, R. Development of EST-PCR markers and monitoring their intrapopulational genetic variation in Picea abies (L.) Karst. Theor. Appl. Genet. 2001, 103, 1223–1231. [Google Scholar]
- Bozhko, M.; Riegel, R.; Schubert, R.; Müller-Starck, G. A cyclophilin gene marker confirming geographical differentiation of Norway spruce populations and indicating viability response on excess soil-born salinity. Mol. Ecol. 2003, 12, 3147–3155. [Google Scholar] [CrossRef] [PubMed]
- Maghuly, F.; Burg, K.; Pinsker, W.; Nittinger, F.; Praznik, W.; Fluch, S. Short Note: Development of mitochondrial markers for population genetics of Norway Spruce [Picea abies (L). Karst]. Silvae Genet. 2008, 57, 41–44. [Google Scholar] [CrossRef]
- Tollefsrud, M.M.; Sønstebø, J.H.; Brochmann, C.; Johnsen, Ø.; Skroppa, T.; Vendramin, G.G. Combined analysis of nuclear and mitochondrial markers provide new insight into the genetic structure of North European. Picea abies. Heredity 2009, 102, 549–562. [Google Scholar]
- Paglia, G.P.; Olivieri, A.M.; Morgante, M. Towards second-generation STS (sequence-tagged sites) linkage maps in conifers: A genetic map of Norway spruce (Picea abies K.). Mol. Gen. Genet. 1998, 258, 466–478. [Google Scholar]
- Perry, D.J.; Isabel, N.; Bousquet, J. Sequence-tagged-site (STS) markers of arbitrary genes: The amount and nature of variation revealed in Norway spruce. Heredity 1999, 83, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Acheré, V.; Favre, J.M.; Besnard, G.; Jeandroz, S. Genomic organization of molecular differentiation in Norway spruce (Picea abies). Mol. Ecol. 2005, 14, 3191–3201. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Uebbing, S.; Gyllenstrand, N.; Lagercrantz, U.; Lascoux, M.; Källman, T. Sequencing of the needle transcriptome from Norway spruce (Picea abies Karst. L.) reveals lower substitution rates, but similar selective constraints in gymnosperms and angiosperms. BMC Genom. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Romšáková, I.; Foffová, E.; Kmeť, J.; Longauer, R.; Pacalaj, M.; Gömöry, D. Nucleotide polymorphisms related to altitude and physiological traits in contrasting provenances of Norway spruce (Picea abies). Biologia 2012, 67, 909–916. [Google Scholar] [CrossRef]
- Pfeiffer, A.M.; Oliviery, A.M.; Morgante, M. Identification and characterization of microsatellites in Norway spruce (Picea abies K.). Genome 1997, 40, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Scotti, I.; Magni, F.; Fink, R.; Powell, W.; Binelli, G.; Hedley, P.E. Microsatellite repeats are not randomly distributed within Norway spruce (Picea abies K.) expressed sequences. Genome 2000, 43, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Melnikova, M.N.; Petrov, N.B.; Lomov, A.A.; la Porta, N.; Politov, D.V. Testing of Microsatellite Primers with Different Populations of Eurasian Spruces Picea abies (L.) Karst. and Picea obovata Ledeb. Rus. J. Genet. 2012, 48, 562–566. [Google Scholar] [CrossRef]
- Rungis, D.; Bérubé, Y.; Zhang, J.; Ralph, S.; Ritland, C.E.; Ellis, B.E.; Douglas, C.; Bohlmann, J.; Ritland, K. Robust simple sequence repeat markers for spruce (Picea spp.) from expressed sequence tags. Theor. Appl. Genet. 2004, 109, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Unger, G.M.; Konrad, H.; Geburek, T. Does spatial genetic structure increase with altitude? An answer from Picea abies in Tyrol, Austria. Plant Syst. Evol. 2011, 292, 133–141. [Google Scholar]
- Pastorelli, R.; Smulders, M.J.M.; VAN’T Westende, W.P.C.; Vosman, B.; Giannini, R.; Vettori, C.; Vendramin, G.G. Characterization of microsatellite markers in Fagus sylvatica L. and Fagus orientalis Lipsky. Mol. Ecol. 2003, 3, 76–78. [Google Scholar] [CrossRef]
- Scotti, I.; Paglia, G.; Magni, F.; Morgante, M. Population genetics (Picea abies Karst.) at regional scale: Sensitivity of different microsatellite motif classes in detecting differentiation. Ann. For. Sci. 2006, 63, 485–491. [Google Scholar] [CrossRef]
- Nystedt, B.; Street, N.R.; Wetterborn, A.; Zuccolo, A.; Lin, Y.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koskela, J.; Lefèvre, F.; Schueler, S. Translating conservation genetics into management: Pan-European minimum requirements for dynamic conservation units of forest tree genetic diversity. Biol. Conserv. 2013, 157, 39–49. [Google Scholar] [CrossRef]
- Van Oosterhout, C.V.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. Micro-Checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. The interpretation of population structure by F-statistics with special regard to systems of mating. Evolution 1965, 19, 395–420. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Raymond, M.; Rousset, F. GENEPOP (version 1.2): Population genetics software for exact tests and ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Rousset, F. Genepop’007: A complete reimplementation of the Genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Nei, M. Genetic distance between populations. Am. Nat. 1972, 106, 283–392. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. 2007, 7, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Harfouche, A.; Meilan, R.; Altman, A. Molecular and physiological responses to abiotic stress in forest trees and their relevance to tree improvement. Tree Physiol. 2014, 34, 1181–1198. [Google Scholar]
- Nowakowska, J.A. Mitochondrial and nuclear DNA differentiation of Picea abies populations in Poland. Dendrobiology 2009, 61, 119–129. [Google Scholar]
- Nascimento de Sousa, S.; Finkeldey, R.; Gailing, O. Experimental verification of microsatellite null alleles in Norway spruce (Picea abies [L.] Karst.): Implications for population genetic studies. Plant Mol. Biol. Rep. 2005, 23, 113–119. [Google Scholar] [CrossRef]
- Cvjetković, B.; Konnert, M.; Fussi, B.; Mataruga, M.; Šijačić-Nikolić, M.; Daničić, V.; Lučić, A. Norway spruce (Picea abies Karst.) variability in progeny tests in Bosnia and Herzegovina. Genetika 2017, 49, 259–272. [Google Scholar] [CrossRef]
- Gömöry, D.; Ditmarová, L.; Hrivnák, M.; Jamnická, G.; Kmet, J.; Krajmerová, D.; Kurjak, D. Differentiation in phenological and physiological traits in European beech (Fagus sylvatica L.). Eur. J. For. Res. 2015, 134, 1075–1085. [Google Scholar] [CrossRef]
- Meloni, M.; Perini, D.; Binelli, G. The distribution of genetic variation in Norway spruce (Picea abies [L.] Karst.) populations in the western Alps. J. Biogeogr. 2007, 34, 929–938. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.J.W.; Sherman-Broyles, S.L. Factors influencing levels of genetic diversity in woody plant species. New For. 1992, 6, 95–124. [Google Scholar] [CrossRef]
- Svoboda, P. Lesní dřeviny a jejich porosty. Část I. [Forest Tree Species and Their Stands. Part I]; Státní zemědělské nakladatelství: Prague, Czech Republic, 1953. [Google Scholar]
- Aravanopoulos, F.A. Genetic monitoring in natural perennial plant populations. Botany 2011, 89, 75–81. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Units | Geographic Coordinates | Altitude | Natural Origin | |
|---|---|---|---|---|
| N | E | m. | ||
| from–to | from–to | |||
| SM 01: Hurst ecotype population (Středočeská pahorkatina) | 49°56’43”–49°58’43” | 14°46’10”–14°48’39 | 400–500 | National nature reserve |
| SM 05: Hurst ecotype population (Středočeská pahorkatina) | 49°51’12”–49°51’54” | 14°35’31”–14°35’7” | 300–400 | Gene conservation unit |
| SM 07: Mountain ecotype of Beskydy population | 49°26’48”–49°31’38” | 18°26’0”–18°29’22” | 700–800 | Gene conservation unit |
| SM 08: Autochthonous highland ecotype population (Českomoravská vrchovina) | 49°30’41”–49°31’50” | 15°22’5”–15°23’56” | 550–600 | Gene conservation unit |
| SM 09: Alpine ecotype population (Hrubý Jeseník) | 50°4’13”–50°4’44” | 17°14’10”–17°15’35” | 1100–1350 | National nature reserve protected landscape area |
| SM 10: Alpine ecotype of Beskydy population (Moravskoslezské Beskydy) | 49°32’23”–49°32’58” | 18°26’42”–18°50’20” | 530–1200 | Gene conservation unit |
| SM 11: Alpine ecotype population (Šumava) | 49°4’35”–49°4’48” | 13°28’26”–13°28’49” | 825–840 | National park |
| SM 12: Alpine ecotype population (Krkonoše) | 50°44’28”–50°46’17” | 15°32’47”–15°36’19” | 980–1280 | National park |
| SM S1: Mountain ecotype (Orlické hory–Šerlich) subpopulation | 50°19’28”–50°19’46” | 16°22’21”–16°22’37” | 850–980 | Gene conservation unit |
| SM S2: Mountain ecotype (Orlické hory–Šerlich) subpopulation | 50°20’21”–50°20’33” | 16°21’36”–16°22’6” | 970–1020 | Gene conservation unit |
| SM S4: Mountain ecotype (Orlické hory–Šerlich) subpopulation | 50°19’39”–50°19’46” | 16°22’20”–16°22’41” | 860–970 | Gene conservation unit |
| SM T1: Mountain ecotype (Orlické hory–Trčkov) subpopulation | 50°18’47”–50°18’55” | 16°24’51”–16°25’7” | 780–830 | Gene conservation unit |
| SM T2: Mountain ecotype (Orlické hory–Trčkov) subpopulation | 50°19’3”–50°19’12” | 16°24’46”–16°24’59” | 780–900 | Gene conservation unit |
| SM T4: Mountain ecotype (Orlické hory–Trčkov) subpopulation | 50°18’43”–50°18’ 51” | 16°24’53”–16°25’9” | 780–870 | Gene conservation unit |
| SM V1: Mountain ecotype (Orlické hory–Vrchmezí) subpopulation | 50°21’25”–50°21’32” | 16°21’1”–16°21’47” | 900–950 | Gene conservation unit |
| SM V2: Mountain ecotype (Orlické hory–Vrchmezí) subpopulation | 50°21’7”–50°21’10” | 16°20’44”–16°21’3” | 820–880 | Gene conservation unit |
| SM V4: Mountain ecotype (Orlické hory–Vrchmezí) subpopulation | 50°21’26”–50°21’32” | 16°21’34”–16°21’45” | 920–960 | Gene conservation unit |
| Locus | Primer Sequence (5′–3′) | PCR Product Size Range (bp) | Na | I | Ho | He | F | F (Null) |
|---|---|---|---|---|---|---|---|---|
| PAAC23 | F: TGTGGCCCCACTTACTAATATCAG R: CGGGCATTGGTTTACAAGAGTTGC | 266–314 | 23 | 1.66 | 0.67 | 0.71 | 0.04 | 0.0322 |
| PAAC19 | F: ATGGGCTCAAGGATGAATG R: AACTCCAAACGATTGATTTCC | 141–237 | 37 | 2.52 | 0.53 | 0.90 | 0.41 *** | 0.2129 |
| SpAGD1 | F: GTCAACCAACTTGTAAAGCCA R: ACTTGTTTGGCATTTTCCC | 110–188 | 38 | 2.86 | 0.65 | 0.93 | 0.31 *** | 0.1555 |
| WS00716.F13 | F: tcaagtaatggacaaacgataca R: tttccaatagaatggtggattt | 206–288 | 25 | 2.60 | 0.86 | 0.91 | 0.06 * | 0.0397 |
| WS0092.A19 | F: gatgttgcaggcattcagag R: gcaccagcatcgattgacta | 207–247 | 7 | 0.41 | 0.22 | 0.20 | 0.02 | −0.0116 |
| WS0022.B15 | F: tttgtaggtgctgcagagatg R: tggctttttattccagcaaga | 166–214 | 24 | 2.27 | 0.84 | 0.86 | 0.02 | 0.0194 |
| WS0073.H08 | F: tgctctcttattcgggcttc R: aagaacaaggcttcccaatg | 182–216 | 9 | 1.24 | 0.69 | 0.67 | –0.03 | −0.0034 |
| WS00111.K13 | F: gactgaagatgccgatatgc R: ggccatatcatctcaaaataaagaa | 209–271 | 34 | 2.82 | 0.94 | 0.93 | –0.013 | 0.0074 |
| WS0023.B03 | F: agcagctggggtcaaagtt R: aaagaaagcatgcatatgactcag | 162–236 | 37 | 2.75 | 0.97 | 0.91 | –0.075 *** | −0.022 |
| Characteristic/Populations | N | Na | Ne | I | Priv. Alleles | Ho | He | F |
|---|---|---|---|---|---|---|---|---|
| SM S1 | 35 | 14.6 | 9.1 | 2.15 | 0.33 | 0.68 | 0.79 | 0.109 *** |
| SM S2 | 35 | 15.1 | 9.3 | 2.16 | 0.11 | 0.69 | 0.78 | 0.101 *** |
| SM S4 | 35 | 15.4 | 9.6 | 2.17 | 0 | 0.68 | 0.78 | 0.104 *** |
| SM T1 | 35 | 15.3 | 9.5 | 2.18 | 0.44 | 0.67 | 0.78 | 0.116 *** |
| SM T2 | 35 | 15.4 | 9.4 | 2.18 | 0 | 0.69 | 0.78 | 0.142 *** |
| SM T4 | 35 | 16.2 | 9.9 | 2.19 | 0 | 0.71 | 0.78 | 0.091 *** |
| SM V1 | 35 | 13.4 | 8.9 | 2.09 | 0 | 0.65 | 0.77 | 0.199 *** |
| SM V2 | 35 | 14.9 | 9.5 | 2.14 | 0 | 0.68 | 0.77 | 0.095 *** |
| SM V4 | 35 | 14.7 | 8.6 | 2.11 | 0.44 | 0.73 | 0.77 | 0.038 *** |
| SM 01 | 32 | 14.2 | 7.9 | 2.09 | 0.67 | 0.68 | 0.78 | 0.121 *** |
| SM 05 | 40 | 14.7 | 7.9 | 2.10 | 0.44 | 0.72 | 0.78 | 0.059 *** |
| SM 07 | 30 | 14.3 | 8.5 | 2.10 | 0.11 | 0.74 | 0.77 | 0.011 *** |
| SM 08 | 24 | 12.2 | 6.5 | 1.93 | 0.11 | 0.72 | 0.74 | 0.051 *** |
| SM 09 | 60 | 15.8 | 8.8 | 2.19 | 0.33 | 0.79 | 0.81 | −0.002 *** |
| SM 10 | 30 | 14 | 8.9 | 2.16 | 0.22 | 0.73 | 0.80 | 0.054 *** |
| SM 11 | 30 | 13.2 | 7.8 | 2.04 | 0.11 | 0.67 | 0.77 | 0.096 *** |
| SM 12 | 30 | 13.6 | 9.0 | 2.16 | 0 | 0.80 | 0.81 | −0.004 *** |
| Mean | 34.7 | 14.5 | 8.8 | 2.13 | 0.19 | 0.71 | 0.78 | 0.081 *** |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Máchová, P.; Trčková, O.; Cvrčková, H. Use of Nuclear Microsatellite Loci for Evaluating Genetic Diversity of Selected Populations of Picea abies (L.) Karsten in the Czech Republic. Forests 2018, 9, 92. https://doi.org/10.3390/f9020092
Máchová P, Trčková O, Cvrčková H. Use of Nuclear Microsatellite Loci for Evaluating Genetic Diversity of Selected Populations of Picea abies (L.) Karsten in the Czech Republic. Forests. 2018; 9(2):92. https://doi.org/10.3390/f9020092
Chicago/Turabian StyleMáchová, Pavlína, Olga Trčková, and Helena Cvrčková. 2018. "Use of Nuclear Microsatellite Loci for Evaluating Genetic Diversity of Selected Populations of Picea abies (L.) Karsten in the Czech Republic" Forests 9, no. 2: 92. https://doi.org/10.3390/f9020092
APA StyleMáchová, P., Trčková, O., & Cvrčková, H. (2018). Use of Nuclear Microsatellite Loci for Evaluating Genetic Diversity of Selected Populations of Picea abies (L.) Karsten in the Czech Republic. Forests, 9(2), 92. https://doi.org/10.3390/f9020092