De Novo Sequencing and Assembly Analysis of Transcriptome in Pinus bungeana Zucc. ex Endl.


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Sample Preparation for Illumina Sequencing
2.2.2. De novo Assembly and Annotation
2.2.3. Differential Expression Analysis and Functional Enrichment
2.2.4. Prediction of Unigene Coding Regions
2.2.5. Detection of SSR
2.2.6. Differential Expression Level Analysis by RT-qPCR
3. Results
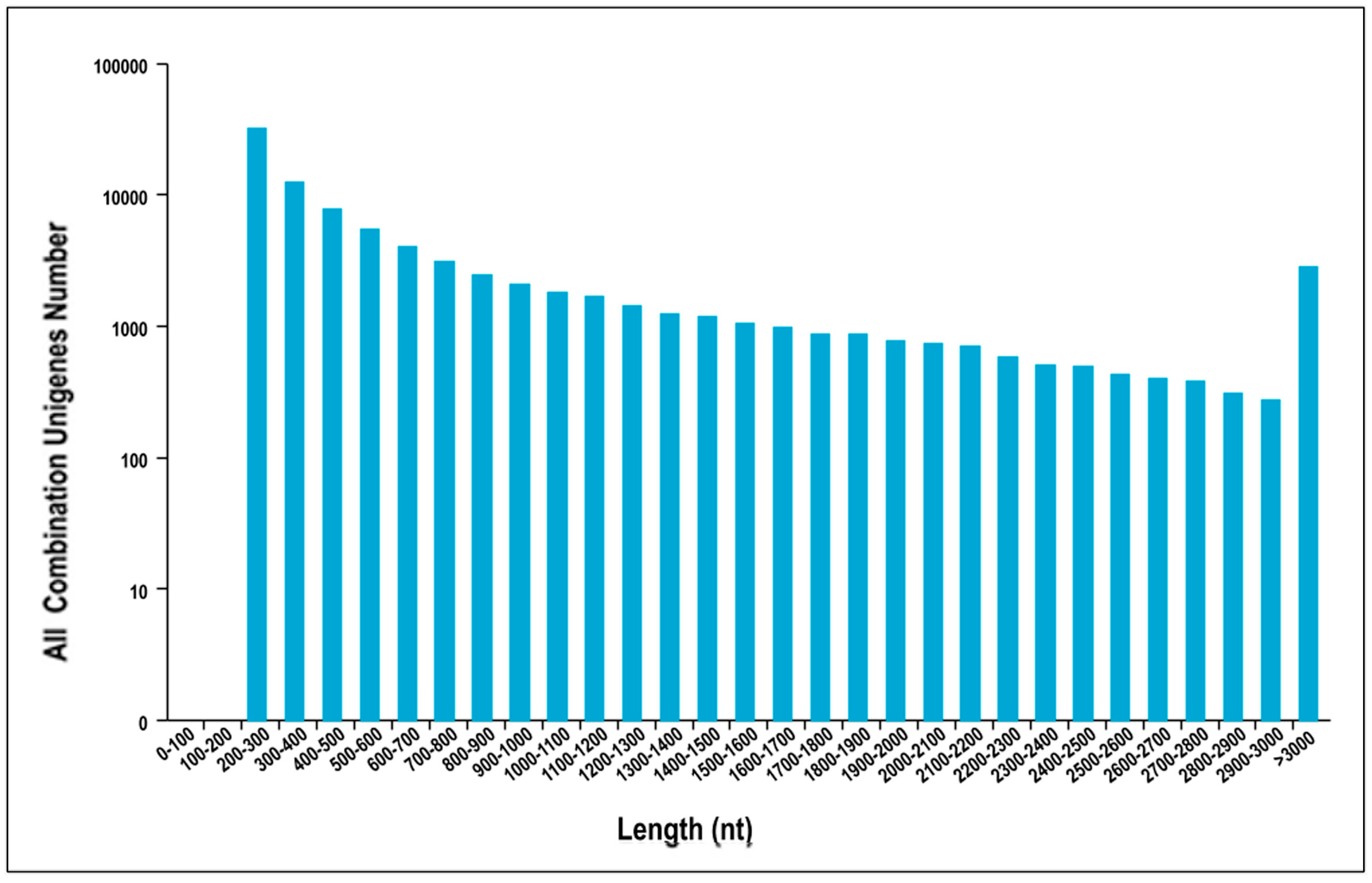
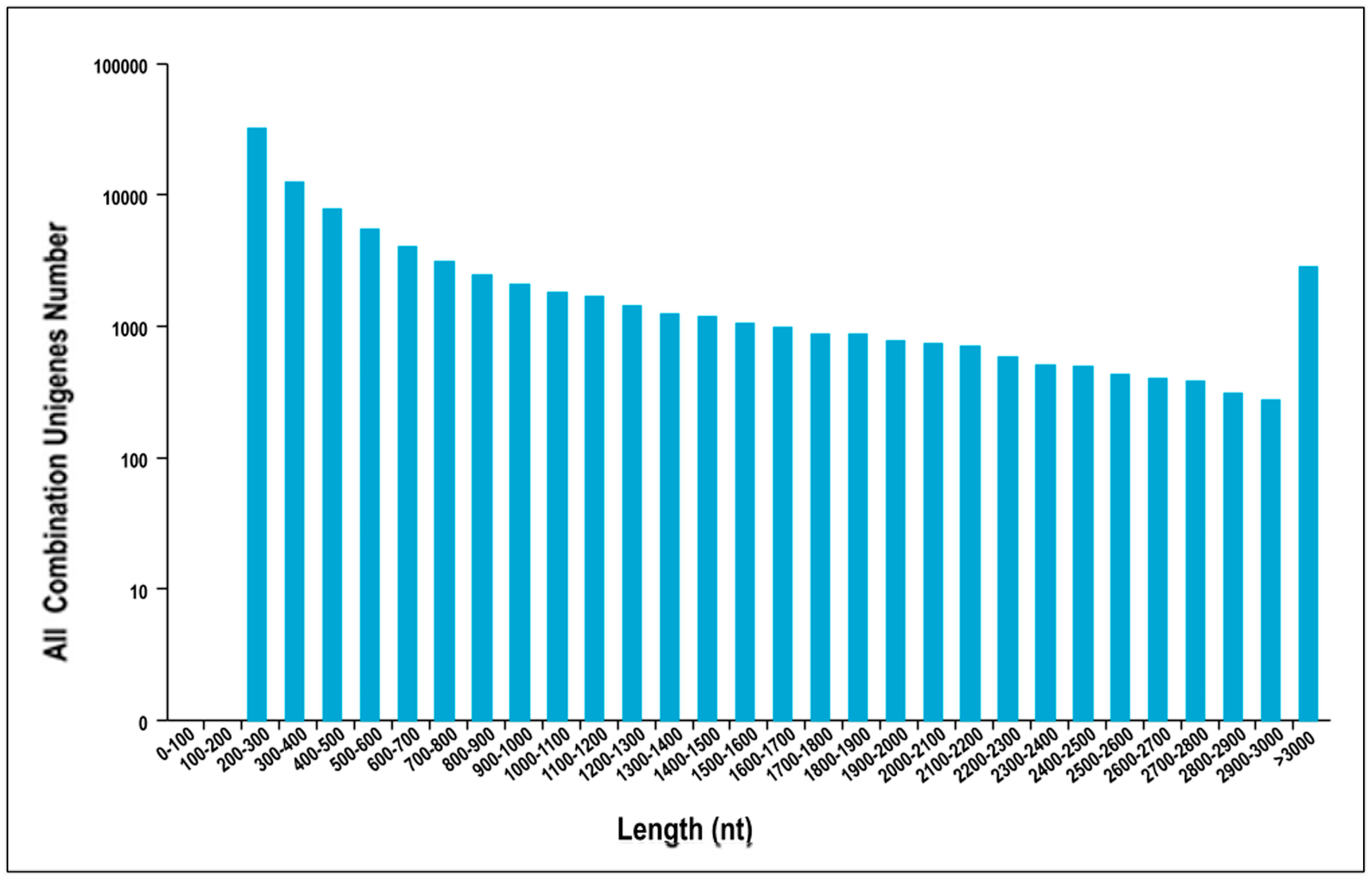
3.1. Sequencing Data Quality and Assembly of Reads
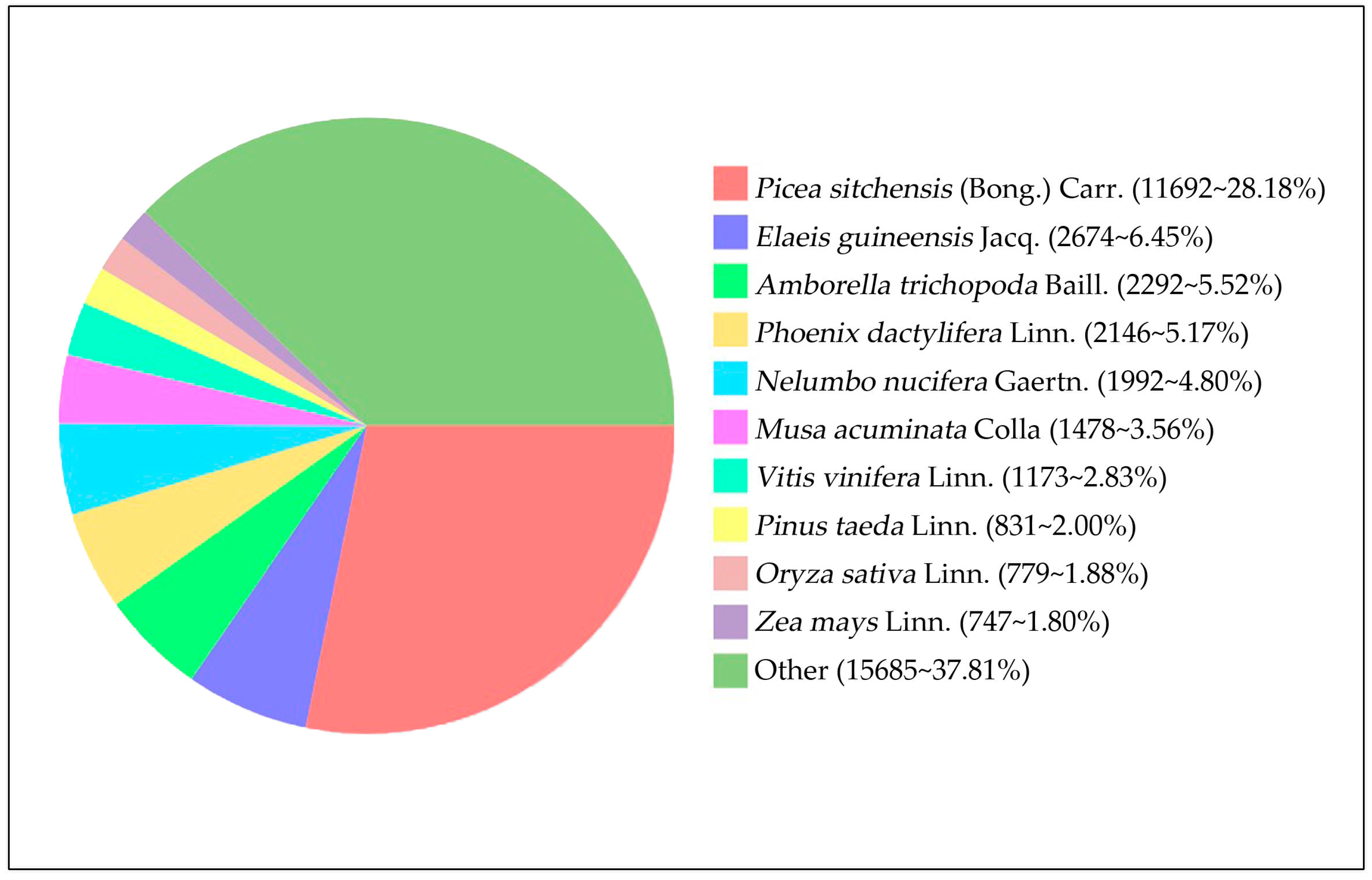
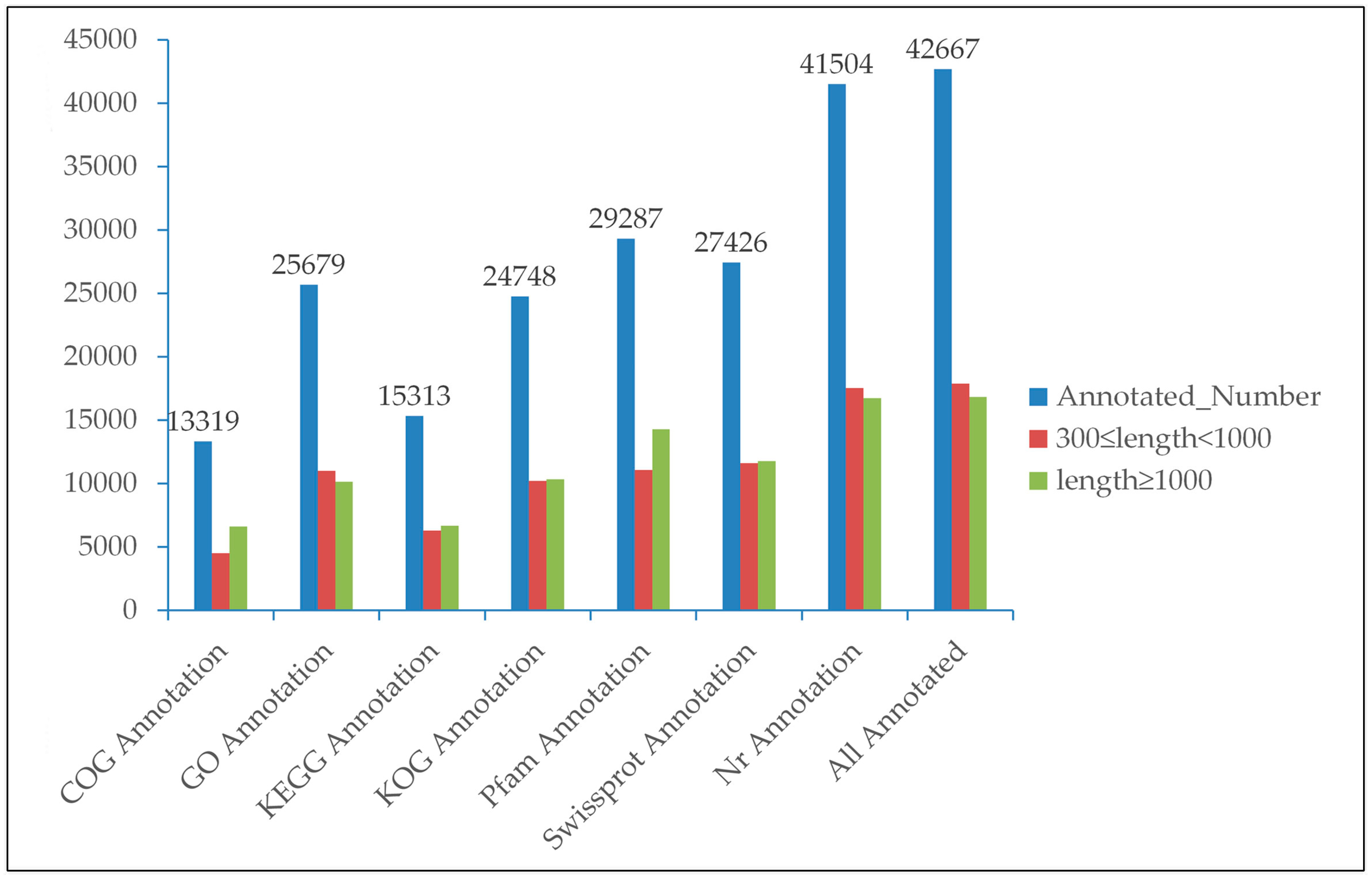
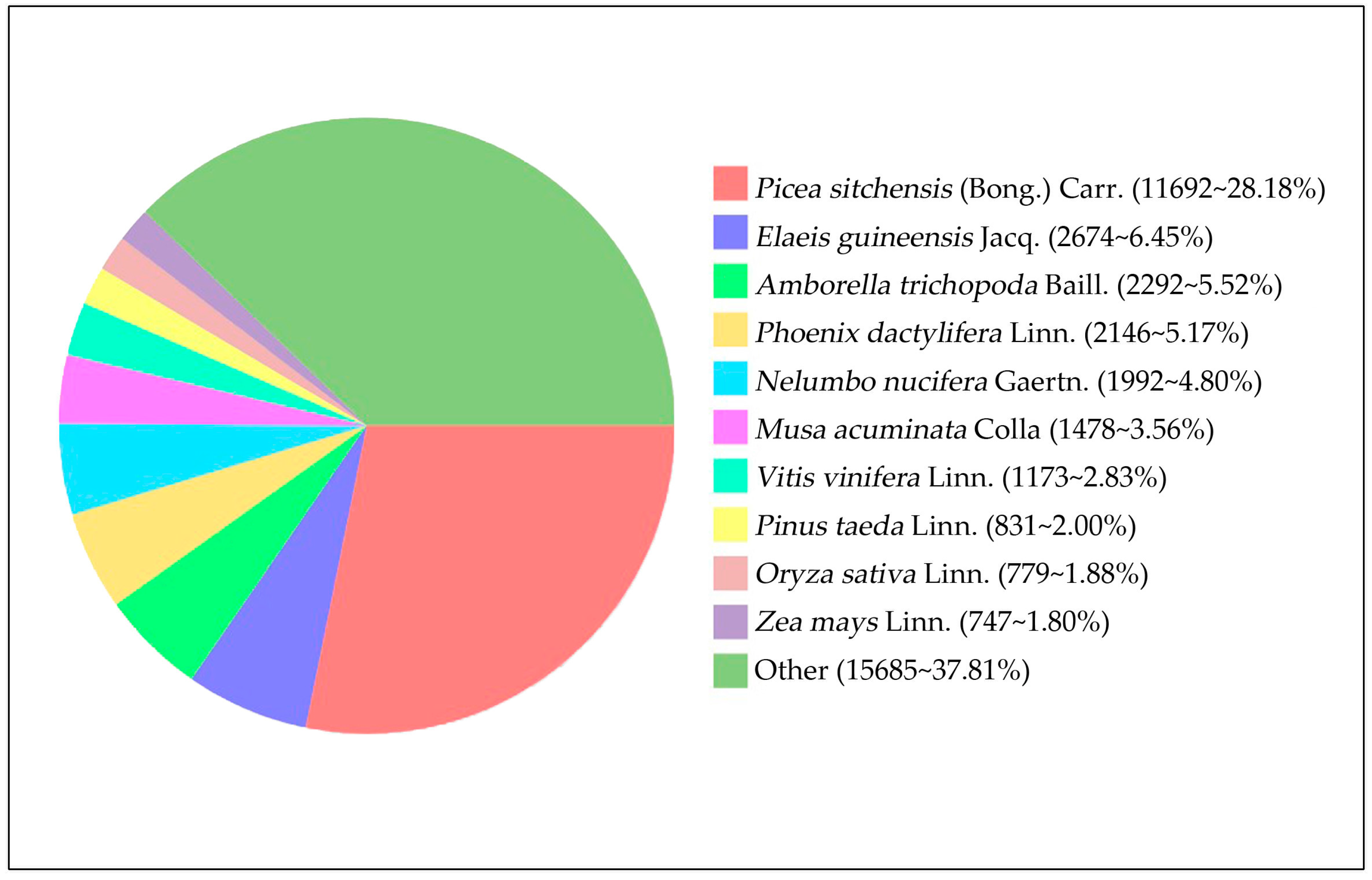
3.2. Functional Annotation
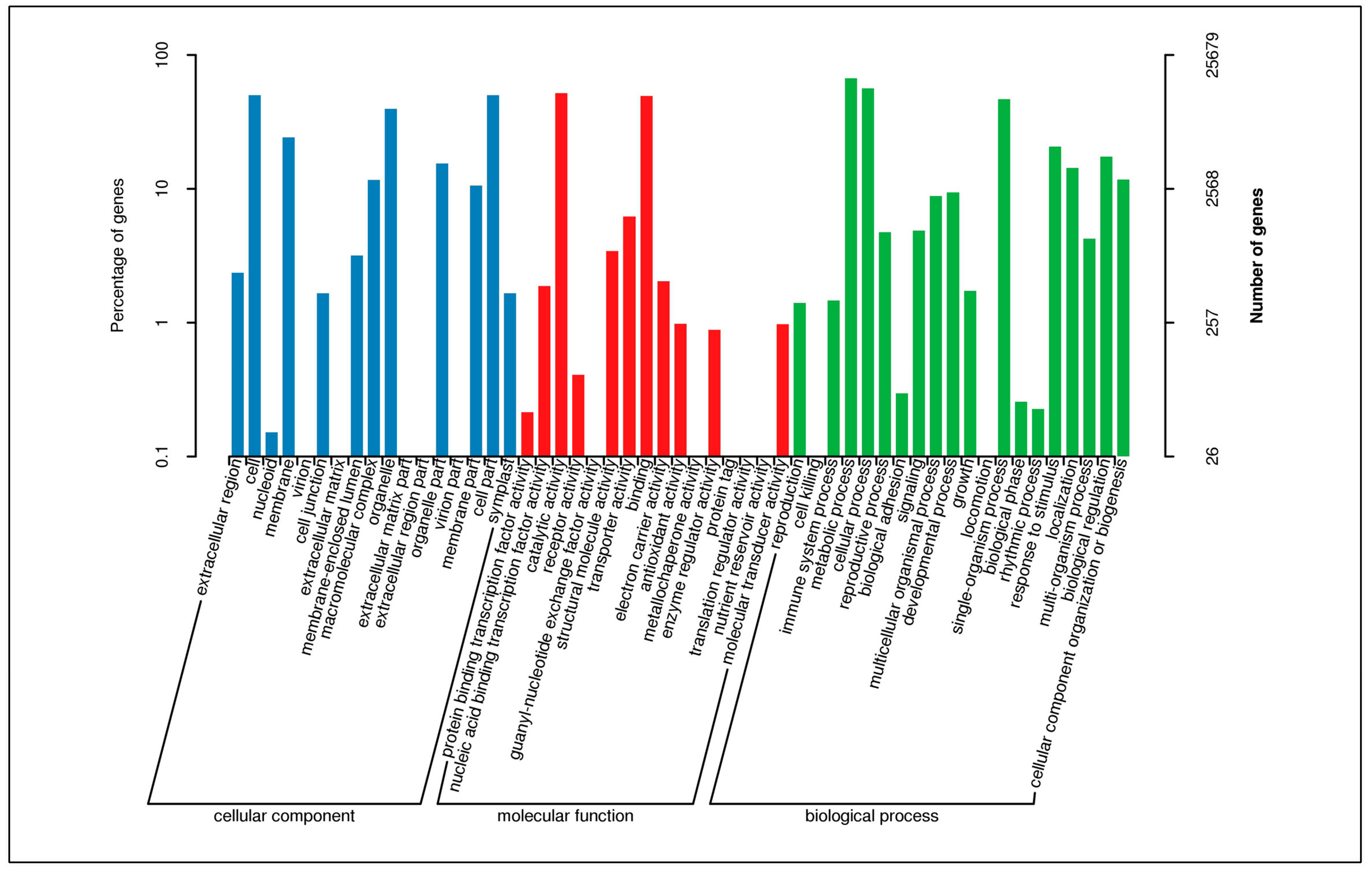
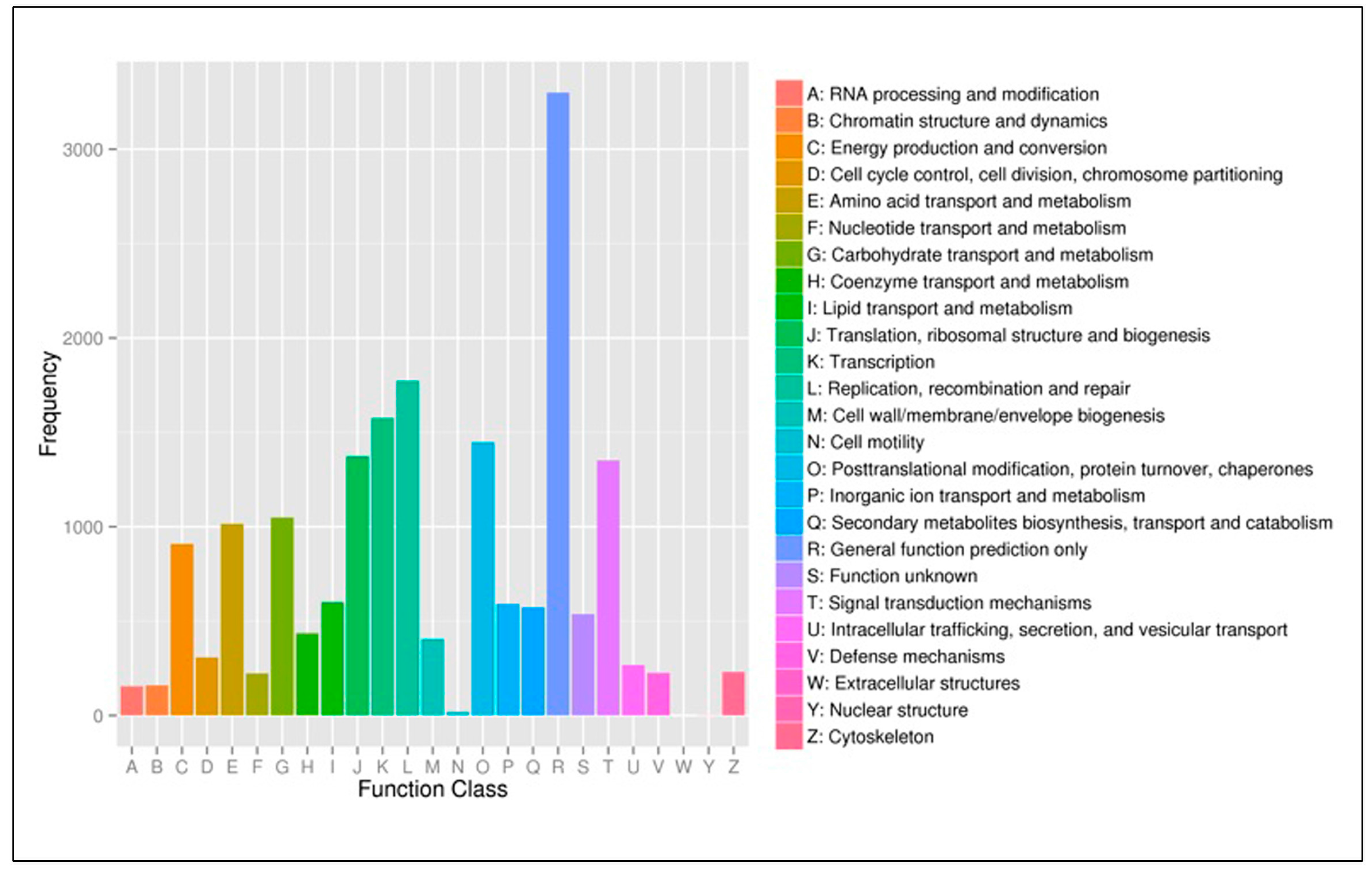
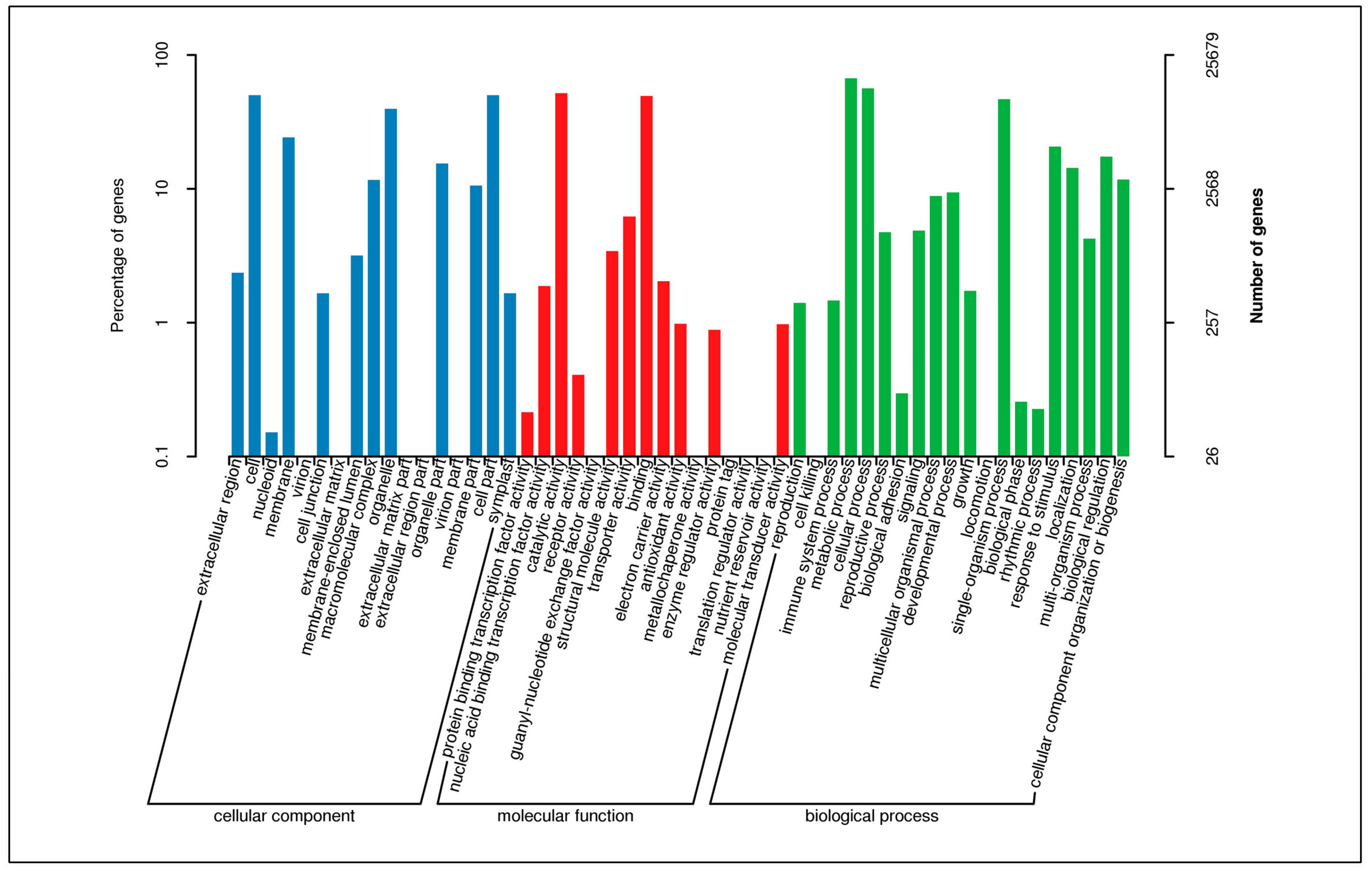
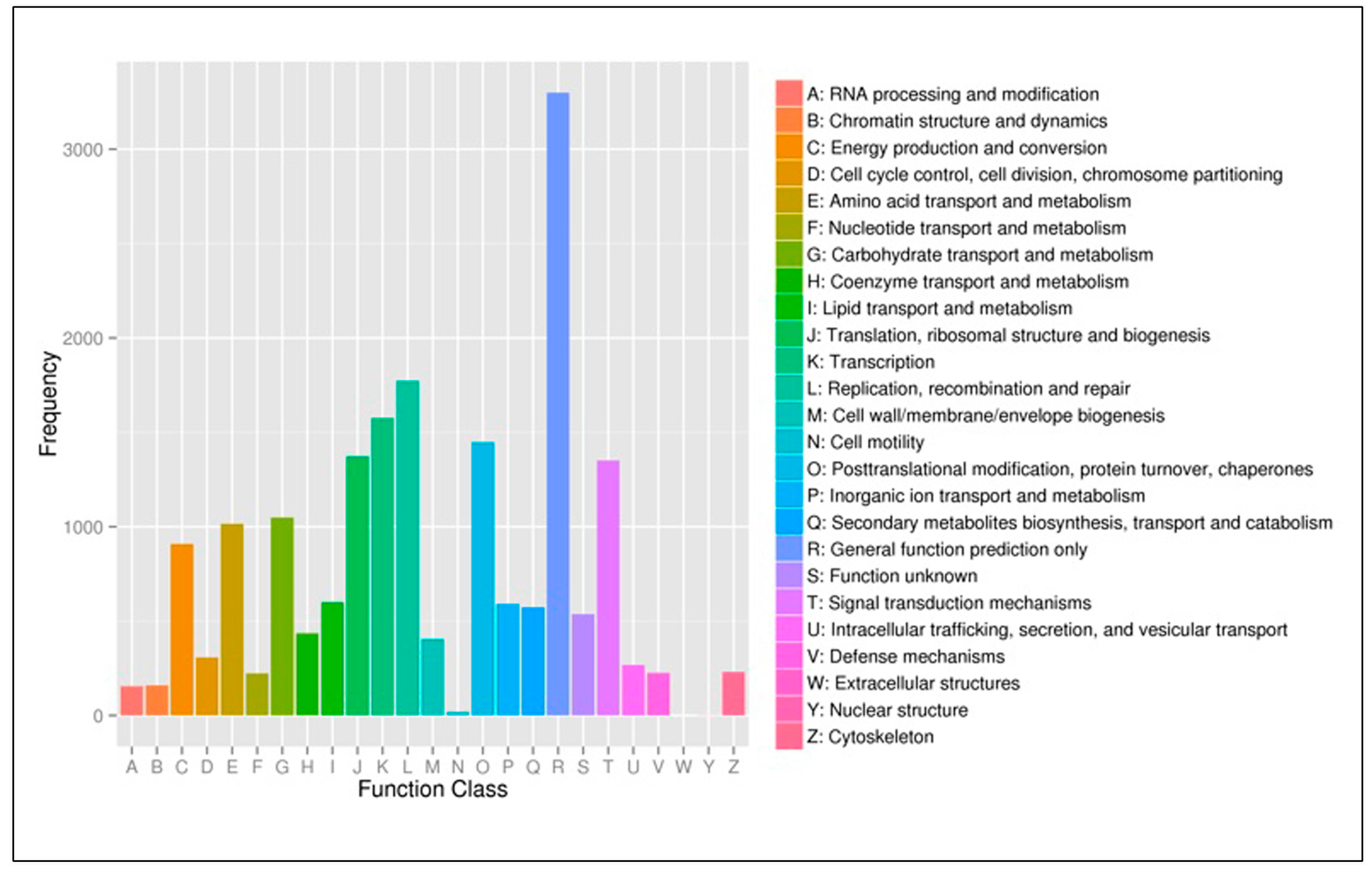
3.3. Functional Classification
3.4. Simple Sequence Repeats (SSR) Loci Discovery
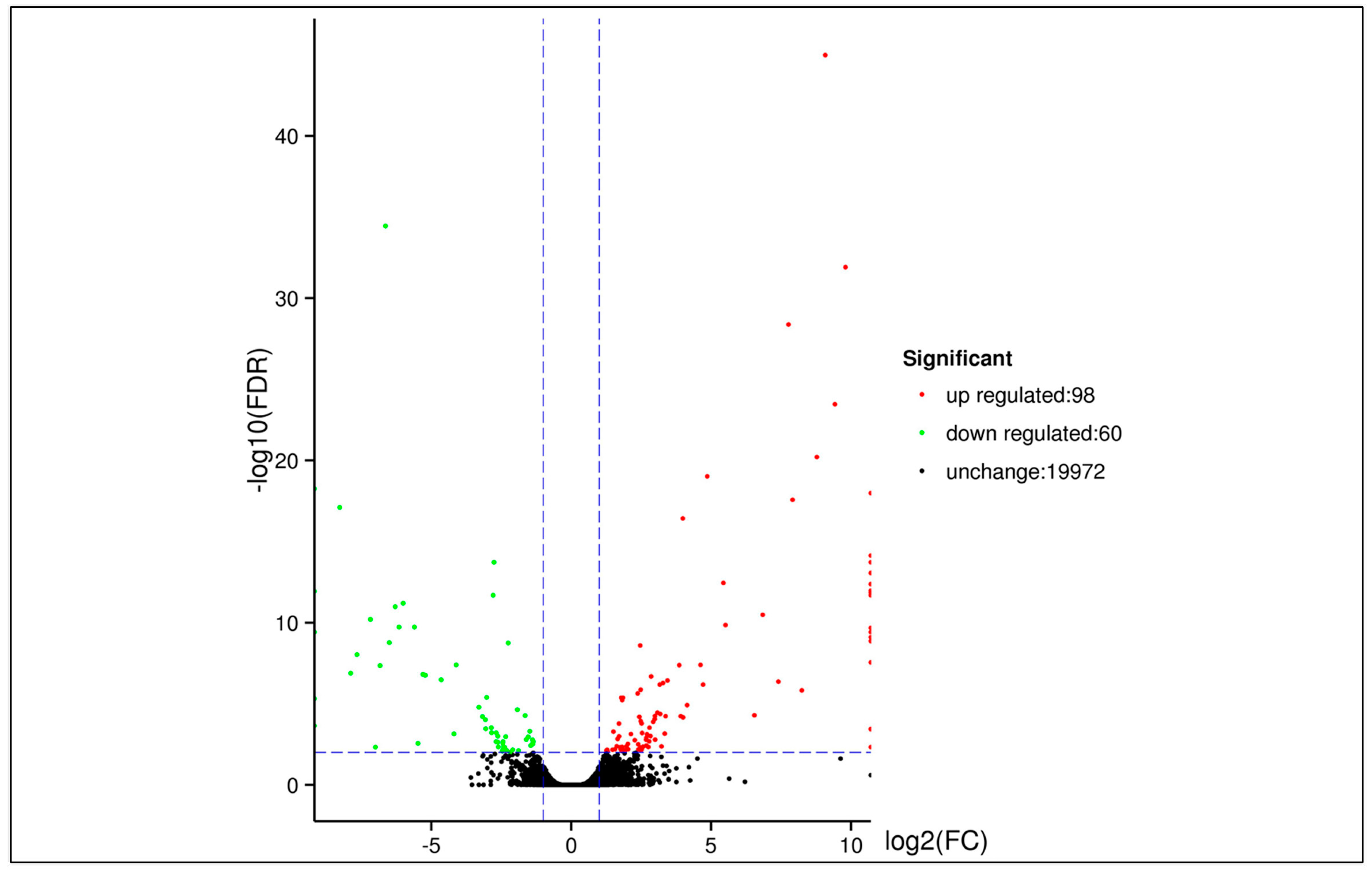
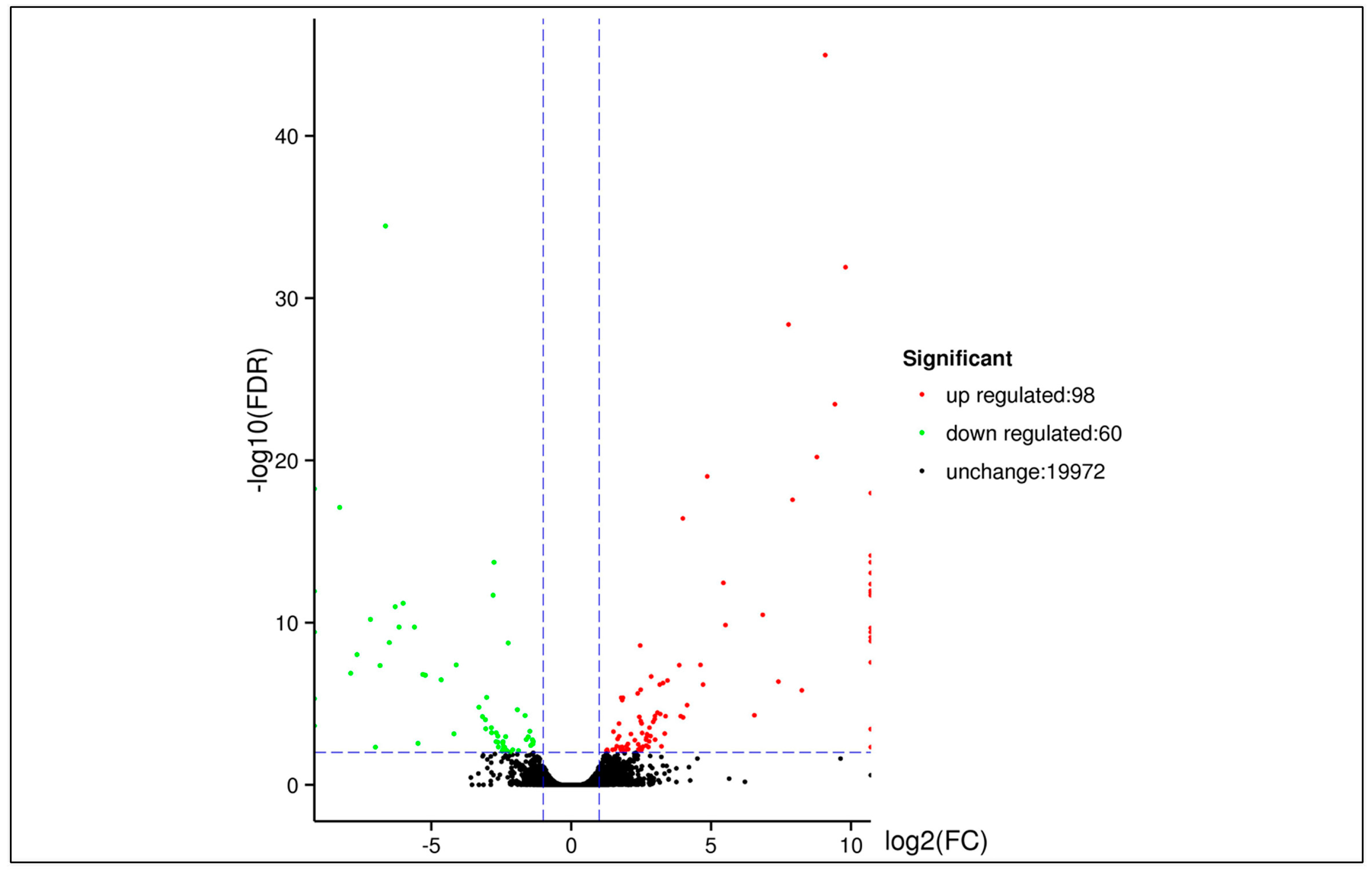
3.5. DEGs between Two Samples
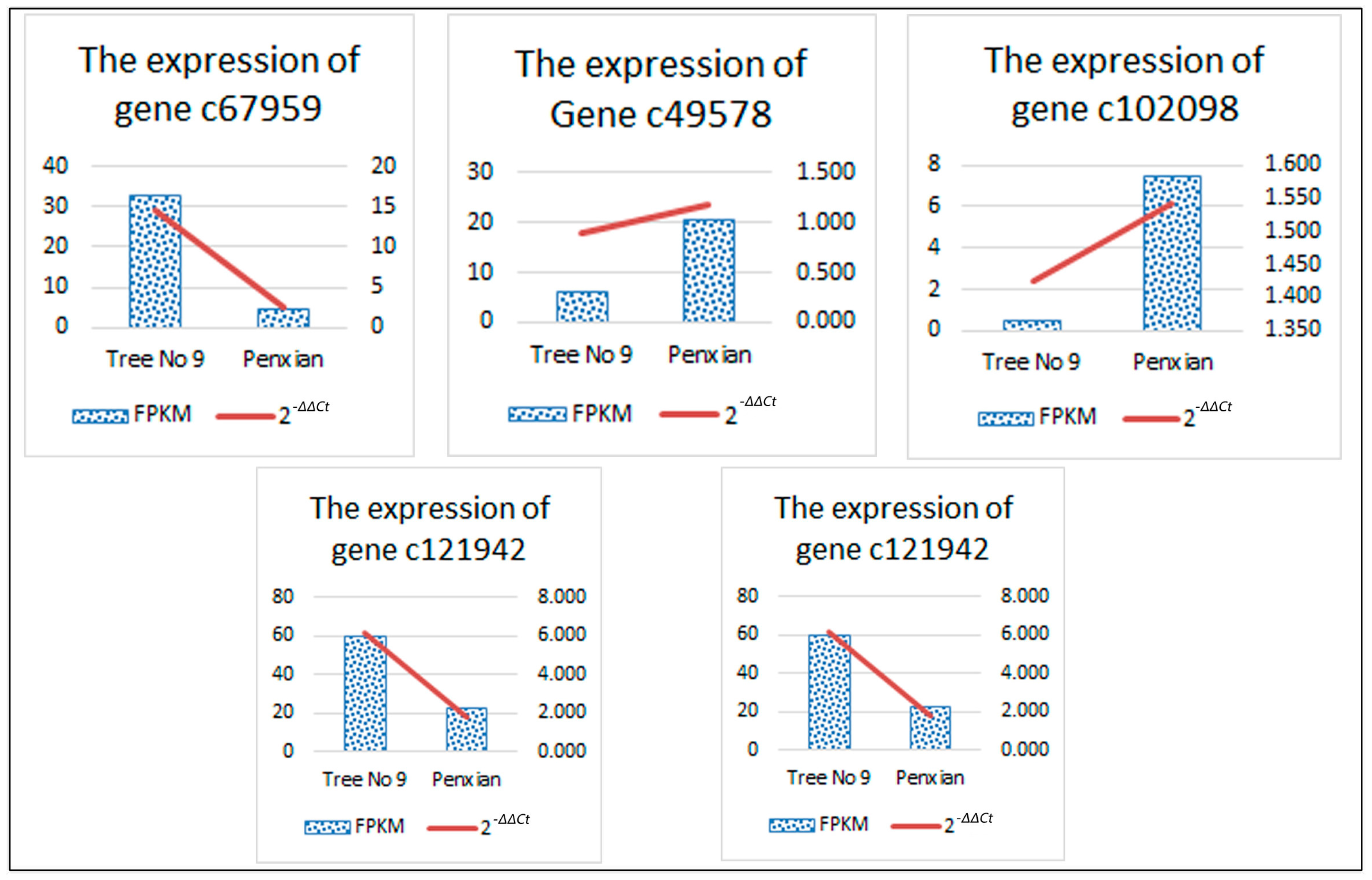
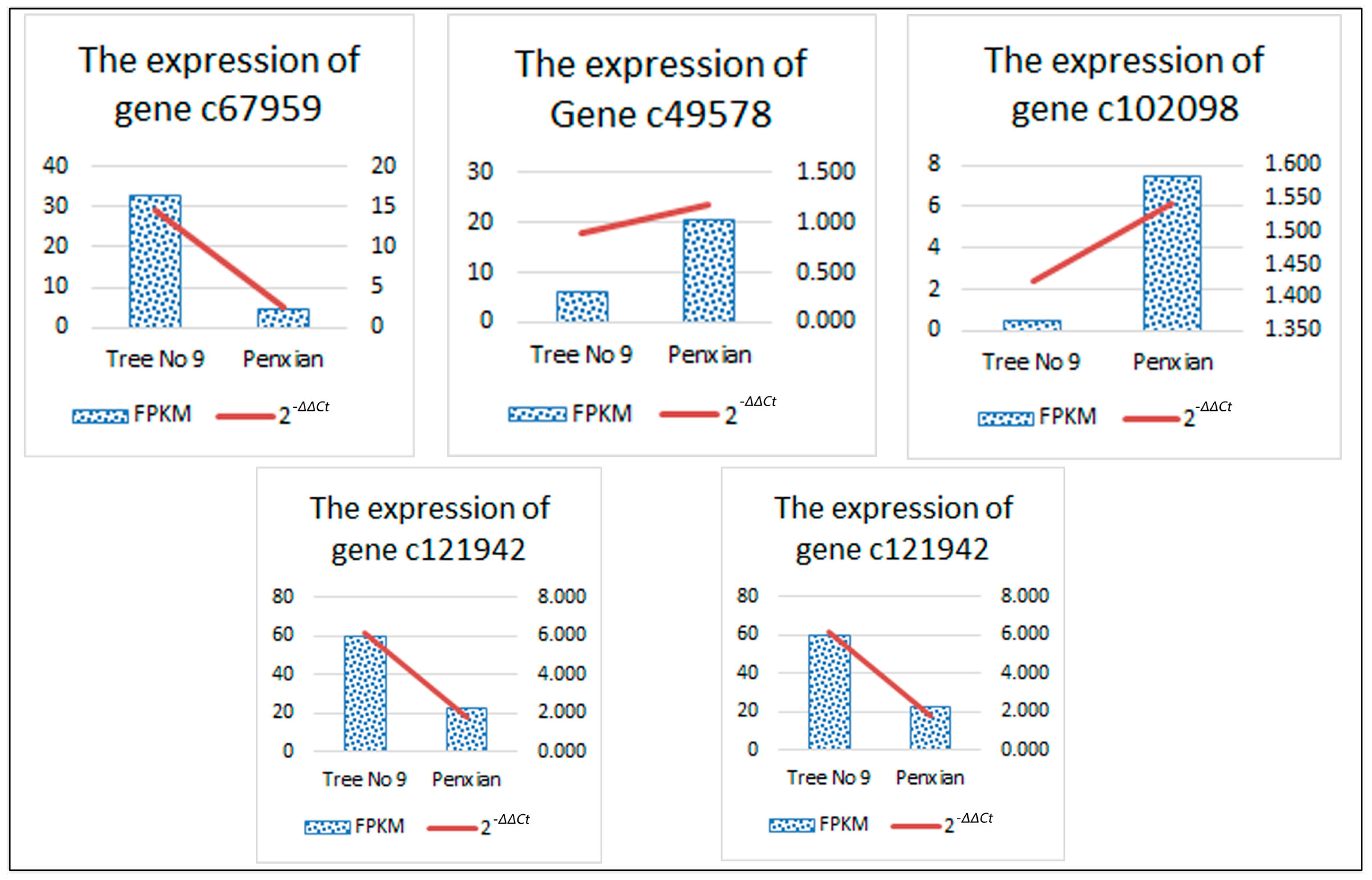
3.6. Confirmation of Solexa Expression Patterns by RT-qPCR
4. Discussion
4.1. The Description of Pinus Bungeana Zucc. ex Endl. Transcriptome
4.2. Transcriptome Assembly and Gene Annotation
4.3. Identification of Transcription Factor Involved in Pinus Bungeana Zucc. ex Endl. Architecture Regulation
4.4. DEGs between the Two Varieties
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stinchcombe, J.R.; Hoekstra, H.E. Combining population genomics and quantitative genetics: Finding the genes underlying ecologically important traits. Heredity 2008, 100, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.R.; Lubberstedt, T. Functional markers in plants. Trends Plant Sci. 2003, 8, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Novaes, E.; Drost, D.R.; Farmerie, W.G.; Pappas, G.J., Jr.; Grattapaglia, D.; Sederoff, R.R.; Kirst, M. High-throughput gene and snp discovery in eucalyptus grandis, an uncharacterized genome. BMC Genom. 2008, 9, 312. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Moon, J.F.; Lim, S.H.; Kim, J.C. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity 2011, 107, 1–15. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. Rna-seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by rna-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Cline, M. Concepts and terminology of apical dominance. Am. J. Bot. 1997, 84, 1064. [Google Scholar] [CrossRef] [PubMed]
- Cline, M.G. Execution of the auxin replacement apical dominance experiment in temperate woody species. Am. J. Bot. 2000, 87, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Takei, K.; Kojima, M.; Sakakibara, H.; Mori, H. Auxin controls local cytokinin biosynthesis in the nodal stem in apical dominance. Plant J. Cell Mol. Biol. 2006, 45, 1028–1036. [Google Scholar] [CrossRef] [PubMed]
- Reig, C.; Farina, V.; Mesejo, C.; Martínez-Fuentes, A.; Barone, F.; Agustí, M. Fruit regulates bud sprouting and vegetative growth in field-grown loquat trees (Eriobotrya japonica lindl.): Nutritional and hormonal changes. J. Plant Growth Regul. 2014, 33, 222–232. [Google Scholar] [CrossRef]
- Dun, E.A.; Ferguson, B.J.; Beveridge, C.A. Apical dominance and shoot branching. Divergent opinions or divergent mechanisms? Plant Physiol. 2006, 142, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Booker, J.; Chatfield, S.; Leyser, O. Auxin acts in xylem-associated or medullary cells to mediate apical dominance. Plant Cell 2003, 15, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Tamas, I.A. Hormonal Regulation of Apical Dominance; Springer: Dordrecht, Netherlands, 1995; pp. 393–410. ISBN 978-0-7923-2985-5. [Google Scholar] [CrossRef]
- Werner, T.; Motyka, V.; Strnad, M.; Schmulling, T. Regulation of plant growth by cytokinin. Proc. Natl. Acad. Sci. USA 2001, 98, 10487–10492. [Google Scholar] [CrossRef] [PubMed]
- Werner, T.; Nehnevajova, E.; Kollmer, I.; Novak, O.; Strnad, M.; Kramer, U.; Schmulling, T. Root-specific reduction of cytokinin causes enhanced root growth, drought tolerance, and leaf mineral enrichment in arabidopsis and tobacco. Plant Cell 2010, 22, 3905–3920. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Jianqi, L.I.; Songfeng, W.U.; Zhu, Y.; Chen, Y.; Fuchu, H.E. Integrated nr database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–72. [Google Scholar]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiqer, E.; Huang, H.; Lopez, R.; Maqrane, M.; et al. Uniprot: The universal protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The cog database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped blast and psi-blast: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2003, 32, D277. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2go: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.; Davis, A.; Dolinski, K.; Dwight, S.; Eppig, J.; et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. Kobas 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Mao, X.; Cai, T.; Luo, J.; Wei, L. Kobas server: A web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006, 34, W720–W724. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. Emboss: The european molecular biology open software suite. Trends Genet. TIG 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2012, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xu, M.; Luo, Q.; Wang, J.; Li, H. De novo transcriptome analysis of Liriodendron chinense petals and leaves by illumina sequencing. Gene 2014, 534, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ming, R.; Zhang, J.; Tao, A.; Fang, P.; Qi, J. De novo transcriptome sequence and identification of major bast-related genes involved in cellulose biosynthesis in jute (Corchorus capsularis L.). BMC Genom. 2015, 16, 1062. [Google Scholar] [CrossRef] [PubMed]
- Mudalkar, S.; Golla, R.; Ghatty, S.; Reddy, A.R. De novo transcriptome analysis of an imminent biofuel crop, Camelina sativa L. Using illumina GAIIX sequencing platform and identification of SSR markers. Plant Mol. Biol. 2014, 84, 159. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Puniya, B.L.; Kulshreshtha, D.; Kumar, S.; Kaur, J.; Ramachandran, S.; Singh, K. De novo transcriptome sequencing reveals important molecular networks and metabolic pathways of the plant, Chlorophytum borivilianum. PLoS ONE 2013, 8, e83336. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.X.; Wang, M.L.; Liu, Z.L.; Zhu, J.; Yan, M.Y.; Li, Z.H. Nucleotide polymorphism and phylogeographic history of an endangered conifer species Pinus bungeana. Biochem. Syst. Ecol. 2016, 64, 89–96. [Google Scholar] [CrossRef]
- Fu, N.; Wang, Q.; Shen, H.L. De novo assembly, gene annotation and marker development using illumina paired-end transcriptome sequences in celery (Apium graveolens L.). PLoS ONE 2013, 8, e57686. [Google Scholar] [CrossRef] [PubMed]
- LI, B.; Meng, Q.; Li, Y.; Dong, W.; Zheng, Y. Identification and evaluation of germplasm resources of Pinus bungeana. Hunan For. Sci. Technol. 2016, 43, 1–7. [Google Scholar] [CrossRef]
- Galuszka, P.; Frebort, I.; Sebela, M.; Sauer, P.; Jacobsen, S.; Pec, P. Cytokinin oxidase or dehydrogenase? Mechanism of cytokinin degradation in cereals. Eur. J. Biochem. 2001, 268, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Ashikari, M.; Sakakibara, H.; Lin, S.; Yamamoto, T.; Takashi, T.; Nishimura, A.; Angeles, E.R.; Qian, Q.; Kitano, H.; Matsuoka, M. Cytokinin oxidase regulates rice grain production. Science 2005, 309, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Werner, T.; Motyka, V.; Laucou, V.; Smets, R.; Van, O.H.; Schmülling, T. Cytokinin-deficient transgenic arabidopsis plants show multiple developmental alterations indicating opposite functions of cytokinins in the regulation of shoot and root meristem activity. Plant Cell 2003, 15, 2532–2550. [Google Scholar] [CrossRef] [PubMed]
- To, J.P.; Haberer, G.; Ferreira, F.J.; Deruere, J.; Mason, M.G.; Schaller, G.E.; Alonso, J.M.; Ecker, J.R.; Kieber, J.J. Type-a arabidopsis response regulators are partially redundant negative regulators of cytokinin signaling. Plant Cell 2004, 16, 658–671. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.J.; Beveridge, C.A. Roles for auxin, cytokinin, and strigolactone in regulating shoot branching. Plant Physiol. 2009, 149, 1929–1944. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Read Number | Base Number | GC Content | % ≥ Q30 |
|---|---|---|---|---|
| Tree No. 9-1 | 24,713,913 | 6,226,575,673 | 45.77 | 91.77 |
| Tree No. 9-2 | 25,402,758 | 6,399,644,060 | 45.78 | 91.68 |
| Tree No. 9-3 | 24,731,097 | 6,230,626,717 | 45.64 | 91.45 |
| Penxian-1 | 24,088,230 | 6,068,870,102 | 46.48 | 91.26 |
| Penxian-2 | 25,067,325 | 6,315,681,345 | 45.88 | 91.68 |
| Penxian-3 | 23,679,887 | 5,966,576,473 | 45.53 | 91.51 |
| Total/average | 147,683,210 | 37,207,974,370 | 45.85 | 91.56 |
| Unigenes Length | Total Number | Percentage |
|---|---|---|
| 200–300 | 31,783 | 36.08% |
| 300–500 | 20,020 | 22.73% |
| 500–1000 | 16,917 | 19.20% |
| 1000–2000 | 11,784 | 13.38% |
| 2000+ | 7588 | 8.61% |
| Total Number | 88,092 | |
| Total Length | 66,791,509 | |
| N50 Length | 1357 | |
| Mean Length | 758 |
| Motif Types | Repeat Number | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 5 | 6 | 7 | 8 | 9 | 10 | >10 | Total | Percent | |
| Dinucleotide | 0 | 248 | 144 | 84 | 67 | 69 | 34 | 646 | 40.2% |
| Trinucleotide | 562 | 197 | 86 | 16 | 1 | 0 | 0 | 862 | 53.7% |
| Tetra | 66 | 8 | 0 | 0 | 2 | 0 | 0 | 76 | 4.7% |
| Penta | 11 | 0 | 0 | 3 | 0 | 0 | 0 | 14 | 0.9% |
| Hexa | 6 | 1 | 0 | 1 | 0 | 0 | 0 | 8 | 0.5% |
| Total | 645 | 454 | 230 | 104 | 70 | 69 | 34 | 1606 | 100.0% |
| Percent | 40.2% | 28.3% | 14.3% | 6.5% | 4.4% | 4.3% | 2.1% | 100.0% | |
| DEG Set | All DEG | Up-Regulated | Down-Regulated |
|---|---|---|---|
| Tree No.9 vs. Penxian | 158 | 98 | 60 |
| Unigene Id | Pathway | Entry | Annotated Gene Name | Regulated | log2FC |
|---|---|---|---|---|---|
| c67959 | ko00908 | K00279 | CKX | DOWN | −2.759074002 |
| c49578 | ko04075 | K14492 | ARR-A | UP | 1.709013026 |
| c102098 | ko04075 | K14490 | AHP | UP | 3.914710727 |
| c121942 | K09264 | MADS-box transcription factor | DOWN | −1.385954738 | |
| c91025 | −5.475015097 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, Q.; Li, B.; Lin, F.; Huang, P.; Guo, W.; Zheng, Y. De Novo Sequencing and Assembly Analysis of Transcriptome in Pinus bungeana Zucc. ex Endl. Forests 2018, 9, 156. https://doi.org/10.3390/f9030156
Cai Q, Li B, Lin F, Huang P, Guo W, Zheng Y. De Novo Sequencing and Assembly Analysis of Transcriptome in Pinus bungeana Zucc. ex Endl. Forests. 2018; 9(3):156. https://doi.org/10.3390/f9030156
Chicago/Turabian StyleCai, Qifei, Bin Li, Furong Lin, Ping Huang, Wenying Guo, and Yongqi Zheng. 2018. "De Novo Sequencing and Assembly Analysis of Transcriptome in Pinus bungeana Zucc. ex Endl." Forests 9, no. 3: 156. https://doi.org/10.3390/f9030156