Differential Virulence and Pathogenesis of West Nile Viruses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
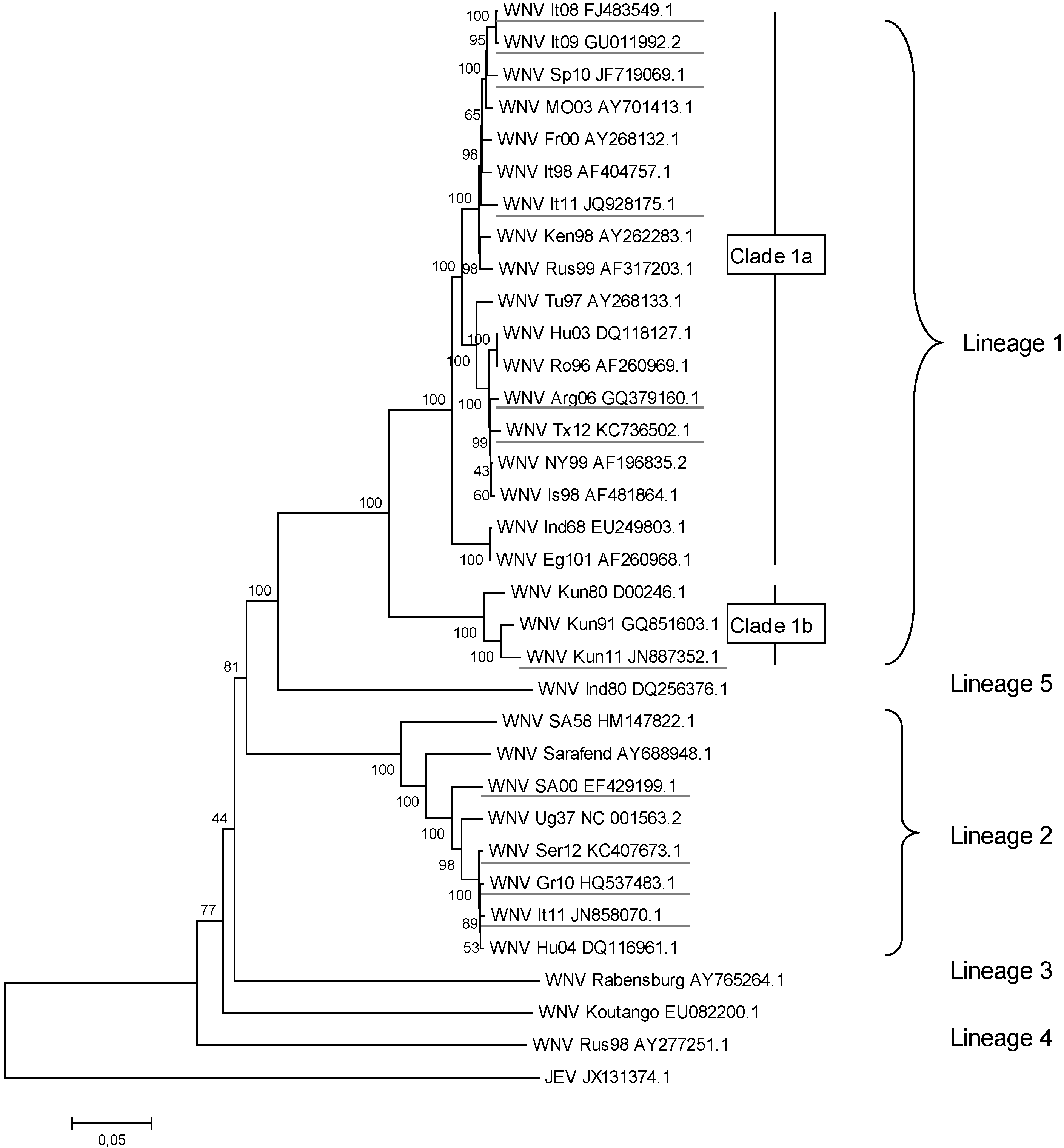
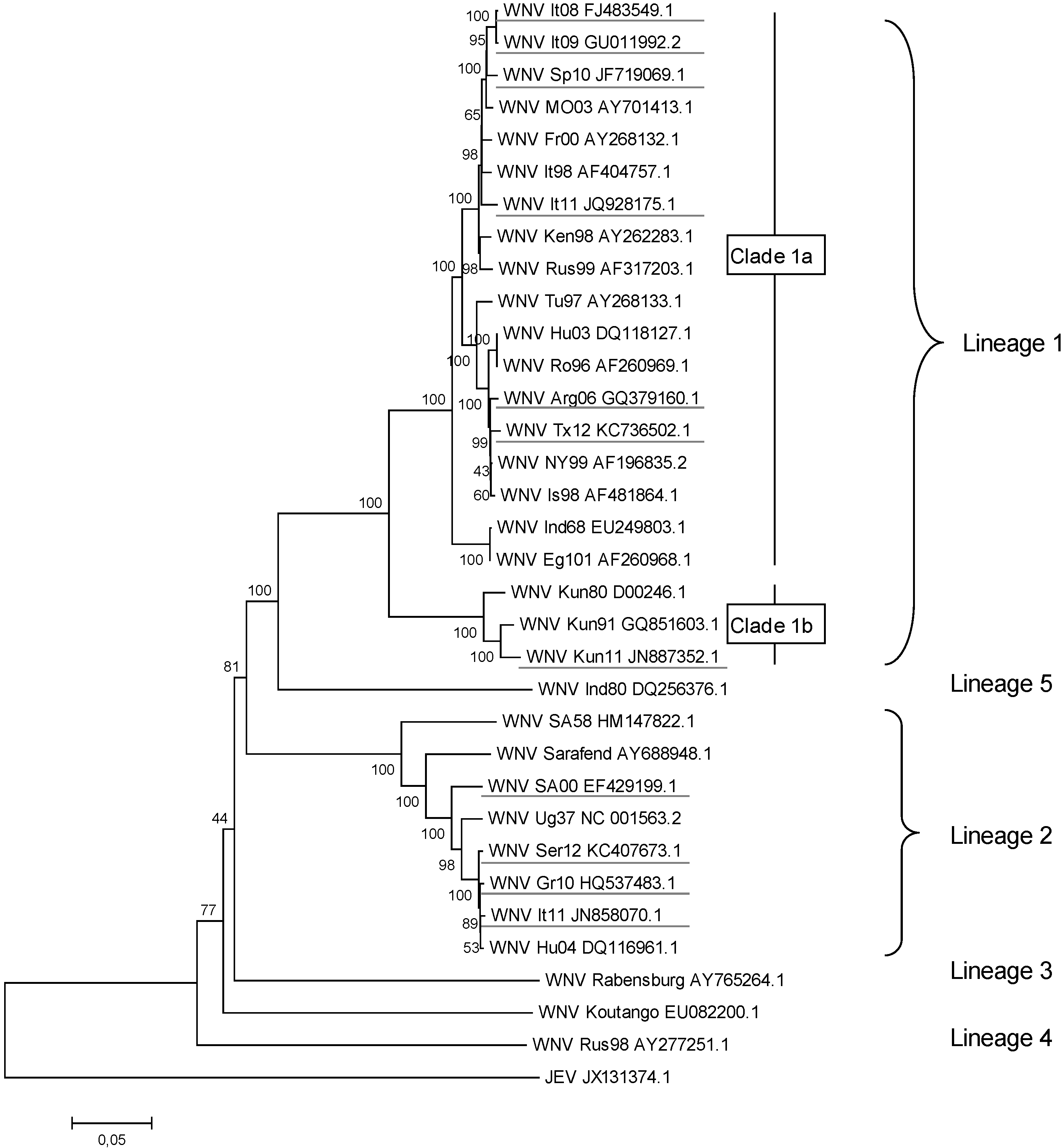
2. WNV Genetic Diversity
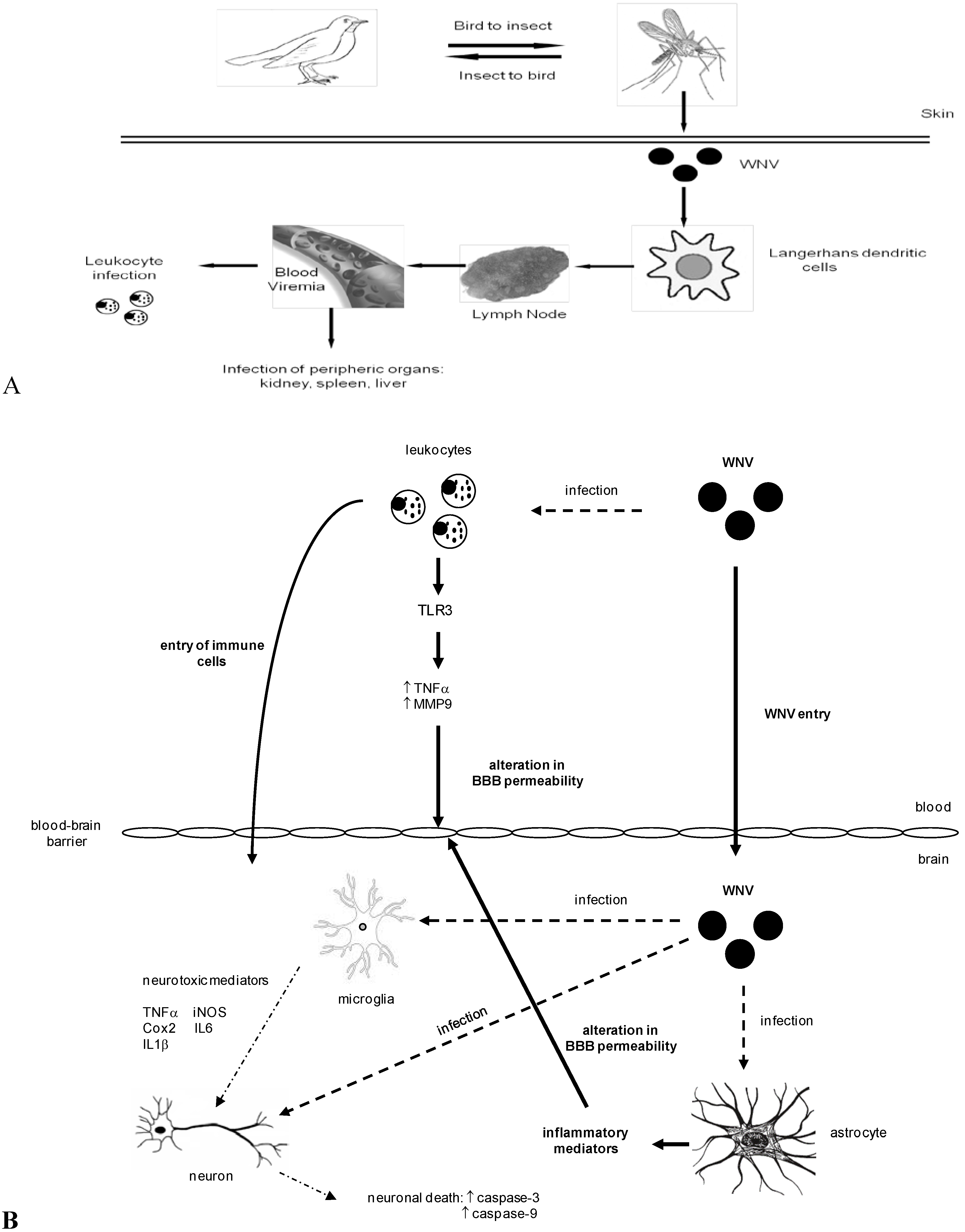
3. WNV Dissemination and Entry into the Central Nervous System
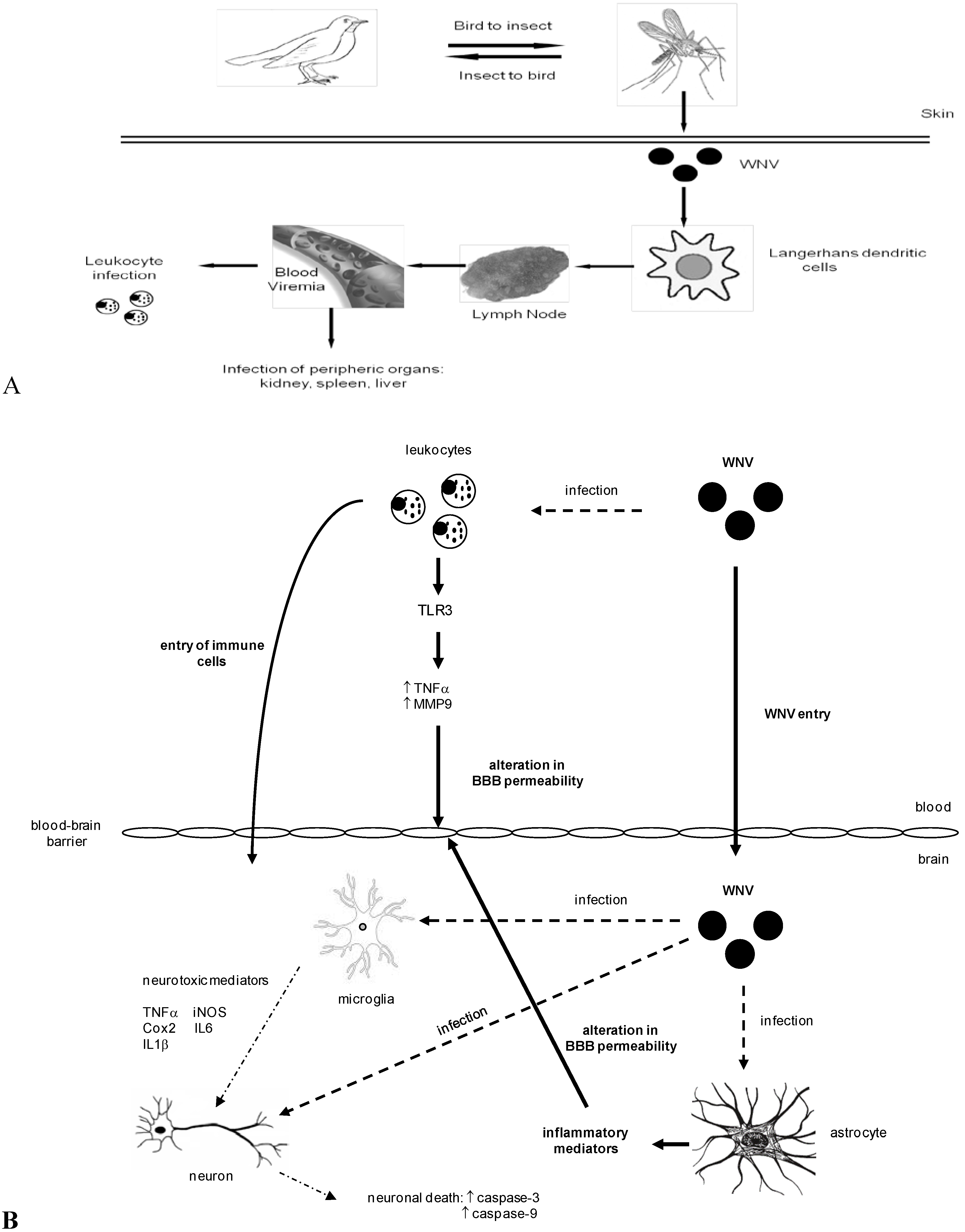
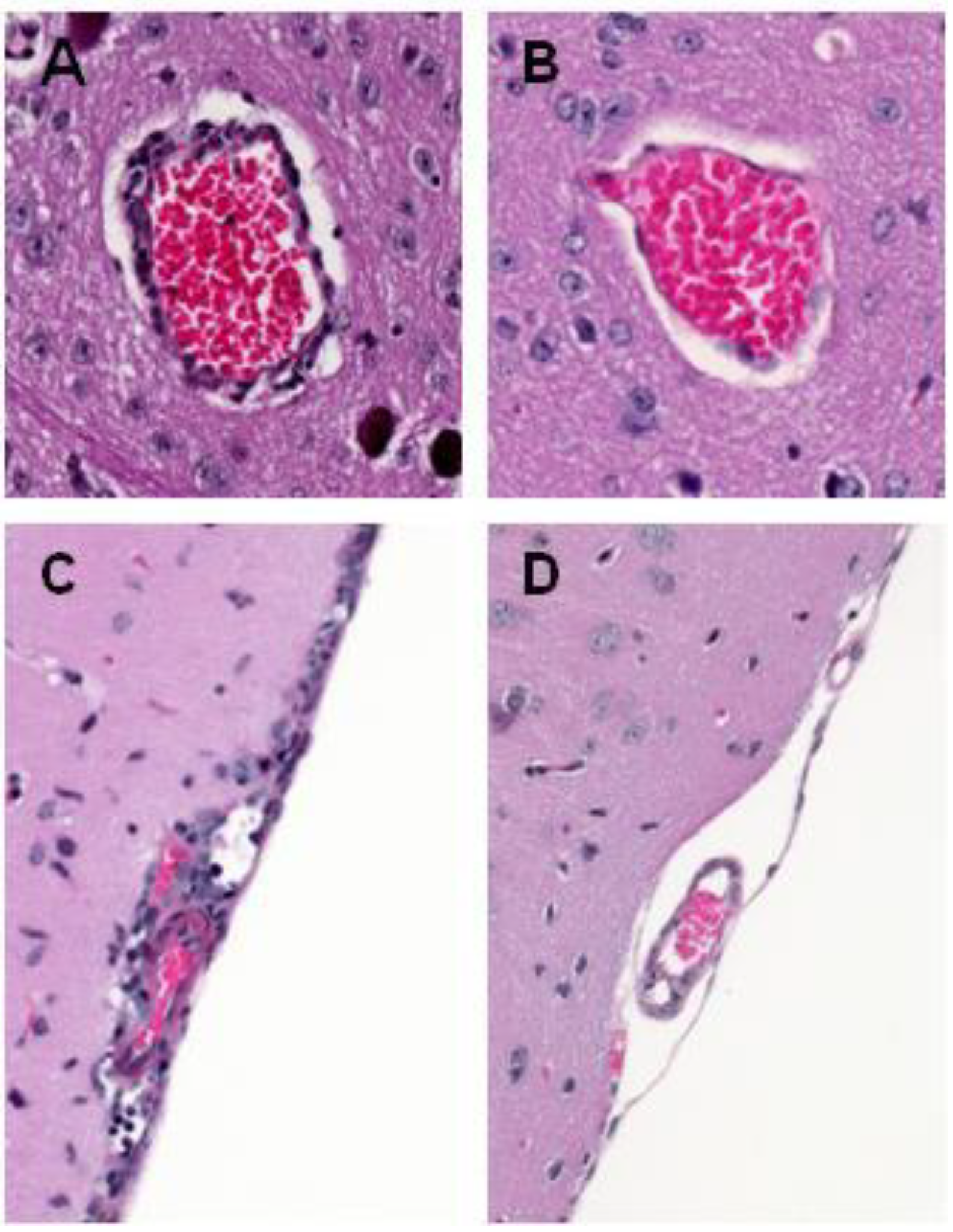
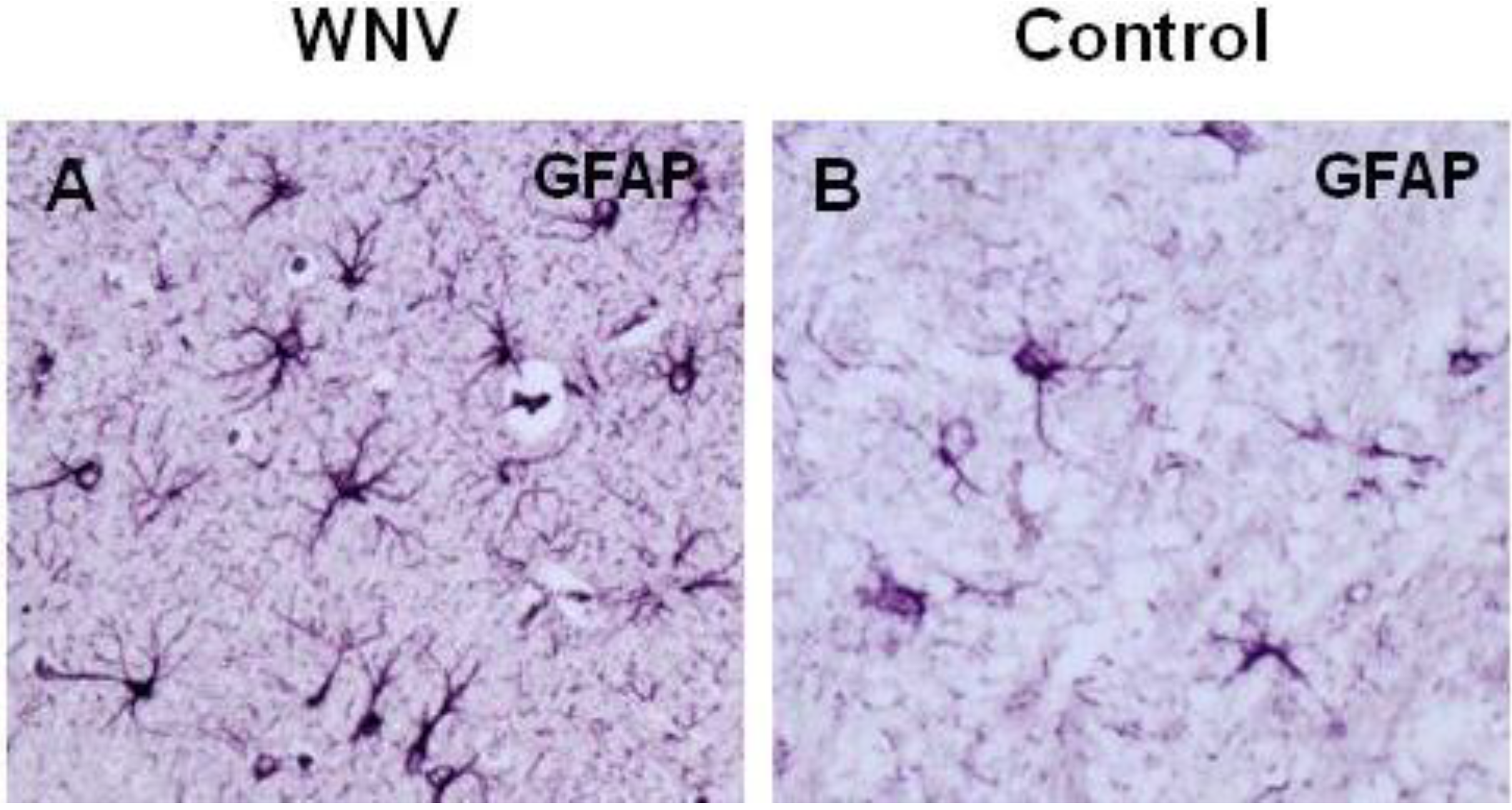
4. WNV Neuropathogenesis
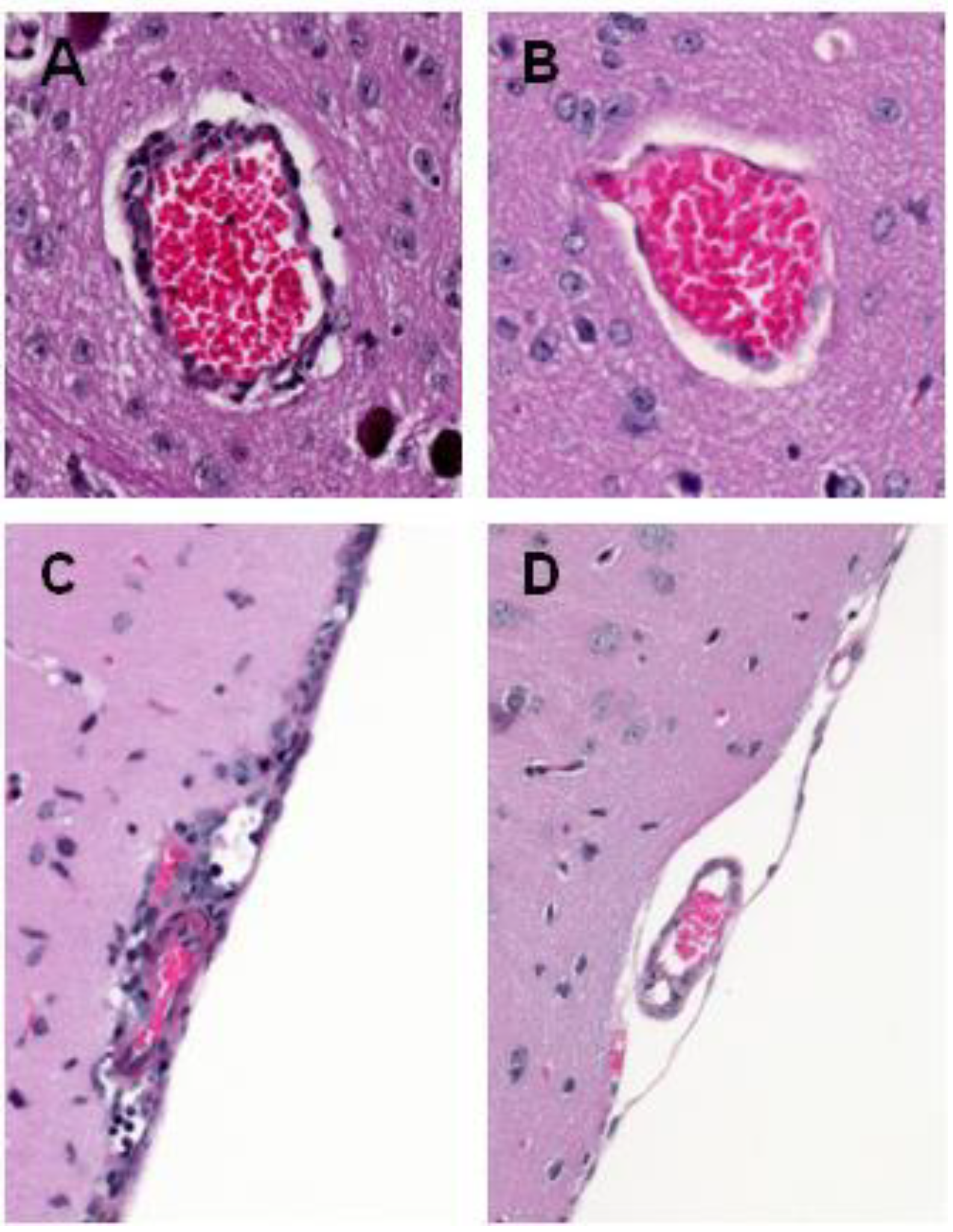
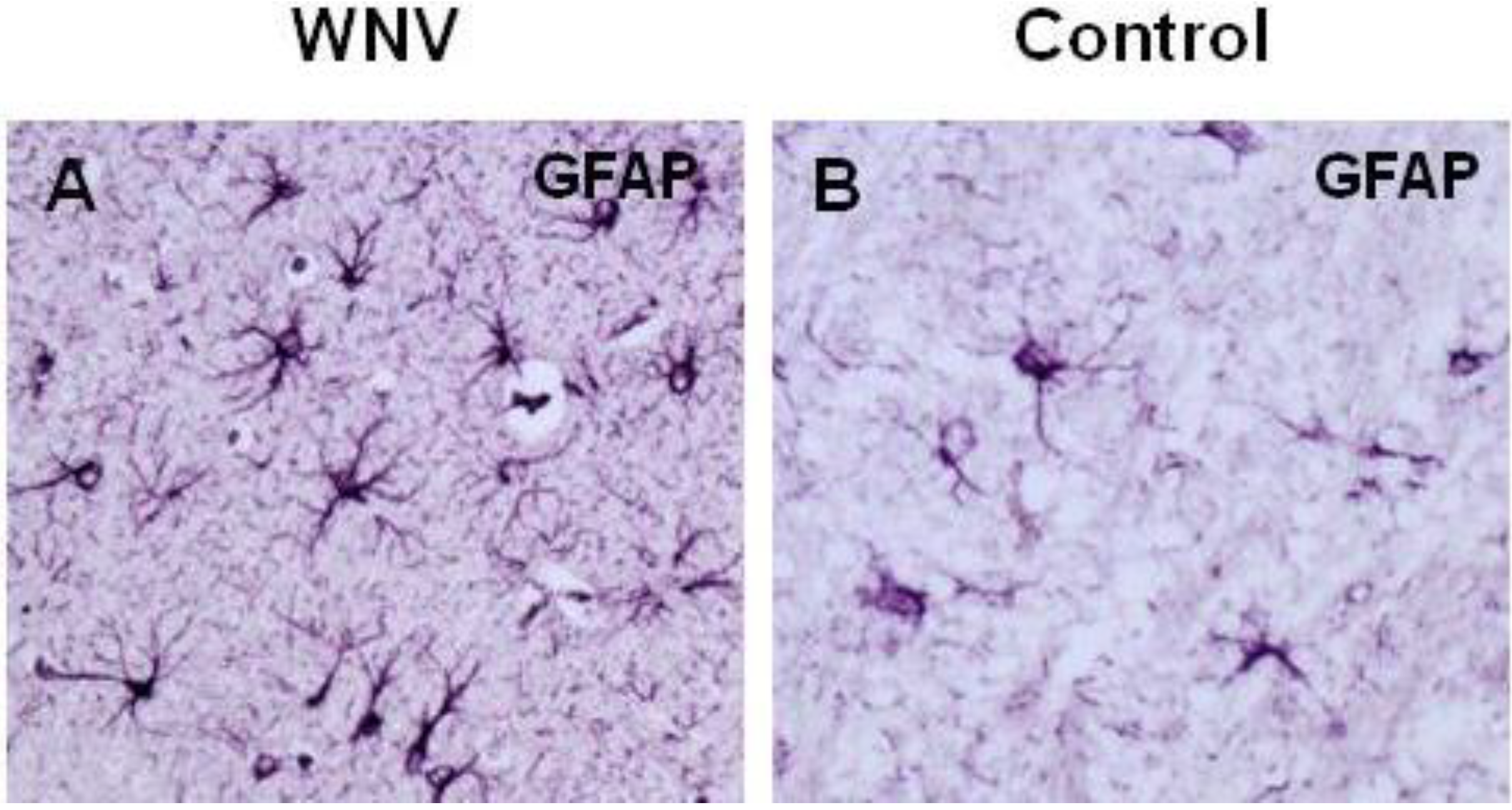
5. WNV Clearance from the Brain
6. Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Kramer, L.D.; Li, J.; Shi, P.Y. West Nile virus. Lancet Neurol. 2007, 6, 171–181. [Google Scholar] [CrossRef]
- Smithburn, K.C.; Hughes, T.P.; Burke, A.W.; Paul, J.H. A neurotropic virus isolated from the blood of a native of Uganda. Am. J. Trop. Med. Hyg. 1940, 20, 471–492. [Google Scholar]
- Hayes, E.B.; Komar, N.; Nasci, R.S.; Montgomery, S.P.; O’Leary, D.R.; Campbell, G.L. Epidemiology and transmission dynamics of West Nile virus disease. Emerg. Infect. Dis. 2005, 11, 1167–1173. [Google Scholar]
- Hayes, E.B.; Gubler, D.J. West Nile virus: Epidemiology and clinical features of an emerging epidemic in the United States. Ann. Rev. Med. 2006, 57, 181–194. [Google Scholar] [CrossRef]
- Leis, A.A.; Fratkin, J.; Stokic, D.S.; Harrington, T.; Webb, R.M.; Slavinski, S.A. West Nile poliomyelitis. Lancet Infect. Dis. 2003, 3, 9–10. [Google Scholar] [CrossRef]
- Petersen, L.R.; Marfin, A.A. West Nile virus: A primer for the clinician. Ann. Intern. Med. 2002, 137, 173–179. [Google Scholar] [CrossRef]
- Cao, N.J.; Ranganathan, C.; Kupsky, W.J.; Li, J. Recovery and prognosticators of paralysis in West Nile virus infection. J. Neurol. Sci. 2005, 236, 73–80. [Google Scholar]
- Sejvar, J.J.; Haddad, M.B.; Tierney, B.C.; Campbell, G.L.; Marfin, A.A.; van Gerpen, J.A.; Fleischauer, A.; Leis, A.A.; Stokic, D.S.; Petersen, L.R. Neurologic manifestations and outcome of West Nile virus infection. J. Am. Med. Assoc. 2003, 290, 511–515. [Google Scholar] [CrossRef]
- Klee, A.L.; Maidin, B.; Edwin, B.; Poshni, I.; Mostashari, F.; Fine, A.; Layton, M.; Nash, D. Long-term prognosis for clinical West Nile virus infection. Emerg. Infect. Dis. 2004, 10, 1405–1411. [Google Scholar] [CrossRef]
- Sejvar, J.J.; Bode, A.V.; Marfin, A.A.; Campbell, G.L.; Pape, J.; Biggerstaff, B.J.; Petersen, L.R. West Nile Virus-associated flaccid paralysis outcome. Emerg. Infect. Dis. 2006, 12, 514–516. [Google Scholar]
- Sejvar, J.J. The long-term outcomes of human West Nile virus infection. Clin. Infect. Dis. 2007, 44, 1617–1624. [Google Scholar] [CrossRef]
- De Filette, M.; Ulbert, S.; Diamond, M.; Sanders, N.N. Recent progress in West Nile virus diagnosis and vaccination. Vet. Res. 2012, 43. [Google Scholar] [CrossRef] [Green Version]
- Zeller, H.G.; Schuffenecker, I. West Nile virus: An overview of its spread in Europe and the Mediterranean basin in contrast to its spread in the Americas. Eur. J. Clin. Microbiol. Infect. Dis. 2004, 23, 147–156. [Google Scholar] [CrossRef]
- Calistri, P.; Giovannini, A.; Hubalek, Z.; Ionescu, A.; Monaco, F.; Savini, G.; Lelli, R. Epidemiology of west nile in europe and in the mediterranean basin. Open Virol. J. 2010, 4, 29–37. [Google Scholar]
- Bin, H.; Grossman, Z.; Pokamunski, S.; Malkinson, M.; Weiss, L.; Duvdevani, P.; Banet, C.; Weisman, Y.; Annis, E.; Gandaku, D.; et al. West Nile fever in Israel 1999–2000: From geese to humans. Ann. NY Acad. Sci. 2001, 951, 127–142. [Google Scholar]
- Kopel, E.; Amitai, Z.; Bin, H.; Shulman, L.M.; Mendelson, E.; Sheffer, R. Surveillance of West Nile virus disease, Tel Aviv district, Israel, 2005 to 2010. Available online: http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=19894 (accessed at 19 August 2013).
- Bakonyi, T.; Ferenczi, E.; Erdelyi, K.; Kutasi, O.; Csorgo, T.; Seidel, B.; Weissenbock, H.; Brugger, K.; Ban, E.; Nowotny, N. Explosive spread of a neuroinvasive lineage 2 West Nile virus in Central Europe, 2008/2009. Vet. Microbiol. 2013, 165, 61–70. [Google Scholar] [CrossRef]
- Pradier, S.; Lecollinet, S.; Leblond, A. West Nile virus epidemiology and factors triggering change in its distribution in Europe. Rev. Sci. Tech. 2012, 31, 829–844. [Google Scholar]
- Sirbu, A.; Ceianu, C.S.; Panculescu-Gatej, R.I.; Vazquez, A.; Tenorio, A.; Rebreanu, R.; Niedrig, M.; Nicolescu, G.; Pistol, A. Outbreak of West Nile virus infection in humans, Romania, July to October 2010. Available online: http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=19762 (accessed at 14 August 2013).
- Danis, K.; Papa, A.; Theocharopoulos, G.; Dougas, G.; Athanasiou, M.; Detsis, M.; Baka, A.; Lytras, T.; Mellou, K.; Bonovas, S.; et al. Outbreak of West Nile virus infection in Greece, 2010. Emerg. Infect. Dis. 2011, 17, 1868–1872. [Google Scholar]
- Platonov, A.E.; Karan, L.S.; Shopenskaia, T.A.; Fedorova, M.V.; Koliasnikova, N.M.; Rusakova, N.M.; Shishkina, L.V.; Arshba, T.E.; Zhuravlev, V.I.; Govorukhina, M.V.; et al. Genotyping of West Nile fever virus strains circulating in southern Russia as an epidemiological investigation method: Principles and results. Zh. Mikrobiol. Epidemiol. immunobiol. 2011, 2011, 29–37. [Google Scholar]
- Frost, M.J.; Zhang, J.; Edmonds, J.H.; Prow, N.A.; Gu, X.; Davis, R.; Hornitzky, C.; Arzey, K.E.; Finlaison, D.; Hick, P.; et al. Characterization of virulent West Nile virus Kunjin strain, Australia, 2011. Emerg. Infect. Dis. 2012, 18, 792–800. [Google Scholar]
- Venter, M.; Human, S.; Zaayman, D.; Gerdes, G.H.; Williams, J.; Steyl, J.; Leman, P.A.; Paweska, J.T.; Setzkorn, H.; Rous, G.; et al. Lineage 2 west nile virus as cause of fatal neurologic disease in horses, South Africa. Emerg. Infect. Dis. 2009, 15, 877–884. [Google Scholar] [CrossRef]
- Venter, M.; Swanepoel, R. West Nile virus lineage 2 as a cause of zoonotic neurological disease in humans and horses in southern Africa. Vector Borne Zoonotic Dis. 2010, 10, 659–664. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Ciccozzi, M.; Peletto, S.; Cella, E.; Giovanetti, M.; Lai, A.; Gabanelli, E.; Acutis, P.L.; Modesto, P.; Rezza, G.; Platonov, A.E.; et al. Epidemiological history and phylogeography of West Nile virus lineage 2. Infect. Genet. Evol. 2013, 17, 46–50. [Google Scholar]
- Blitvich, B.J. Transmission dynamics and changing epidemiology of West Nile virus. Anim. HealthRes. Rev. 2008, 9, 71–86. [Google Scholar] [CrossRef]
- LaDeau, S.L.; Kilpatrick, A.M.; Marra, P.P. West Nile virus emergence and large-scale declines of North American bird populations. Nature 2007, 447, 710–713. [Google Scholar] [CrossRef]
- Chung, W.M.; Buseman, C.M.; Joyner, S.N.; Hughes, S.M.; Fomby, T.B.; Luby, J.P.; Haley, R.W. The 2012 West Nile encephalitis epidemic in Dallas, Texas. J. Am. Med. Assoc. 2013, 310, 297–307. [Google Scholar]
- Duggal, N.K.; D’Anton, M.; Xiang, J.; Seiferth, R.; Day, J.; Nasci, R.; Brault, A.C. Sequence analyses of 2012 west nile virus isolates from Texas fail to associate viral genetic factors with outbreak magnitude. Am. J. Trop. Medi. Hyg. 2013, 89, 205–210. [Google Scholar] [CrossRef]
- Cho, H.; Diamond, M.S. Immune responses to West Nile virus infection in the central nervous system. Viruses 2012, 4, 3812–3830. [Google Scholar]
- Lim, S.M.; Koraka, P.; Osterhaus, A.D.; Martina, B.E. West Nile virus: Immunity and pathogenesis. Viruses 2011, 3, 811–828. [Google Scholar]
- Suthar, M.S.; Diamond, M.S.; Gale, M., Jr. West Nile virus infection and immunity. Nat. Rev. Microbiol. 2013, 11, 115–128. [Google Scholar] [CrossRef]
- Samuel, M.A.; Diamond, M.S. Pathogenesis of West Nile Virus infection: A balance between virulence, innate and adaptive immunity, and viral evasion. J. Virol. 2006, 80, 9349–9360. [Google Scholar] [CrossRef]
- Diamond, M.S.; Gale, M., Jr. Cell-intrinsic innate immune control of West Nile virus infection. Trends. Immunol. 2012, 33, 522–530. [Google Scholar] [CrossRef]
- Ulbert, S. West Nile virus: The complex biology of an emerging pathogen. Intervirology 2011, 54, 171–184. [Google Scholar] [CrossRef]
- Guarner, J.; Shieh, W.J.; Hunter, S.; Paddock, C.D.; Morken, T.; Campbell, G.L.; Marfin, A.A.; Zaki, S.R. Clinicopathologic study and laboratory diagnosis of 23 cases with West Nile virus encephalomyelitis. Hum. Pathol. 2004, 35, 983–990. [Google Scholar] [CrossRef]
- Kleinschmidt-DeMasters, B.K.; Marder, B.A.; Levi, M.E.; Laird, S.P.; McNutt, J.T.; Escott, E.J.; Everson, G.T.; Tyler, K.L. Naturally acquired West Nile virus encephalomyelitis in transplant recipients: Clinical, laboratory, diagnostic, and neuropathological features. Arch. Neurol. 2004, 61, 1210–1220. [Google Scholar] [CrossRef]
- Samuel, M.A.; Morrey, J.D.; Diamond, M.S. Caspase 3-dependent cell death of neurons contributes to the pathogenesis of West Nile virus encephalitis. J. Virol. 2007, 81, 2614–2623. [Google Scholar] [CrossRef]
- Bondre, V.P.; Jadi, R.S.; Mishra, A.C.; Yergolkar, P.N.; Arankalle, V.A. West Nile virus isolates from India: Evidence for a distinct genetic lineage. J. Gen. Virol. 2007, 88, 875–884. [Google Scholar] [CrossRef]
- Erdelyi, K.; Ursu, K.; Ferenczi, E.; Szeredi, L.; Ratz, F.; Skare, J.; Bakonyi, T. Clinical and pathologic features of lineage 2 West Nile virus infections in birds of prey in Hungary. Vector Borne Zoonotic Dis. 2007, 7, 181–188. [Google Scholar]
- Kutasi, O.; Bakonyi, T.; Lecollinet, S.; Biksi, I.; Ferenczi, E.; Bahuon, C.; Sardi, S.; Zientara, S.; Szenci, O. Equine encephalomyelitis outbreak caused by a genetic lineage 2 West Nile virus in Hungary. J. Vet. Intern. Med. 2011, 25, 586–591. [Google Scholar] [CrossRef] [Green Version]
- Magurano, F.; Remoli, M.E.; Baggieri, M.; Fortuna, C.; Marchi, A.; Fiorentini, C.; Bucci, P.; Benedetti, E.; Ciufolini, M.G.; Rizzo, C.; et al. Circulation of West Nile virus lineage 1 and 2 during an outbreak in Italy. Clin. Microbiol. Infect. 2012, 18, E545–E547. [Google Scholar]
- Bakonyi, T.; Hubalek, Z.; Rudolf, I.; Nowotny, N. Novel flavivirus or new lineage of West Nile virus, central Europe. Emerg. Infect. Dis. 2005, 11, 225–231. [Google Scholar] [CrossRef]
- Lvov, D.K.; Butenko, A.M.; Gromashevsky, V.L.; Kovtunov, A.I.; Prilipov, A.G.; Kinney, R.; Aristova, V.A.; Dzharkenov, A.F.; Samokhvalov, E.I.; Savage, H.M.; et al. West Nile virus and other zoonotic viruses in Russia: Examples of emerging-reemerging situations. Arch. Virol. Suppl. 2004, 18, 85–96. [Google Scholar]
- Scherret, J.H.; Poidinger, M.; Mackenzie, J.S.; Broom, A.K.; Deubel, V.; Lipkin, W.I.; Briese, T.; Gould, E.A.; Hall, R.A. The relationships between West Nile and Kunjin viruses. Emerg. Infect. Dis. 2001, 7, 697–705. [Google Scholar]
- Vazquez, A.; Sanchez-Seco, M.P.; Ruiz, S.; Molero, F.; Hernandez, L.; Moreno, J.; Magallanes, A.; Tejedor, C.G.; Tenorio, A. Putative new lineage of west nile virus, Spain. Emerg. Infect. Dis. 2010, 16, 549–552. [Google Scholar] [CrossRef]
- Traore-Lamizana, M.; Fontenille, D.; Diallo, M.; Ba, Y.; Zeller, H.G.; Mondo, M.; Adam, F.; Thonon, J.; Maiga, A. Arbovirus surveillance from 1990 to 1995 in the Barkedji area (Ferlo) of Senegal, a possible natural focus of Rift Valley fever virus. J. Med. Entomol. 2001, 38, 480–492. [Google Scholar] [CrossRef]
- May, F.J.; Davis, C.T.; Tesh, R.B.; Barrett, A.D. Phylogeography of West Nile virus: From the cradle of evolution in Africa to Eurasia, Australia, and the Americas. J. Virol. 2011, 85, 2964–2974. [Google Scholar] [CrossRef]
- Monini, M.; Falcone, E.; Busani, L.; Romi, R.; Ruggeri, F.M. West nile virus: Characteristics of an african virus adapting to the third millennium world. Open Virol. J. 2010, 4, 42–51. [Google Scholar]
- Rappole, J.H.; Compton, B.W.; Leimgruber, P.; Robertson, J.; King, D.I.; Renner, S.C. Modeling movement of West Nile virus in the Western hemisphere. Vector Borne Zoonotic Dis. 2006, 6, 128–139. [Google Scholar] [CrossRef]
- Rappole, J.H.; Derrickson, S.R.; Hubalek, Z. Migratory birds and spread of West Nile virus in the Western Hemisphere. Emerg. Infect. Dis. 2000, 6, 319–328. [Google Scholar] [CrossRef]
- Zehender, G.; Ebranati, E.; Bernini, F.; Lo Presti, A.; Rezza, G.; Delogu, M.; Galli, M.; Ciccozzi, M. Phylogeography and epidemiological history of West Nile virus genotype 1a in Europe and the Mediterranean basin. Infect. Genet. Evol. 2011, 11, 646–653. [Google Scholar] [CrossRef]
- Besselaar, T.G.; Blackburn, N.K. Antigenic analysis of West Nile virus strains using monoclonal antibodies. Arch. Virol. 1988, 99, 75–88. [Google Scholar] [CrossRef]
- Morvan, J.; Besselaar, T.; Fontenille, D.; Coulanges, P. Antigenic variations in West Nile virus strains isolated in Madagascar since 1978. Res. Virol. 1990, 141, 667–676. [Google Scholar] [CrossRef]
- Beasley, D.W.; Barrett, A.D. Identification of neutralizing epitopes within structural domain III of the West Nile virus envelope protein. J. Virol. 2002, 76, 13097–13100. [Google Scholar] [CrossRef]
- Beasley, D.W.; Li, L.; Suderman, M.T.; Barrett, A.D. Mouse neuroinvasive phenotype of West Nile virus strains varies depending upon virus genotype. Virology 2002, 296, 17–23. [Google Scholar] [CrossRef]
- Brault, A.C.; Langevin, S.A.; Bowen, R.A.; Panella, N.A.; Biggerstaff, B.J.; Miller, B.R.; Komar, N. Differential virulence of West Nile strains for American crows. Emerg. Infect. Dis. 2004, 10, 2161–2168. [Google Scholar] [CrossRef]
- Langevin, S.A.; Brault, A.C.; Panella, N.A.; Bowen, R.A.; Komar, N. Variation in virulence of West Nile virus strains for house sparrows (Passer domesticus). Am. J. Trop. Med. Hyg. 2005, 72, 99–102. [Google Scholar]
- Adams, S.C.; Broom, A.K.; Sammels, L.M.; Hartnett, A.C.; Howard, M.J.; Coelen, R.J.; Mackenzie, J.S.; Hall, R.A. Glycosylation and antigenic variation among Kunjin virus isolates. Virology 1995, 206, 49–56. [Google Scholar] [CrossRef]
- Berthet, F.X.; Zeller, H.G.; Drouet, M.T.; Rauzier, J.; Digoutte, J.P.; Deubel, V. Extensive nucleotide changes and deletions within the envelope glycoprotein gene of Euro-African West Nile viruses. J. Gen. Virol 1997, 78, 2293–2297. [Google Scholar]
- Chambers, T.J.; Halevy, M.; Nestorowicz, A.; Rice, C.M.; Lustig, S. West Nile virus envelope proteins: nucleotide sequence analysis of strains differing in mouse neuroinvasiveness. J. Gen. Virol. 1998, 79, 2375–2380. [Google Scholar]
- Beasley, D.W.; Whiteman, M.C.; Zhang, S.; Huang, C.Y.; Schneider, B.S.; Smith, D.R.; Gromowski, G.D.; Higgs, S.; Kinney, R.M.; Barrett, A.D. Envelope protein glycosylation status influences mouse neuroinvasion phenotype of genetic lineage 1 West Nile virus strains. J. Virol. 2005, 79, 8339–8347. [Google Scholar] [CrossRef]
- Murata, R.; Eshita, Y.; Maeda, A.; Maeda, J.; Akita, S.; Tanaka, T.; Yoshii, K.; Kariwa, H.; Umemura, T.; Takashima, I. Glycosylation of the West Nile Virus envelope protein increases in vivo and in vitro viral multiplication in birds. Am. J. Trop. Med. Hyg. 2010, 82, 696–704. [Google Scholar] [CrossRef]
- Langevin, S.A.; Bowen, R.A.; Ramey, W.N.; Sanders, T.A.; Maharaj, P.D.; Fang, Y.; Cornelius, J.; Barker, C.M.; Reisen, W.K.; Beasley, D.W.; et al. Envelope and pre-membrane protein structural amino acid mutations mediate diminished avian growth and virulence of a Mexican West Nile virus isolate. J. Gen. Virol. 2011, 92, 2810–2820. [Google Scholar] [CrossRef]
- Moudy, R.M.; Zhang, B.; Shi, P.Y.; Kramer, L.D. West Nile virus envelope protein glycosylation is required for efficient viral transmission by Culex vectors. Virology 2009, 387, 222–228. [Google Scholar] [CrossRef]
- Brault, A.C.; Huang, C.Y.; Langevin, S.A.; Kinney, R.M.; Bowen, R.A.; Ramey, W.N.; Panella, N.A.; Holmes, E.C.; Powers, A.M.; Miller, B.R. A single positively selected West Nile viral mutation confers increased virogenesis in American crows. Nat. Genet. 2007, 39, 1162–1166. [Google Scholar]
- Audsley, M.; Edmonds, J.; Liu, W.; Mokhonov, V.; Mokhonova, E.; Melian, E.B.; Prow, N.; Hall, R.A.; Khromykh, A.A. Virulence determinants between New York 99 and Kunjin strains of West Nile virus. Virology 2011, 414, 63–73. [Google Scholar] [CrossRef]
- Liu, W.J.; Wang, X.J.; Clark, D.C.; Lobigs, M.; Hall, R.A.; Khromykh, A.A. A single amino acid substitution in the West Nile virus nonstructural protein NS2A disables its ability to inhibit alpha/beta interferon induction and attenuates virus virulence in mice. J. Virol. 2006, 80, 2396–2404. [Google Scholar]
- Botha, E.M.; Markotter, W.; Wolfaardt, M.; Paweska, J.T.; Swanepoel, R.; Palacios, G.; Nel, L.H.; Venter, M. Genetic determinants of virulence in pathogenic lineage 2 West Nile virus strains. Emerg. Infect. Dis. 2008, 14, 222–230. [Google Scholar]
- Borisevich, V.; Seregin, A.; Nistler, R.; Mutabazi, D.; Yamshchikov, V. Biological properties of chimeric West Nile viruses. Virology 2006, 349, 371–381. [Google Scholar] [CrossRef]
- Pogodina, V.V.; Frolova, M.P.; Malenko, G.V.; Fokina, G.I.; Koreshkova, G.V.; Kiseleva, L.L.; Bochkova, N.G.; Ralph, N.M. Study on West Nile virus persistence in monkeys. Arch. Virol. 1983, 75, 71–86. [Google Scholar] [CrossRef]
- Xiao, S.Y.; Guzman, H.; Zhang, H.; Travassos da Rosa, A.P.; Tesh, R.B. West Nile virus infection in the golden hamster (Mesocricetus auratus): A model for West Nile encephalitis. Emerg. Infect. Dis. 2001, 7, 714–721. [Google Scholar]
- Bai, F.; Kong, K.F.; Dai, J.; Qian, F.; Zhang, L.; Brown, C.R.; Fikrig, E.; Montgomery, R.R. A paradoxical role for neutrophils in the pathogenesis of West Nile virus. J. Infect. Dis. 2010, 202, 1804–1812. [Google Scholar]
- Lim, P.Y.; Behr, M.J.; Chadwick, C.M.; Shi, P.Y.; Bernard, K.A. Keratinocytes are cell targets of West Nile virus in vivo. J. Virol. 2011, 85, 5197–5201. [Google Scholar]
- Welte, T.; Reagan, K.; Fang, H.; Machain-Williams, C.; Zheng, X.; Mendell, N.; Chang, G.J.; Wu, P.; Blair, C.D.; Wang, T. Toll-like receptor 7-induced immune response to cutaneous West Nile virus infection. J. Gen. Virol. 2009, 90, 2660–2668. [Google Scholar] [CrossRef]
- Byrne, S.N.; Halliday, G.M.; Johnston, L.J.; King, N.J. Interleukin-1beta but not tumor necrosis factor is involved in West Nile virus-induced Langerhans cell migration from the skin in C57BL/6 mice. J. Invest. Dermatol. 2001, 117, 702–709. [Google Scholar] [CrossRef]
- Johnston, L.J.; Halliday, G.M.; King, N.J. Langerhans cells migrate to local lymph nodes following cutaneous infection with an arbovirus. J. Invest. Dermatol. 2000, 114, 560–568. [Google Scholar] [CrossRef]
- McCandless, E.E.; Zhang, B.; Diamond, M.S.; Klein, R.S. CXCR4 antagonism increases T cell trafficking in the central nervous system and improves survival from West Nile virus encephalitis. Proc. Natl. Acad. Sci. USA 2008, 105, 11270–11275. [Google Scholar] [CrossRef]
- Ramos, H.J.; Lanteri, M.C.; Blahnik, G.; Negash, A.; Suthar, M.S.; Brassil, M.M.; Sodhi, K.; Treuting, P.M.; Busch, M.P.; Norris, P.J.; et al. IL-1beta signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog. 2012, 8, e1003039. [Google Scholar] [CrossRef]
- Wang, S.; Welte, T.; McGargill, M.; Town, T.; Thompson, J.; Anderson, J.F.; Flavell, R.A.; Fikrig, E.; Hedrick, S.M.; Wang, T. Drak2 contributes to West Nile virus entry into the brain and lethal encephalitis. J. Immunol. 2008, 181, 2084–2091. [Google Scholar]
- Samuel, M.A.; Diamond, M.S. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J. Virol. 2005, 79, 13350–13361. [Google Scholar] [CrossRef]
- Anderson, J.F.; Rahal, J.J. Efficacy of interferon alpha-2b and ribavirin against West Nile virus in vitro. Emerg. Infect. Dis. 2002, 8, 107–108. [Google Scholar] [CrossRef]
- Morrey, J.D.; Day, C.W.; Julander, J.G.; Blatt, L.M.; Smee, D.F.; Sidwell, R.W. Effect of interferon-alpha and interferon-inducers on West Nile virus in mouse and hamster animal models. Antiviral Chem. Chemother. 2004, 15, 101–109. [Google Scholar]
- Keller, B.C.; Fredericksen, B.L.; Samuel, M.A.; Mock, R.E.; Mason, P.W.; Diamond, M.S.; Gale, M., Jr. Resistance to alpha/beta interferon is a determinant of West Nile virus replication fitness and virulence. J. Virol. 2006, 80, 9424–9434. [Google Scholar] [CrossRef]
- Liu, W.J.; Chen, H.B.; Wang, X.J.; Huang, H.; Khromykh, A.A. Analysis of adaptive mutations in Kunjin virus replicon RNA reveals a novel role for the flavivirus nonstructural protein NS2A in inhibition of beta interferon promoter-driven transcription. J. Virol. 2004, 78, 12225–12235. [Google Scholar] [CrossRef]
- Munoz-Jordan, J.L.; Laurent-Rolle, M.; Ashour, J.; Martinez-Sobrido, L.; Ashok, M.; Lipkin, W.I.; Garcia-Sastre, A. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J. Virol. 2005, 79, 8004–8013. [Google Scholar] [CrossRef]
- Evans, J.D.; Seeger, C. Differential effects of mutations in NS4B on West Nile virus replication and inhibition of interferon signaling. J. Virol. 2007, 81, 11809–11816. [Google Scholar] [CrossRef]
- Laurent-Rolle, M.; Boer, E.F.; Lubick, K.J.; Wolfinbarger, J.B.; Carmody, A.B.; Rockx, B.; Liu, W.; Ashour, J.; Shupert, W.L.; Holbrook, M.R.; et al. The NS5 protein of the virulent West Nile virus NY99 strain is a potent antagonist of type I interferon-mediated JAK-STAT signaling. J. Virol. 2010, 84, 3503–3515. [Google Scholar] [CrossRef] [Green Version]
- Daffis, S.; Lazear, H.M.; Liu, W.J.; Audsley, M.; Engle, M.; Khromykh, A.A.; Diamond, M.S. The naturally attenuated Kunjin strain of West Nile virus shows enhanced sensitivity to the host type I interferon response. J. Virol. 2011, 85, 5664–5668. [Google Scholar] [CrossRef]
- Diamond, M.S.; Shrestha, B.; Marri, A.; Mahan, D.; Engle, M. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J. Virol. 2003, 77, 2578–2586. [Google Scholar] [CrossRef]
- Vargin, V.V.; Semenov, B.F. Changes of natural killer cell activity in different mouse lines by acute and asymptomatic flavivirus infections. Acta Virol. 1986, 30, 303–308. [Google Scholar]
- Wang, T.; Scully, E.; Yin, Z.; Kim, J.H.; Wang, S.; Yan, J.; Mamula, M.; Anderson, J.F.; Craft, J.; Fikrig, E. IFN-gamma-producing gamma delta T cells help control murine West Nile virus infection. J. Immunol. 2003, 171, 2524–2531. [Google Scholar]
- Arjona, A.; Wang, P.; Montgomery, R.R.; Fikrig, E. Innate immune control of West Nile virus infection. Cell Microbiol. 2011, 13, 1648–1658. [Google Scholar] [CrossRef]
- Shrestha, B.; Wang, T.; Samuel, M.A.; Whitby, K.; Craft, J.; Fikrig, E.; Diamond, M.S. Gamma interferon plays a crucial early antiviral role in protection against West Nile virus infection. J. Virol. 2006, 80, 5338–5348. [Google Scholar]
- Shrestha, B.; Samuel, M.A.; Diamond, M.S. CD8+ T cells require perforin to clear West Nile virus from infected neurons. J. Virol. 2006, 80, 119–129. [Google Scholar] [CrossRef]
- Fang, H.; Welte, T.; Zheng, X.; Chang, G.J.; Holbrook, M.R.; Soong, L.; Wang, T. Gammadelta T cells promote the maturation of dendritic cells during West Nile virus infection. FEMS Immunol. Med. Microbiol. 2010, 59, 71–80. [Google Scholar]
- Kulkarni, A.B.; Mullbacher, A.; Blanden, R.V. Functional analysis of macrophages, B cells and splenic dendritic cells as antigen-presenting cells in West Nile virus-specific murine T lymphocyte proliferation. Immunol. Cell Biol. 1991, 69, 71–80. [Google Scholar] [CrossRef]
- Lin, Y.L.; Huang, Y.L.; Ma, S.H.; Yeh, C.T.; Chiou, S.Y.; Chen, L.K.; Liao, C.L. Inhibition of Japanese encephalitis virus infection by nitric oxide: antiviral effect of nitric oxide on RNA virus replication. J. Virol. 1997, 71, 5227–5235. [Google Scholar]
- Saxena, S.K.; Singh, A.; Mathur, A. Antiviral effect of nitric oxide during Japanese encephalitis virus infection. Int. J. Exp. Pathol. 2000, 81, 165–172. [Google Scholar] [CrossRef]
- Ben-Nathan, D.; Huitinga, I.; Lustig, S.; van Rooijen, N.; Kobiler, D. West Nile virus neuroinvasion and encephalitis induced by macrophage depletion in mice. Arch. Virol. 1996, 141, 459–469. [Google Scholar] [CrossRef]
- Appler, K.K.; Brown, A.N.; Stewart, B.S.; Behr, M.J.; Demarest, V.L.; Wong, S.J.; Bernard, K.A. Persistence of West Nile virus in the central nervous system and periphery of mice. PLoS One 2010, 5, e10649. [Google Scholar]
- Samuel, M.A.; Wang, H.; Siddharthan, V.; Morrey, J.D.; Diamond, M.S. Axonal transport mediates West Nile virus entry into the central nervous system and induces acute flaccid paralysis. Proc. Natl. Acad. Sci. USA 2007, 104, 17140–17145. [Google Scholar]
- Verma, S.; Lo, Y.; Chapagain, M.; Lum, S.; Kumar, M.; Gurjav, U.; Luo, H.; Nakatsuka, A.; Nerurkar, V.R. West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: Transmigration across the in vitro blood-brain barrier. Virology 2009, 385, 425–433. [Google Scholar]
- Pardridge, W.M. Brain metabolism: A perspective from the blood-brain barrier. Physiol. Rev. 1983, 63, 1481–1535. [Google Scholar]
- Muldoon, L.L.; Alvarez, J.I.; Begley, D.J.; Boado, R.J.; Del Zoppo, G.J.; Doolittle, N.D.; Engelhardt, B.; Hallenbeck, J.M.; Lonser, R.R.; Ohlfest, J.R.; et al. Immunologic privilege in the central nervous system and the blood-brain barrier. J. Cereb. blood Flow Metab. 2013, 33, 13–21. [Google Scholar] [CrossRef]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood-brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef]
- Shen, J.; SS, T.T.; Schrieber, L.; King, N.J. Early E-selectin, VCAM-1, ICAM-1, and late major histocompatibility complex antigen induction on human endothelial cells by flavivirus and comodulation of adhesion molecule expression by immune cytokins. J. Virol. 1997, 71, 9323–9332. [Google Scholar]
- King, N.J.; Shrestha, B.; Kesson, A.M. Immune modulation by flaviviruses. Adv. Virus Res. 2003, 60, 121–155. [Google Scholar] [CrossRef]
- Dai, J.; Wang, P.; Bai, F.; Town, T.; Fikrig, E. Icam-1 participates in the entry of west nile virus into the central nervous system. J. Virol. 2008, 82, 4164–4168. [Google Scholar] [CrossRef]
- Drevets, D.A.; Leenen, P.J. Leukocyte-facilitated entry of intracellular pathogens into the central nervous system. Microbes Infect. 2000, 2, 1609–1618. [Google Scholar] [CrossRef]
- Greenwood, J.; Etienne-Manneville, S.; Adamson, P.; Couraud, P.O. Lymphocyte migration into the central nervous system: Implication of ICAM-1 signalling at the blood-brain barrier. Vascul. Pharmacol. 2002, 38, 315–322. [Google Scholar] [CrossRef]
- Hubbard, A.K.; Rothlein, R. Intercellular adhesion molecule-1 (ICAM-1) expression and cell signaling cascades. Free Radic. Biol. Med. 2000, 28, 1379–1386. [Google Scholar] [CrossRef]
- Morrey, J.D.; Olsen, A.L.; Siddharthan, V.; Motter, N.E.; Wang, H.; Taro, B.S.; Chen, D.; Ruffner, D.; Hall, J.O. Increased blood-brain barrier permeability is not a primary determinant for lethality of West Nile virus infection in rodents. J. Gen. Virol. 2008, 89, 467–473. [Google Scholar] [CrossRef]
- Daffis, S.; Samuel, M.A.; Suthar, M.S.; Gale, M., Jr.; Diamond, M.S. Toll-like receptor 3 has a protective role against West Nile virus infection. J. Virol. 2008, 82, 10349–10358. [Google Scholar]
- Wang, T.; Town, T.; Alexopoulou, L.; Anderson, J.F.; Fikrig, E.; Flavell, R.A. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat. Med. 2004, 10, 1366–1373. [Google Scholar] [CrossRef]
- Wang, P.; Dai, J.; Bai, F.; Kong, K.F.; Wong, S.J.; Montgomery, R.R.; Madri, J.A.; Fikrig, E. Matrix metalloproteinase 9 facilitates West Nile virus entry into the brain. J. Virol. 2008, 82, 8978–8985. [Google Scholar]
- De Vries, H.E.; Blom-Roosemalen, M.C.; van Oosten, M.; de Boer, A.G.; van Berkel, T.J.; Breimer, D.D.; Kuiper, J. The influence of cytokines on the integrity of the blood-brain barrier in vitro. J. Neuroimmunol. 1996, 64, 37–43. [Google Scholar] [CrossRef]
- Arjona, A.; Foellmer, H.G.; Town, T.; Leng, L.; McDonald, C.; Wang, T.; Wong, S.J.; Montgomery, R.R.; Fikrig, E.; Bucala, R. Abrogation of macrophage migration inhibitory factor decreases West Nile virus lethality by limiting viral neuroinvasion. J. Clin. Invest. 2007, 117, 3059–3066. [Google Scholar] [CrossRef]
- Sultana, H.; Neelakanta, G.; Foellmer, H.G.; Montgomery, R.R.; Anderson, J.F.; Koski, R.A.; Medzhitov, R.M.; Fikrig, E. Semaphorin 7A contributes to West Nile virus pathogenesis through TGF-beta1/Smad6 signaling. J. Immunol. 2012, 189, 3150–3158. [Google Scholar] [CrossRef]
- Wilson, J.R.; de Sessions, P.F.; Leon, M.A.; Scholle, F. West Nile virus nonstructural protein 1 inhibits TLR3 signal transduction. J. Virol. 2008, 82, 8262–8271. [Google Scholar] [CrossRef]
- Kong, K.F.; Delroux, K.; Wang, X.; Qian, F.; Arjona, A.; Malawista, S.E.; Fikrig, E.; Montgomery, R.R. Dysregulation of TLR3 impairs the innate immune response to West Nile virus in the elderly. J. Virol. 2008, 82, 7613–7623. [Google Scholar] [CrossRef]
- Venter, M.; Myers, T.G.; Wilson, M.A.; Kindt, T.J.; Paweska, J.T.; Burt, F.J.; Leman, P.A.; Swanepoel, R. Gene expression in mice infected with West Nile virus strains of different neurovirulence. Virology 2005, 342, 119–140. [Google Scholar] [CrossRef]
- Chambers, T.J.; Diamond, M.S. Pathogenesis of flavivirus encephalitis. Adv. Virus Res. 2003, 60, 273–342. [Google Scholar] [CrossRef]
- Hunsperger, E.A.; Roehrig, J.T. Temporal analyses of the neuropathogenesis of a West Nile virus infection in mice. J. Neurovirol. 2006, 12, 129–139. [Google Scholar] [CrossRef]
- Morrey, J.D.; Siddharthan, V.; Wang, H.; Hall, J.O.; Skirpstunas, R.T.; Olsen, A.L.; Nordstrom, J.L.; Koenig, S.; Johnson, S.; Diamond, M.S. West Nile virus-induced acute flaccid paralysis is prevented by monoclonal antibody treatment when administered after infection of spinal cord neurons. J. Neurovirol. 2008, 14, 152–163. [Google Scholar] [CrossRef]
- Donadieu, E.; Lowenski, S.; Severly, J.-L.; Laloy, E.; Lilin, T.; Nowotny, N.; Richardson, J.; Zientara, S.; Lecollinet, S.; Coulpier, M. Comparison of the neuropathogenesis induced by two West Nile virus strains of lineage 1. PLoS One. submitted for publication.
- Lucas, M.; Frenkiel, M.P.; Mashimo, T.; Guenet, J.L.; Deubel, V.; Despres, P.; Ceccaldi, P.E. The Israeli strain IS-98-ST1 of West Nile virus as viral model for West Nile encephalitis in the old world. Virol. J. 2004, 1. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lobigs, M.; Lee, E.; Mullbacher, A. CD8+ T cells mediate recovery and immunopathology in West Nile virus encephalitis. J. Virol. 2003, 77, 13323–13334. [Google Scholar] [CrossRef]
- Lim, P.Y.; Louie, K.L.; Styer, L.M.; Shi, P.Y.; Bernard, K.A. Viral pathogenesis in mice is similar for West Nile virus derived from mosquito and mammalian cells. Virology 2010, 400, 93–103. [Google Scholar] [CrossRef]
- Eldadah, A.H.; Nathanson, N. Pathogenesis of West Nile Virus encepahlitis in mice and rats. II. Virus multiplication, evolution of immunofluorescence, and development of histological lesions in the brain. Am. J. Epidemiol. 1967, 86, 776–790. [Google Scholar]
- Cheeran, M.C.; Hu, S.; Sheng, W.S.; Rashid, A.; Peterson, P.K.; Lokensgard, J.R. Differential responses of human brain cells to West Nile virus infection. J. Neurovirol. 2005, 11, 512–524. [Google Scholar] [CrossRef]
- Shrestha, B.; Gottlieb, D.; Diamond, M.S. Infection and injury of neurons by West Nile encephalitis virus. J. Virol. 2003, 77, 13203–13213. [Google Scholar] [CrossRef]
- Sitati, E.; McCandless, E.E.; Klein, R.S.; Diamond, M.S. CD40-CD40 ligand interactions promote trafficking of CD8+ T cells into the brain and protection against West Nile virus encephalitis. J. Virol. 2007, 81, 9801–9811. [Google Scholar] [CrossRef]
- Kelley, T.W.; Prayson, R.A.; Ruiz, A.I.; Isada, C.M.; Gordon, S.M. The neuropathology of West Nile virus meningoencephalitis—A report of two cases and review of the literature. Am. J. Clin. Pathol. 2003, 119, 749–753. [Google Scholar]
- Parquet, M.C.; Kumatori, A.; Hasebe, F.; Morita, K.; Igarashi, A. West Nile virus-induced bax-dependent apoptosis. FEBS Lett. 2001, 500, 17–24. [Google Scholar] [CrossRef]
- Medigeshi, G.R.; Lancaster, A.M.; Hirsch, A.J.; Briese, T.; Lipkin, W.I.; Defilippis, V.; Fruh, K.; Mason, P.W.; Nikolich-Zugich, J.; Nelson, J.A. West Nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J. Virol. 2007, 81, 10849–10860. [Google Scholar] [CrossRef]
- Ramanathan, M.P.; Chambers, J.A.; Pankhong, P.; Chattergoon, M.; Attatippaholkun, W.; Dang, K.; Shah, N.; Weiner, D.B. Host cell killing by the West Nile Virus NS2B-NS3 proteolytic complex: NS3 alone is sufficient to recruit caspase-8-based apoptotic pathway. Virology 2006, 345, 56–72. [Google Scholar]
- Yang, J.S.; Ramanathan, M.P.; Muthumani, K.; Choo, A.Y.; Jin, S.H.; Yu, Q.C.; Hwang, D.S.; Choo, D.K.; Lee, M.D.; Dang, K.; et al. Induction of inflammation by West Nile virus capsid through the caspase-9 apoptotic pathway. Emerg. Infect. Dis. 2002, 8, 1379–1384. [Google Scholar] [CrossRef]
- Shieh, W.J.; Guarner, J.; Layton, M.; Fine, A.; Miller, J.; Nash, D.; Campbell, G.L.; Roehrig, J.T.; Gubler, D.J.; Zaki, S.R. The role of pathology in an investigation of an outbreak of West Nile encephalitis in New York, 1999. Emerg. Infect. Dis. 2000, 6, 370–372. [Google Scholar] [CrossRef]
- Chu, J.J.; Ng, M.L. The mechanism of cell death during West Nile virus infection is dependent on initial infectious dose. J. Gen. Virol. 2003, 84, 3305–3314. [Google Scholar] [CrossRef]
- Kumar, M.; Verma, S.; Nerurkar, V.R. Pro-inflammatory cytokines derived from West Nile virus (WNV)-infected SK-N-SH cells mediate neuroinflammatory markers and neuronal death. J. Neuroinflammation 2010, 7. [Google Scholar] [CrossRef]
- Benarroch, E.E. Neuron-astrocyte interactions: Partnership for normal function and disease in the central nervous system. Mayo Clin. Proc. 2005, 80, 1326–1338. [Google Scholar] [CrossRef]
- Liu, Y.; King, N.; Kesson, A.; Blanden, R.V.; Mullbacher, A. West Nile virus infection modulates the expression of class I and class II MHC antigens on astrocytes in vitro. Ann. NY Acad. Sci. 1988, 540, 483–485. [Google Scholar] [CrossRef]
- Bradl, M.; Hohlfeld, R. Molecular pathogenesis of neuroinflammation. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1364–1370. [Google Scholar] [CrossRef]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef]
- Giulian, D.; Li, J.; Leara, B.; Keenen, C. Phagocytic microglia release cytokines and cytotoxins that regulate the survival of astrocytes and neurons in culture. Neurochem. Int. 1994, 25, 227–233. [Google Scholar] [CrossRef]
- Chen, C.J.; Ou, Y.C.; Lin, S.Y.; Raung, S.L.; Liao, S.L.; Lai, C.Y.; Chen, S.Y.; Chen, J.H. Glial activation involvement in neuronal death by Japanese encephalitis virus infection. J. Gen. Virol. 2010, 91, 1028–1037. [Google Scholar] [CrossRef] [Green Version]
- Ghoshal, A.; Das, S.; Ghosh, S.; Mishra, M.K.; Sharma, V.; Koli, P.; Sen, E.; Basu, A. Proinflammatory mediators released by activated microglia induces neuronal death in Japanese encephalitis. Glia 2007, 55, 483–496. [Google Scholar] [CrossRef]
- Teeling, J.L.; Perry, V.H. Systemic infection and inflammation in acute CNS injury and chronic neurodegeneration: underlying mechanisms. Neuroscience 2009, 158, 1062–1073. [Google Scholar] [CrossRef]
- Hussmann, K.L.; Samuel, M.A.; Kim, K.S.; Diamond, M.S.; Fredericksen, B.L. Differential replication of pathogenic and nonpathogenic strains of West Nile virus within astrocytes. J. Virol. 2013, 87, 2814–2822. [Google Scholar]
- Shueb, R.H.; Papadimitriou, J.; Urosevic, N. Fatal persistence of West Nile virus subtype Kunjin in the brains of flavivirus resistant mice. Virus Res. 2011, 155, 455–461. [Google Scholar] [CrossRef]
- Morrey, J.D.; Siddharthan, V.; Wang, H.; Hall, J.O. Respiratory insufficiency correlated strongly with mortality of rodents infected with West Nile virus. PLoS One 2012, 7, e38672. [Google Scholar]
- Tesh, R.B.; Siirin, M.; Guzman, H.; Travassos da Rosa, A.P.; Wu, X.; Duan, T.; Lei, H.; Nunes, M.R.; Xiao, S.Y. Persistent West Nile virus infection in the golden hamster: studies on its mechanism and possible implications for other flavivirus infections. J. Infect. Dis. 2005, 192, 287–295. [Google Scholar] [CrossRef]
- Murray, K.; Walker, C.; Herrington, E.; Lewis, J.A.; McCormick, J.; Beasley, D.W.; Tesh, R.B.; Fisher-Hoch, S. Persistent infection with West Nile virus years after initial infection. J. Infect. Dis. 2010, 201, 2–4. [Google Scholar] [CrossRef]
- Durrant, D.M.; Robinette, M.L.; Klein, R.S. IL-1R1 is required for dendritic cell-mediated T cell reactivation within the CNS during West Nile virus encephalitis. J. Exp. Med. 2013, 210, 503–516. [Google Scholar]
- Lazear, H.M.; Pinto, A.K.; Vogt, M.R.; Gale, M., Jr.; Diamond, M.S. Beta interferon controls West Nile virus infection and pathogenesis in mice. J. Virol. 2011, 85, 7186–7194. [Google Scholar] [CrossRef]
- Szretter, K.J.; Daffis, S.; Patel, J.; Suthar, M.S.; Klein, R.S.; Gale, M., Jr.; Diamond, M.S. The innate immune adaptor molecule MyD88 restricts West Nile virus replication and spread in neurons of the central nervous system. J. Virol. 2010, 84, 12125–12138. [Google Scholar] [CrossRef]
- Shrestha, B.; Diamond, M.S. Role of CD8+ T cells in control of West Nile virus infection. J. Virol. 2004, 78, 8312–8321. [Google Scholar] [CrossRef]
- Glass, W.G.; Lim, J.K.; Cholera, R.; Pletnev, A.G.; Gao, J.L.; Murphy, P.M. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J. Exp. Med. 2005, 202, 1087–1098. [Google Scholar] [CrossRef]
- Glass, W.G.; McDermott, D.H.; Lim, J.K.; Lekhong, S.; Yu, S.F.; Frank, W.A.; Pape, J.; Cheshier, R.C.; Murphy, P.M. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J. Exp. Med. 2006, 203, 35–40. [Google Scholar] [CrossRef]
- Klein, R.S.; Lin, E.; Zhang, B.; Luster, A.D.; Tollett, J.; Samuel, M.A.; Engle, M.; Diamond, M.S. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J. Virol. 2005, 79, 11457–11466. [Google Scholar] [CrossRef]
- Lim, J.K.; McDermott, D.H.; Lisco, A.; Foster, G.A.; Krysztof, D.; Follmann, D.; Stramer, S.L.; Murphy, P.M. CCR5 deficiency is a risk factor for early clinical manifestations of West Nile virus infection but not for viral transmission. J. Infect. Dis. 2010, 201, 178–185. [Google Scholar]
- Brien, J.D.; Uhrlaub, J.L.; Nikolich-Zugich, J. West Nile virus-specific CD4 T cells exhibit direct antiviral cytokine secretion and cytotoxicity and are sufficient for antiviral protection. J. Immunol. 2008, 181, 8568–8575. [Google Scholar]
- Sitati, E.M.; Diamond, M.S. CD4+ T-cell responses are required for clearance of West Nile virus from the central nervous system. J. Virol. 2006, 80, 12060–12069. [Google Scholar] [CrossRef]
- Lanteri, M.C.; O’Brien, K.M.; Purtha, W.E.; Cameron, M.J.; Lund, J.M.; Owen, R.E.; Heitman, J.W.; Custer, B.; Hirschkorn, D.F.; Tobler, L.H.; et al. Tregs control the development of symptomatic West Nile virus infection in humans and mice. J. Clin. Invest. 2009, 119, 3266–3277. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Donadieu, E.; Bahuon, C.; Lowenski, S.; Zientara, S.; Coulpier, M.; Lecollinet, S. Differential Virulence and Pathogenesis of West Nile Viruses. Viruses 2013, 5, 2856-2880. https://doi.org/10.3390/v5112856
Donadieu E, Bahuon C, Lowenski S, Zientara S, Coulpier M, Lecollinet S. Differential Virulence and Pathogenesis of West Nile Viruses. Viruses. 2013; 5(11):2856-2880. https://doi.org/10.3390/v5112856
Chicago/Turabian StyleDonadieu, Emilie, Céline Bahuon, Steeve Lowenski, Stéphan Zientara, Muriel Coulpier, and Sylvie Lecollinet. 2013. "Differential Virulence and Pathogenesis of West Nile Viruses" Viruses 5, no. 11: 2856-2880. https://doi.org/10.3390/v5112856
APA StyleDonadieu, E., Bahuon, C., Lowenski, S., Zientara, S., Coulpier, M., & Lecollinet, S. (2013). Differential Virulence and Pathogenesis of West Nile Viruses. Viruses, 5(11), 2856-2880. https://doi.org/10.3390/v5112856