2.1. Crystal Structure of Sudan Virus Boniface GP1,2
The crystal structure of SUDV-Bon GP
1,2 bound to antibody 16F6 was determined to a resolution of 3.35 Å using molecular replacement. The crystallographic asymmetric unit contains one GP
1,2 monomer bound to one 16F6 Fab fragment. The final model contains residues 32-191, 213-285, 300-311 and 510-614 of GP
1,2 and residues 1-220 and 1-212 of the heavy and light chains of 16F6, respectively. A disulfide bond between C53 and C609 covalently links the GP
1 and GP
2 subunits together, and is only visible in structures of SUDV, but not EBOV GP
1,2. In all three ebolavirus GP
1,2 structures, GP
1 and GP
2 bury a surface area of ~2400 Å
2 on each other and are held together by numerous hydrogen bonds and non-bonded contacts. Although GP
1,2 was treated with peptide-N-glycosidase F (PNGase F), clear electron density is still observed for the first two monosaccharides of glycans attached to N257 (NAG 850) in GP
1 and N563 (NAG 901) in GP
2 indicating that these sites (in both SUDV and EBOV GP
1,2) are resistant to PNGaseF digestion (
Figure 1). The trimeric form of the GP
1,2 is obtained by application of 3-fold crystal symmetry, and has an approximate dimension of 95 Å × 95 Å × 90 Å.
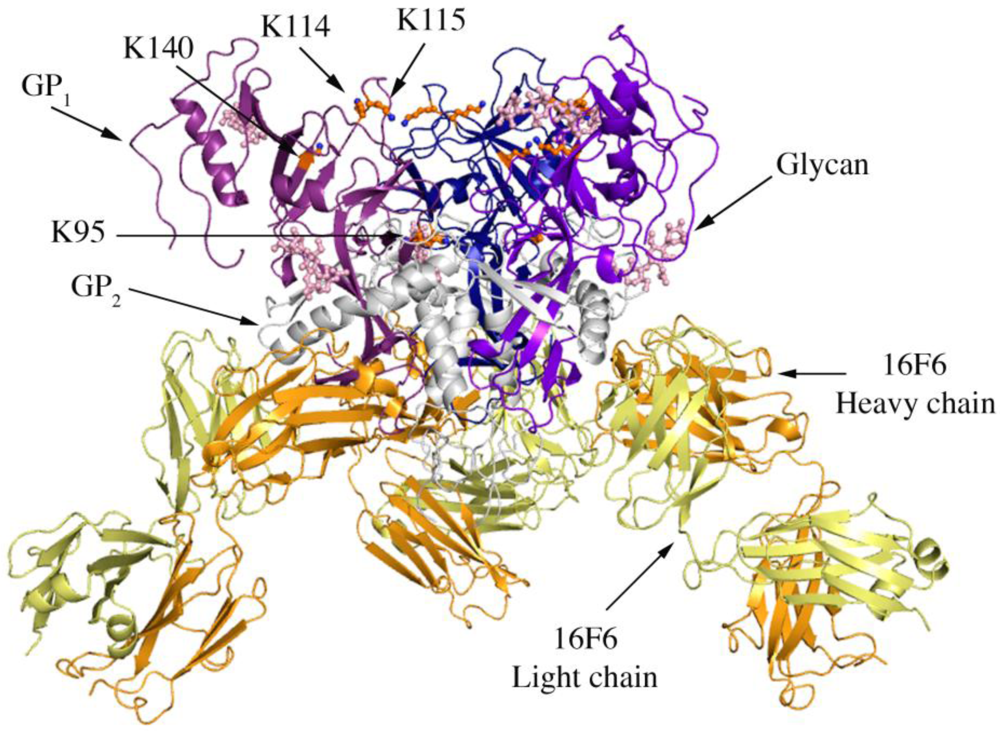
Figure 1.
Cartoon representation of trimer of SUDV-Bon GP1,2 bound to 16F6. The GP1 chains are colored in various shades of blue and GP2 chains are colored in white. The light chain of 16F6 is shown in pale yellow and the heavy chain of 16F6 is colored bright orange. The sugar residues on the glycoprotein are shown in ball and colored cyan. Lysine residues critical for receptor binding are shown in ball and stick and colored orange.
Figure 1.
Cartoon representation of trimer of SUDV-Bon GP1,2 bound to 16F6. The GP1 chains are colored in various shades of blue and GP2 chains are colored in white. The light chain of 16F6 is shown in pale yellow and the heavy chain of 16F6 is colored bright orange. The sugar residues on the glycoprotein are shown in ball and colored cyan. Lysine residues critical for receptor binding are shown in ball and stick and colored orange.
Four lysines (95, 114, 115, and 140) in the putative receptor-binding head of GP
1 have been identified as critical for attachment of EBOV to target cells [
21]. Three of these (114, 115, and 140) are strictly conserved among all known ebolaviruses, while position 95 is occupied by glutamine in some variants of SUDV (but is lysine in the SUDV-Bon and SUDV-Gul sequences crystallized). In all three ebolavirus GP
1,2 structures, K95 is buried deep in the chalice bowl of GP
1 and projects into GP
2 rather than out toward the target cell and receptor (
Figure 1). Inside GP
1,2, K95 forms a hydrogen bond with the main-chain carbonyl of T576 and forms van der Waals interactions with L573, which is located in the loop between HR1
B and HR1
C of GP
2. Hence the crystal structures support biochemical predictions [
21] that the observed functional importance of K95 may not be in direct receptor engagement, but rather in proper maintenance or springing of the prefusion GP
1,2 assembly. Alternatively, K95 may become better exposed for interaction with host factor(s) in the endosome if any conformational changes occur as a result of receptor binding. K115 also points down toward the viral membrane, but is solvent exposed and could potentially reorient upon receptor engagement. K114 and K140 project into solvent towards the target cell and may serve as direct contacts for receptor.
In the prefusion conformation of ebolavirus GP1,2, the first heptad repeat of GP2 is broken into four structural segments (HR1A-D) that wrap around GP1. We observe in all three structures (SUDV-Gul, SUDV-Bon, and EBOV-May) that residues R580-T581 of HR1C form a short β strand that assembles into a continuous β sheet with the K95-S99 β strand of the GP1 receptor-binding head. These residues are completely conserved among all ebolaviruses and marburgviruses.
Other structural details not previously reported include the noncovalent assembly by which GP
1 is anchored to GP
2. Five β strands of the GP
1 base and two antiparallel β strands of the GP
2 internal fusion loop combine to form a continuous seven-stranded twisted β sheet. The GP
1 component of that sheet and a second small β sheet of the GP
1 base together assemble a semicircular GP
1 horseshoe surface that clamps the first heptad repeat of GP
2 in its metastable, prefusion structure (
Figure 2 A,B). In order for GP
2 to rearrange into its post-fusion six-helix bundle structure [
13,
15] both the continuous GP
1-GP
2β sheet and the horseshoe clamp must be broken. The C-terminal heptad repeat 2 is disordered in all three SUDV-Bon, SUDV-Gul and EBOV-May structures, which may result from the functional mobility of this region as well as the lack of the transmembrane regions that tether GP
1,2 on the viral surface.
2.3 Implications for fusion
A CX
6CC motif in GP
2 that is conserved among all ebolaviruses is visible in both structures of SUDV GP
1,2. This motif contains the disulfide bond that anchors GP
1 to GP
2 (C53-C609) and a second disulfide bond within GP
2 (C601-C608). The glycoproteins of some retroviruses have a CXXC motif within the receptor-binding subunit that isomerizes to release the disulfide bond between the receptor-binding and fusion subunits [
20]. Filoviruses, however, lack such a motif in GP
1, and it is currently unknown if the GP
1-GP
2 disulfide link, which is encoded by a different motif, is retained or released during fusion. It has been speculated that endosomal thiolreductases could reduce the C53-C609 GP
1-GP
2 bond [
22], and mild reduction of the enzymatically processed 19 kDa GP
1,2 does confer binding to liposomes (an ability presumably attained via conformational change, as unreduced 19 kDa GP
1,2 does not bind liposomes) [
23]. The neighboring, C601-C608 intra-GP
2 disulfide bond however, is observed in pre-fusion as well as post-fusion crystal structures and thus, probably remains intact during fusion. Given the close proximity of both the GP
1-GP
2 and GP
2-GP
2 disulfide bonds, reduction is either very specific for the GP
1-GP
2 disulfide bond or plays no role during fusion. Separate possibilities are that GP
1 simply rotates out of the way, or that continual enzymatic processing of GP
1 by cathepsins digests enough of GP
1 to remove steric hindrance to GP
2 conformational rearrangement [
22].
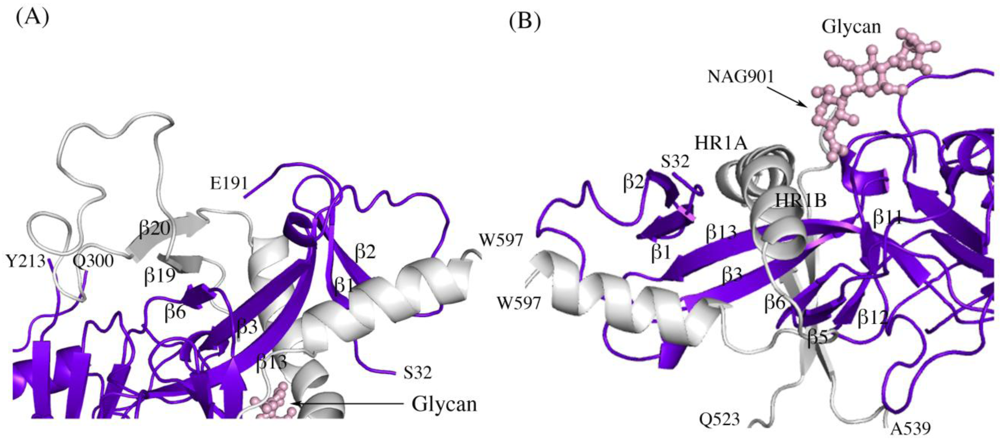
Figure 2.
Secondary structure interactions between SUDV-Bon GP1 (colored blue and purple) and GP2 (colored light grey). (A) Close up of the continuous seven-stranded twisted β sheet formed by five β strands of the GP1 base (β2, β1, β13, β3, and β6 in blue) and two antiparallel β strands of the GP2 internal fusion loop (β19 and β20 in grey). (B) The first heptad repeat of GP2 (which contains the grey coils labeled HR1A and HR1B and the loop afterwards) is clamped by a horseshoe surface formed by five β strands of GP1 base (β12, β5, β6, β3, β13), a second small β sheet of the GP1 base (β1 and β2) and a glycan (pink ball-and-stick). To orient the reader, key residues are labeled.
Figure 2.
Secondary structure interactions between SUDV-Bon GP1 (colored blue and purple) and GP2 (colored light grey). (A) Close up of the continuous seven-stranded twisted β sheet formed by five β strands of the GP1 base (β2, β1, β13, β3, and β6 in blue) and two antiparallel β strands of the GP2 internal fusion loop (β19 and β20 in grey). (B) The first heptad repeat of GP2 (which contains the grey coils labeled HR1A and HR1B and the loop afterwards) is clamped by a horseshoe surface formed by five β strands of GP1 base (β12, β5, β6, β3, β13), a second small β sheet of the GP1 base (β1 and β2) and a glycan (pink ball-and-stick). To orient the reader, key residues are labeled.
A recent study by Delos
et al. has revealed residues in the “chain reversal region” of GP
2 critical for fusion of Ebola virions [
20]. This region comprises a short stretch of hydrophobic residues, a glycine-glycine pair, a CX
6CC motif, and a bulky hydrophobic residue after the motif. Mutational analysis in EBOV GP
1,2 has shown that residues R587, F592, W597, G598, H602, and I610 are critical for viral infectivity. All the above residues except H602 (which is R in SUDV and RESTV) are conserved in SUDV. The better ordered structures of SUDV provide us with newer insights into the role of the conserved residues in the prefusion conformation, and are reported here. Interactions made by the side chains of these residues in the prefusion-form in SUDV and the post-fusion form in analogous EBOV (strain Mayinga, PDB code 1EBO) are shown in
Table 1. In the pre-fusion state, the conserved residues make interactions with GP
1 (hydrogen bonding to T60′ and stacking interaction against L57 and I185 (
Figure 3A); the ′ denotes residues from a 3-fold related monomer). W597 is involved in a stacking interaction with other W597 residues from two 3-fold related monomers, suggesting its role in stabilizing the trimeric form in the heptad repeat region. Definitive density was not seen for the side chains of R602 and I610 in either SUDV-Bon or SUDV-Gul GP
1,2 and we could not assign any interactions of these residues; positions 602 and 610 are thus modeled as alanine. However, in the post-fusion state, the conserved residues make interactions solely with residues in GP
2 (
Figure 3B). In addition, the conserved residues make interactions with different residues in the pre-fusion and post-fusion forms suggesting a greater role of these residues during the fusion process.
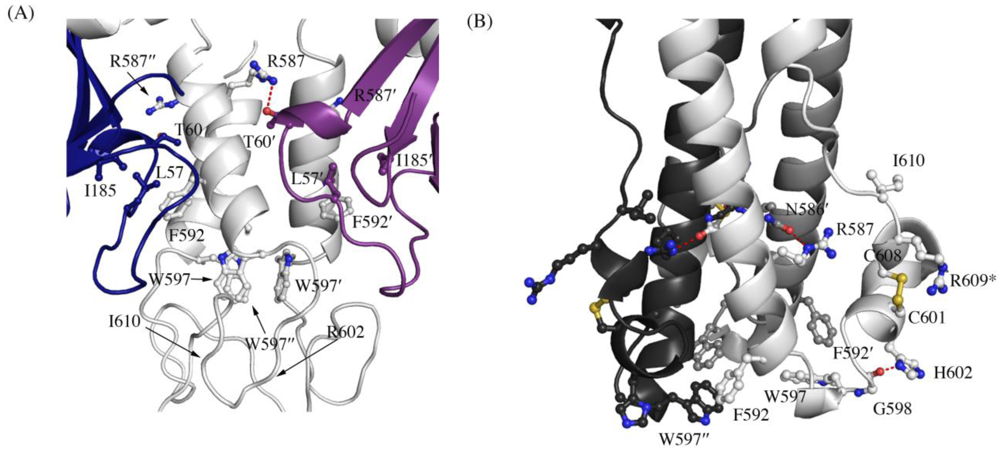
Figure 3.
Interaction of residues in the chain reversal region in (A) the prefusion SUDV-Bon GP1,2 and (B) the post fusion EBOV-May GP2. Prefusion SUDV-Bon is used here as it is better ordered than prefusion EBOV-May. Interacting residues are shown in ball and stick. In (A), different monomers of GP1 are colored blue and purple and the three copies of GP2 are all colored light grey. In (B), the three copies of GP2 are colored different shades of grey. Equivalent residues in the 3-fold related protomers are labeled with ´ and ´´ respectively. Hydrogen bonds are shown as red dashed lines. The residue R609* in the postfusion form is an engineered mutation to replace the cysteine residue (C609) in the native protein that is involved in a disulfide bond with C53 of GP1.
Figure 3.
Interaction of residues in the chain reversal region in (A) the prefusion SUDV-Bon GP1,2 and (B) the post fusion EBOV-May GP2. Prefusion SUDV-Bon is used here as it is better ordered than prefusion EBOV-May. Interacting residues are shown in ball and stick. In (A), different monomers of GP1 are colored blue and purple and the three copies of GP2 are all colored light grey. In (B), the three copies of GP2 are colored different shades of grey. Equivalent residues in the 3-fold related protomers are labeled with ´ and ´´ respectively. Hydrogen bonds are shown as red dashed lines. The residue R609* in the postfusion form is an engineered mutation to replace the cysteine residue (C609) in the native protein that is involved in a disulfide bond with C53 of GP1.
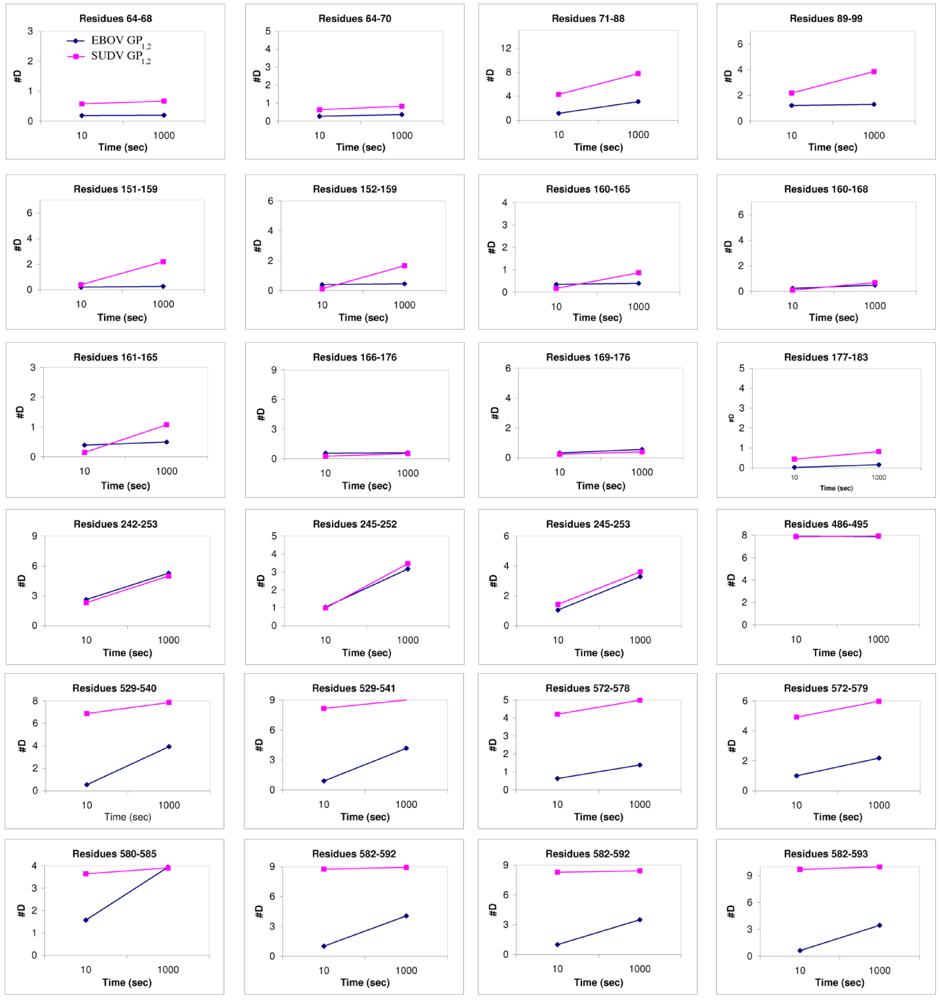
2.5 Thermal Motion in GP
Comparison of B-factor values (an atomic displacement parameter arising from thermal vibration of atoms and static disorder of atoms in different unit cells of the protein crystal) of key portions of GP
1 and GP
2 in SUDV GP
1,2 reveals that motion predominates in the glycan cap regions, the C-terminal half of the fusion loop, and the visible C-terminal regions of GP
2 (
Figure 5). How does SUDV compare to EBOV GP
1,2 in this regard? Deuterium Exchange Mass Spectrometry (DXMS) reveals that although GP
1 of SUDV and EBOV exhibit nearly identical rates of exchange of amide hydrogens with solvent deuterium, all regions of GP
2 of SUDV, including the fusion loop, heptad repeats, disulfide-containing linker and C-terminal regions, are fundamentally more mobile than those of EBOV GP
1,2 [
17] (
Figure 6). Interestingly, the disulfide-containing linker regions of GP
2 are only visible in crystals of SUDV GP
1,2, not EBOV GP
1,2. The unique crystal packing environment of the SUDV I23 unit cell and the acute angle made by the bound 16F6 antibody may have constrained this region.
Figure 5.
Comparison of thermal factors (B-factors) of various regions of SUDV GP1,2 (Panels A and B). Regions of low thermal mobility are deep blue (deep blue color set at ~ 80 Å2), whereas regions of high thermal mobility are red (deep red color set at ~ 220 Å2). The glycan cap and C-terminal “stem” regions of SUDV GP1,2 have higher B-values than the core region, indicating higher motion in those regions.
Figure 5.
Comparison of thermal factors (B-factors) of various regions of SUDV GP1,2 (Panels A and B). Regions of low thermal mobility are deep blue (deep blue color set at ~ 80 Å2), whereas regions of high thermal mobility are red (deep red color set at ~ 220 Å2). The glycan cap and C-terminal “stem” regions of SUDV GP1,2 have higher B-values than the core region, indicating higher motion in those regions.
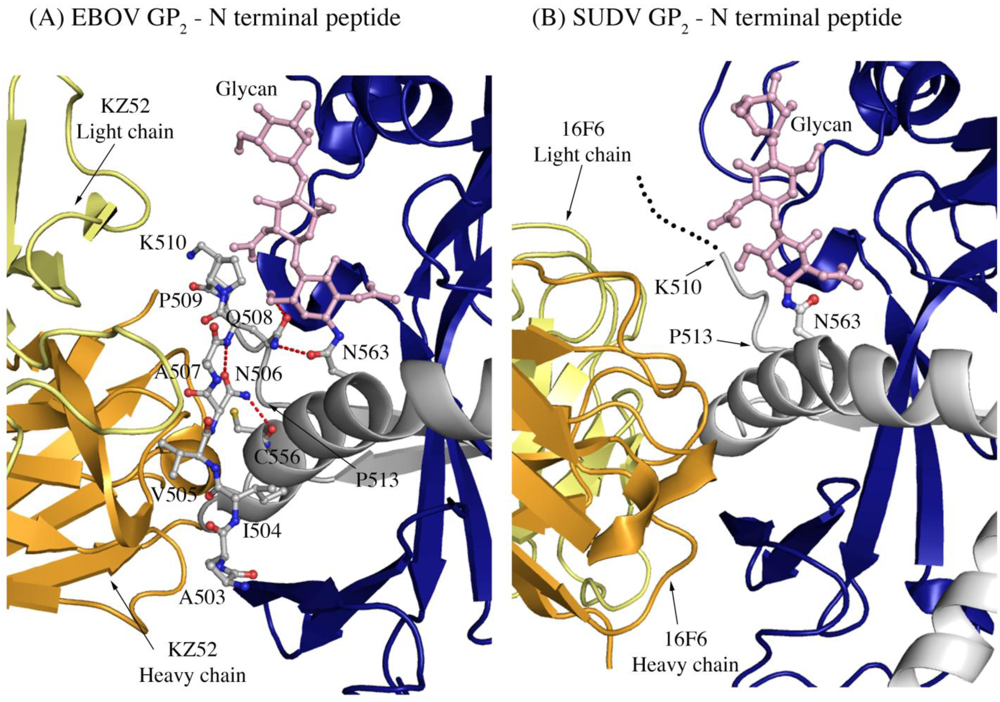
Another difference in mobility between SUDV and EBOV GP
1,2 relevant for antibody binding is the N terminus of GP
2, which is released from GP
1 by furin cleavage. In EBOV GP
1,2, this peptide is hydrogen bonded to the GP
1,2 core, is bound between heavy and light chains in the central region of the KZ52 paratope and forms a significant portion of its epitope. By contrast, in the SUDV structure, the same peptide is disordered and is not bound by 16F6. Instead, 16F6 binds the underlying GP
1,2 core beneath the peptide (
Figure 7).
One explanation could be that the GP2 N terminus is simply tacked down in the EBOV GP1,2 crystal structure by KZ52 binding. A more likely explanation, supported by DXMS, is that the mobility of this region fundamentally differs between SUDV and EBOV, even in the absence of antibody binding. Key differences in sequence between SUDV and EBOV at this site may explain this observation. The anchor point of the GP2 N terminus to the GP1,2 core is residue G509. At position 509, EBOV contains a proline, which may restrict mobility, while SUDV contains a glycine, which may enhance mobility. Further, residues N506 and Q508 of EBOV use both their terminal oxygen and nitrogen atoms to form a network of hydrogen bonds to the EBOV GP1,2 core; specifically, to amino acids as well as the attached glycan of heptad repeat 1. By contrast, position 506 is K in SUDV-Gul and Q in SUDV-Bon, and position 508 is T in both SUDV GP1,2 (Gulu and Boniface). None of those hydrogen bonds made by EBOV are observed for SUDV, and the specific hydrogen bonds made by the side-chain oxygens of N506 and Q508 in EBOV are not possible for the corresponding residues in SUDV. The key differences in sequence and inherent mobility of N-term region of SUDV GP1,2 reflects in a marginally varied epitope for binding of 16F6.
Figure 6.
Plots of number of deuterons
vs. time. These compare the deuteration levels of key peptides of EBOV GP
1,2 (shown in blue) to their counterparts in SUDV-Gul GP
1,2 (shown in magenta), measured over a time scale of 10-1000 sec using deuterium exchange mass spectrometry [
17].
Figure 6.
Plots of number of deuterons
vs. time. These compare the deuteration levels of key peptides of EBOV GP
1,2 (shown in blue) to their counterparts in SUDV-Gul GP
1,2 (shown in magenta), measured over a time scale of 10-1000 sec using deuterium exchange mass spectrometry [
17].
2.6 Comparison of Antibody-GP complexes
The crystal structures reveal that the 16F6 and KZ52 antibodies bind at overlapping epitopes that contain residues from both GP
1 and GP
2. However, the number of interactions made by each antibody with its respective antigen differs (
Table 3). 16F6 interacts with SUDV GP
1,2 via 7 hydrogen bonds and 19 non-bonded contacts. KZ52 interacts with EBOV GP
1,2 via 13 hydrogen bonds and 29 non-bonded contacts. Hence, the binding of KZ52 to EBOV GP
1,2 is expected to be stronger than the binding of SUDV GP
1,2 to 16F6. Accordingly, Surface Plasmon Resonance studies reveal that the K
d of KZ52 binding to recombinant, soluble EBOV GP
1,2 ectodomain is 6.31 ± 3.70 nM while the binding of 16F6 to recombinant, soluble SUDV GP
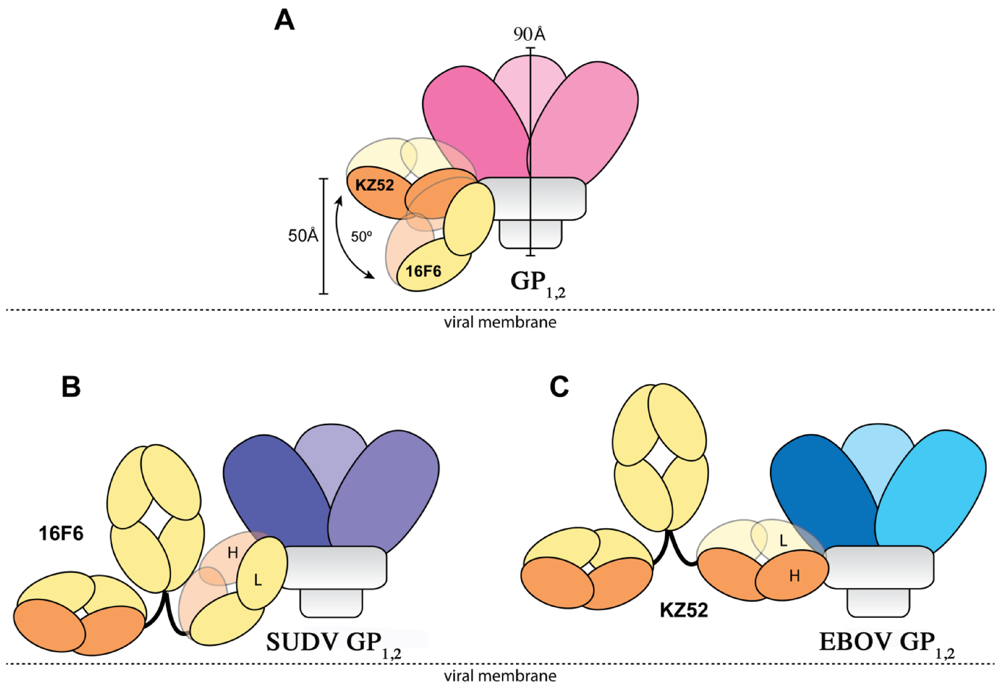
1,2 ectodomain is 1.94 ± 1.40 µM to SUDV-Bon, and 1.59 ± 1.0 µM to SUDV-Gul. Further, the angle the antibodies make when binding against the core of the GP
1,2 differs dramatically. KZ52 binds GP
1,2 at an angle more parallel to the viral membrane, while 16F6 is angled 50° downward toward the membrane, such that the C-terminal, constant regions of 16F6 are 50 Å closer to the viral membrane than those of KZ52 (
Figure 8). The hinge regions attached to the membrane-proximal end of the 16F6 Fab must therefore adopt acute angles so that the other Fab and Fc of the IgG do not penetrate the viral membrane. It is a bit surprising that the 16F6 IgG can reach down to access the epitope from that angle.
Figure 7.
The N-terminal peptide of GP2 in (A) EBOV-May GP1,2 and (B) SUDV-Bon GP1,2. In both panels, GP2 is shown in ball-and-stick and colored grey. GP1 is colored purple and the heavy and light chains of the antibody are colored bright orange and pale yellow, respectively. Note that the first eight residues of GP2 are disordered in SUDV-Bon/-Gul GP2 and are shown as black dots. Hydrogen bonds in (A) are shown as dashed red lines. There are no hydrogen bonds observed in (B) as the N-terminal peptide is disordered. The glycan residue connected to N563 is shown in ball and stick and colored light pink.
Figure 7.
The N-terminal peptide of GP2 in (A) EBOV-May GP1,2 and (B) SUDV-Bon GP1,2. In both panels, GP2 is shown in ball-and-stick and colored grey. GP1 is colored purple and the heavy and light chains of the antibody are colored bright orange and pale yellow, respectively. Note that the first eight residues of GP2 are disordered in SUDV-Bon/-Gul GP2 and are shown as black dots. Hydrogen bonds in (A) are shown as dashed red lines. There are no hydrogen bonds observed in (B) as the N-terminal peptide is disordered. The glycan residue connected to N563 is shown in ball and stick and colored light pink.
Figure 8.
Cartoon illustration of the different modes of antibody binding. (A) KZ52 binds perpendicular to the central axis of GP1,2 (parallel to the viral membrane), while 16F6 recognizes the overlapping epitope from an acute angle. The central axes of the two Fabs make a 50° angle with each other, and the constant portion of the 16F6 Fab is 50 Å lower than that of KZ52. Fab heavy chains are colored orange, while light chains are colored yellow. Approximate location of the viral membrane is indicated by a dotted line. C terminal portions of GP2 that anchor the visualized trimer to the viral membrane are disordered and not drawn here. (B) Model of a 16F6 IgG binding SUDV GP1,2 (purple) based on the 16F6 Fab-SUDV-Bon GP1,2 complex (PDB: 3VE0). (C) Model of a KZ52 IgG binding EBOV-May GP1,2 (blue) based on the KZ52 Fab-EBOV-May GP1,2 complex structure (PDB: 3CSY). The IgG and GP1,2 fragments are drawn to scale in the models.
Figure 8.
Cartoon illustration of the different modes of antibody binding. (A) KZ52 binds perpendicular to the central axis of GP1,2 (parallel to the viral membrane), while 16F6 recognizes the overlapping epitope from an acute angle. The central axes of the two Fabs make a 50° angle with each other, and the constant portion of the 16F6 Fab is 50 Å lower than that of KZ52. Fab heavy chains are colored orange, while light chains are colored yellow. Approximate location of the viral membrane is indicated by a dotted line. C terminal portions of GP2 that anchor the visualized trimer to the viral membrane are disordered and not drawn here. (B) Model of a 16F6 IgG binding SUDV GP1,2 (purple) based on the 16F6 Fab-SUDV-Bon GP1,2 complex (PDB: 3VE0). (C) Model of a KZ52 IgG binding EBOV-May GP1,2 (blue) based on the KZ52 Fab-EBOV-May GP1,2 complex structure (PDB: 3CSY). The IgG and GP1,2 fragments are drawn to scale in the models.
As a result of the higher affinity for the GP
1,2 ectodomain, greater number of contacts, and presumably easier access to its epitope, one might expect KZ52 to neutralize better than 16F6. Surprisingly, however, 16F6 neutralizes SUDV GP
1,2-bearing VSIV better than KZ52 neutralizes EBOV GP
1,2-bearing vesicular stomatitis Indiana virus (VSIV) [
16] (data replotted here as
Figure 9). At all IgG concentrations, a greater portion of virus remains un-neutralized by KZ52 than by 16F6. Even small differences in neutralization capacity may confer significant functional variation
in vivo for filoviruses because a single p.f.u. can be a lethal dose for primates.
Our data suggests that binding affinity and access to the epitope alone do not account completely for antibody neutralization efficacy; the composition of the particular epitopes and the susceptibility of the different GP
1,2, themselves, to be neutralized are also important. For example, although the epitopes overlap, KZ52 is shifted more towards GP
2, burying mostly GP
2 while 16F6 buries more of GP
1 on SUDV GP
1,2 [
16]. Affinity for transmembrane-anchored GP
1,2 could differ somewhat from affinity for the GP
1,2 ectodomain as well, although the epitopes of these antibodies are quite some distance (>50 Å) from the transmembrane anchor.
In addition, the 16F6 and KZ52 Fabs are rotated relative to each other when bound to GP
1,2. KZ52 binds with its heavy chain “down” toward the membrane, while 16F6 binds with its heavy chain “up” toward GP
1. In addition, the crystal structures reveal that the heavy chain of 16F6 interacts directly with both heptad repeat 1 of GP
2 (specifically HR1
B) and the β
2 strand of GP
1. By contrast, KZ52 interacts primarily with the N-terminal peptide of GP
2 and the turn region prior to the HR1 helix. KZ52 is unable to make the same contacts as 16F6 because (1) its heavy and light chain are switched in space relative to those of 16F6 and (2) the N-terminal peptide of GP
2 is nestled into the center of the paratope in EBOV (
Figure 7). The N-terminal peptide of GP
2 is probably less important for GP
1,2 function than are the HR1 helix and GP
1 base strand. Hence, an antibody that anchors a less-important terminal peptide might be expected to be less effective than one that anchors a heptad repeat critical for conformational change to its prefusion GP
1β-sheet clamp. Hence, 16F6 binds more weakly, but may bind more effectively. These antibodies are thought to neutralize by blocking conformational changes during fusion. Perhaps better bridging of GP
1 and GP
2 by 16F6 more effectively anchors the pair in a pre-fusion conformation.
Alternatively, key differences between EBOV and SUDV GP
1,2 may render them differently susceptible to neutralization. GP
2 of SUDV is less stably ordered than GP
2 of EBOV. Perhaps 16F6 has recognized a greater portion of GP
1 because its GP
2 is a “moving target”. Certainly, the increased mobility of the GP
2 N-terminal peptide in SUDV means that more of underlying GP
1,2 structure is available for antibody interaction, antibody anchoring of the prefusion conformation and greater neutralization. Another difference is that EBOV GP
1,2 has a greater requirement for cathepsin cleavage than does SUDV GP
1,2 [
18], and crystal structures reveal that the two GP
1,2 have different electrostatics at their trimer interface. Electrostatics at the trimer interface may influence how the low pH environment of the endosomes determines susceptibility of GP
1,2 to triggering [
16].
In summary, we find by structural and functional analysis, that the antibody of higher affinity and presumably easier access to a shared site is not necessarily the most effective at viral neutralization. Other characteristics of the epitope bound, including which amino acids are contained in the individual epitopes and the functional importance of these amino acids, may confer neutralization susceptibility. A conundrum for filoviruses has been that antibody efficacy in vitro and in vivo do not always correlate. Here, we demonstrate that certain in vitro characteristics of antibody binding strength, epitope access, and in vitro neutralization also do not necessarily correlate. In addition to binding tightly, an effective antibody may need to bind correctly.
Development of effective immunotherapeutics against filoviruses will thus require detailed and coupled structural and functional characterization of candidate antibodies. It will also require production and analysis of larger panels of antibodies against the filoviruses than have been currently described so that we may compare several different antibodies within the same competition group, in order to determine what the most effective antibodies are and why this is so. This knowledge base will also clarify what would constitute an ideal antibody response elicited by vaccination, and will aid in development of vaccines designed to elicit these types of antibodies.
Table 1.
Interactions made by key residues in the “chain reversal region” in pre-fusion and post-fusion forms of ebolavirus GP1,2.
Table 1.
Interactions made by key residues in the “chain reversal region” in pre-fusion and post-fusion forms of ebolavirus GP1,2.
| Residue | Prefusion (SUDV-Bon) | Prefusion (EBOV-May) | Postfusion (EBOV-May) |
|---|
| R587 | Hydrogen bond to T60′ | No interactions | Hydrogen bond to N586′ |
| F592 | Stacks against L57, I185, L594′, R596 | Stacks against V48, L57, L593 | Stacks against L594′, W597′, I603′, |
| W597 | Stacks against W597′, W597′′ | Side chain disordered | Stacks against F592′, L593′, L594, I603, R596 |
| G598 | No interactions | No interactions | Hydrogen bond to H602 |
| H602 (R in SUDV) | Side chain disordered | Residue disordered | Hydrogen bond to G598 |
| I610 | Side chain disordered | Residue disordered | No interactions |
Table 2.
Interactions between SUDV GP1,2 and Fab 16F6.
GP1 to antibody heavy chain*
GP1 to antibody heavy chain*
| 16F6-H | | | GP1 | Interaction |
| Y31 | OH | O | S32 | H-bond |
| Y31 | OH | OE2 | E47 | H-bond |
| Y32 | OH | OE1 | E44 | H-bond |
| R98 | NH2 | OE1 | E44 | H-bond |
| Y31 | OH | C | S32 | Nonbonded |
| Y31 | CE1 | CG | P34 | Nonbonded |
| R98 | NH2 | CD | E44 | Nonbonded |
| R98 | NH2 | OE2 | E44 | Nonbonded |
| Y32 | OH | N | V45 | Nonbonded |
| A28 | CB | O | V45 | Nonbonded |
| Y31 | OH | CG | E47 | Nonbonded |
GP2 to antibody heavy chain
GP2 to antibody heavy chain
| 16F6-H | | | GP2 | Interaction |
| N103 | ND2 | O | A553 | H-bond |
| Y101 | OH | OE1 | E564 | H-bond |
| Y101 | OH | OE2 | E564 | H-bond |
| Y101 | O | CA | G557 | Nonbonded |
| Y101 | CE1 | CB | Q560 | Nonbonded |
| Y101 | CE1 | N | L561 | Nonbonded |
| Y101 | OH | CA | L561 | Nonbonded |
| Y101 | OH | CD | E564 | Nonbonded |
| Y101 | OH | N | L561 | Nonbonded |
GP1 to antibody light chain
GP1 to antibody light chain
| 16F6-L | | | GP1 | Interaction |
| T56 | CG2 | ND2 | N40 | Nonbonded |
GP2 to antibody light chain
GP2 to antibody light chain
| 16F6-L | | | GP2 | Interaction |
| W50 | NE1 | C | N552 | Nonbonded |
| W50 | CD2 | CB | N552 | Nonbonded |
| W50 | NE1 | CB | N552 | Nonbonded |
| W50 | CE2 | CB | N552 | Nonbonded |
| W50 | CD1 | N | N553 | Nonbonded |
Table 3.
Comparison of the 16F6 and KZ52 epitopes on SUDV and EBOV GP1,2, respectively.Residues at the shared intersection of the two epitopes are colored orange. Residues listed are conserved between SUDV and EBOV, unless specified.
Table 3.
Comparison of the 16F6 and KZ52 epitopes on SUDV and EBOV GP1,2, respectively.Residues at the shared intersection of the two epitopes are colored orange. Residues listed are conserved between SUDV and EBOV, unless specified.
| Residues of SUDV GP1,2 bound by 16F6 | Residues of EBOV GP1,2 bound by KZ52 |
|---|
| S32 | |
| P34 | |
| T39 (note: residue 39 is H in EBOV) | |
| N40 | |
| | S41 |
| T42 | V42 |
| L43 | L43 |
| E44 | Q44 |
| V45 | |
| T46 (is S in EBOV) | |
| E47 (is D in EBOV) | |
| Q50 (is K in EBOV) | |
| V52 | |
| | A503 (is T in SUDV) |
| | I504 (is N in SUDV) |
| | V505 (is T in SUDV) |
| | N506 (is K in SUDV) |
| | A507 |
| | Q508 (is T in SUDV) |
| | P509 (is G in SUDV) |
| | K510 |
| | C511 |
| P513 | P513 |
| | N514 |
| | H549 |
| N550 | N550 |
| Q551 | Q551 |
| N552 | D552 |
| A553 | G553 |
| C556 | C556 |
| G557 | |
| Q560 | |
| L561 | |
| E564 | |
Table 4.
Data collection and refinement statistics of SUDV-Bon GP1,2 bound to 16F6.
Table 4.
Data collection and refinement statistics of SUDV-Bon GP1,2 bound to 16F6.
| Beamline | ALS 8.3.1 |
| Space group | I23 |
| Cell parameters (Å) | a=b=c=194.86 |
| Wavelength (Å) | 1.1159 |
| Resolution (Å) (last shell) | 50.00-3.35 (3.47-3.35)a |
| Total observations | 149425 |
| Unique reflections | 17818 |
| I/σ | 13.4 (2.8) |
| Rsym (%)b | 7.9 (77.7) |
| Completeness (%) | 99.9 (100.0) |
| Redundancy | 8.4 |
| Matthews coefficient (VM, A3Da-1) | 3.5, 65% solvent content |
| |
| Rwork (%)c | 21.98 |
| Rfree (%) | 28.44 |
| Number of protein residues/atoms | 782/6003 |
| Number of glycan residues/atoms | 9/111 |
| r.m.s.d. bond length (Å) | 0.012 |
| r.m.s.d. bond angles (°) | 1.561 |
| Average B values (Å2) | |
| Fab 16F6 (chains heavy/light) | 116/124 |
| GP1 | 175 |
| GP2 | 148 |
| Overall | 141 |
| Ramachandran plot (%) | |
| Most favored | 78.8 |
| Allowed | 18.4 |
| Generously allowed | 2.2 |
| Disallowed | 0.6 |
Figure 9.
The ability of 16F6 and KZ52 IgGs to neutralize SUDV GP
1,2-bearing VSIV or EBOV GP
1,2-bearing VSIV, plotted in a log (A) and linear (B) scales. VSIV particles were pre-incubated with the indicated concentrations of antibody for one hour at room temperature, and then exposed to Vero cells at 37° C. Viral infectivity was scored at 16 hours post-infection. The starting titers were 9×10
7 IU/ml for EBOV and 3×10
7 IU/ml for SUDV on Vero cells [
16].
Figure 9.
The ability of 16F6 and KZ52 IgGs to neutralize SUDV GP
1,2-bearing VSIV or EBOV GP
1,2-bearing VSIV, plotted in a log (A) and linear (B) scales. VSIV particles were pre-incubated with the indicated concentrations of antibody for one hour at room temperature, and then exposed to Vero cells at 37° C. Viral infectivity was scored at 16 hours post-infection. The starting titers were 9×10
7 IU/ml for EBOV and 3×10
7 IU/ml for SUDV on Vero cells [
16].










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
