The Variation of the Soil Bacterial and Fungal Community Is Linked to Land Use Types in Northeast China

 , ,
, ,
Abstract
:1. Introduction
2. Material and Methods
2.1. Soil Sampling and Characterization
2.2. Soil DNA Extraction, Illumina Sequencing, and Data Analysis
2.3. Statistical Analysis
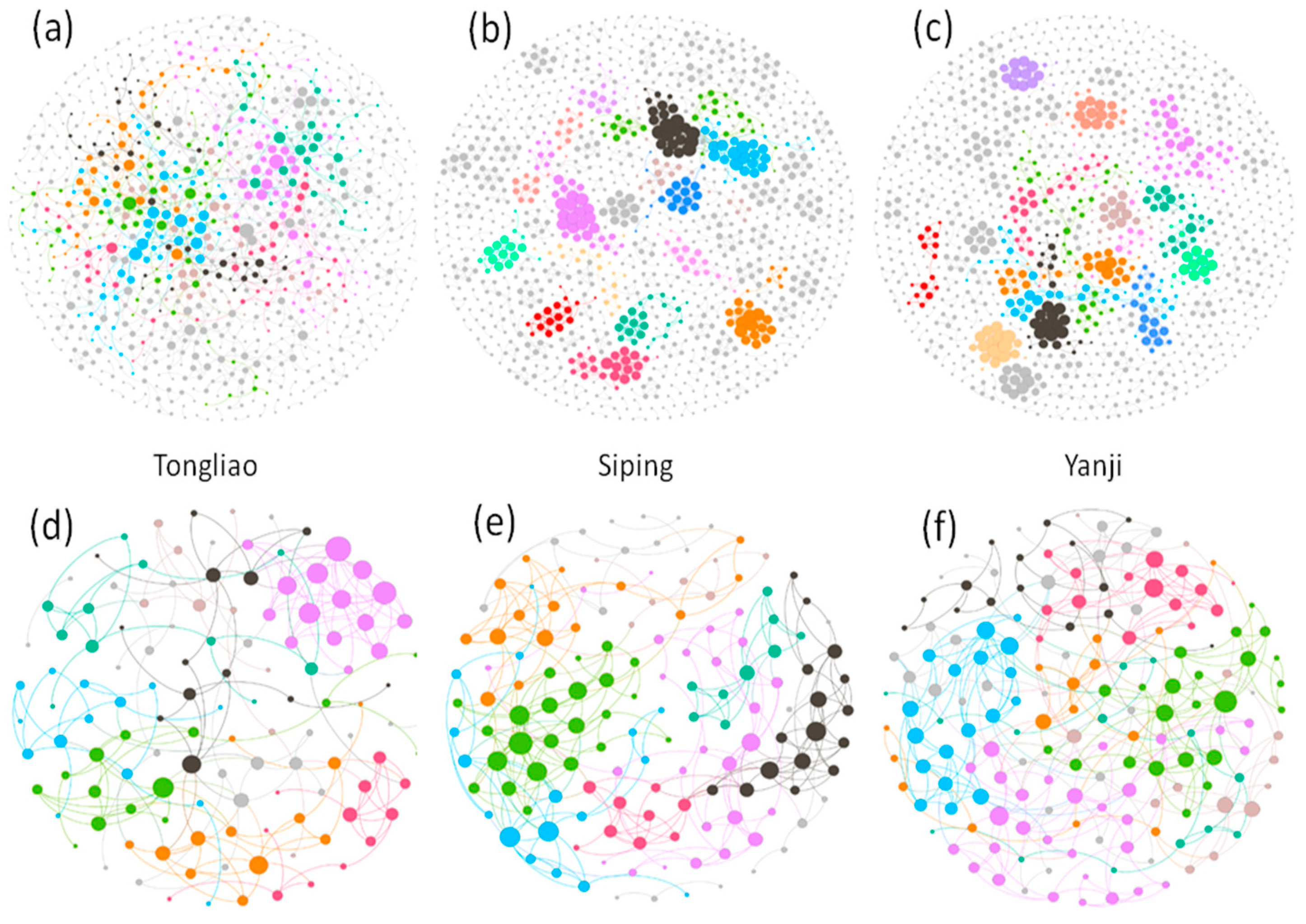
2.4. Network Construction and Statistical Analysis
3. Results
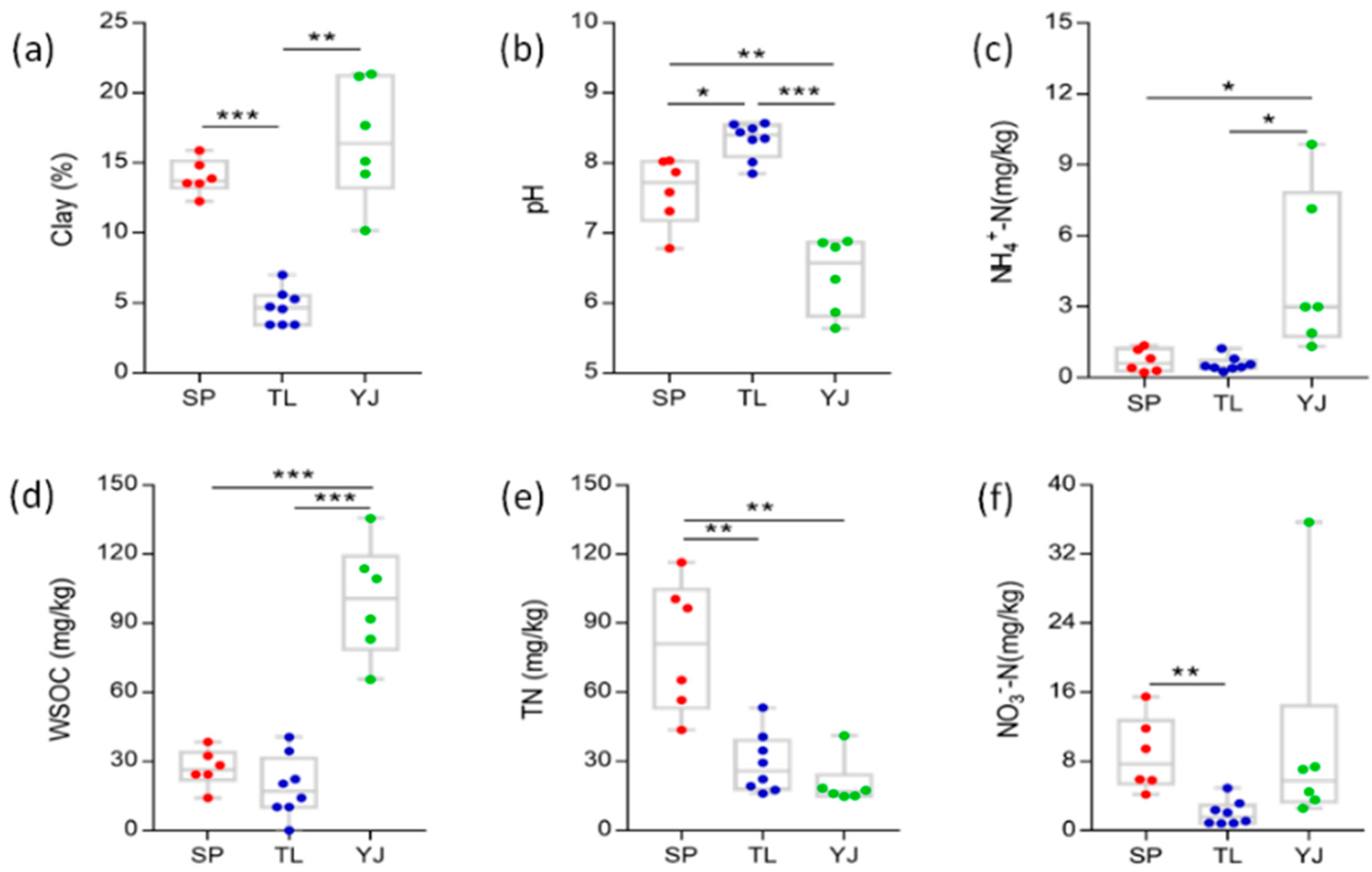
3.1. Soil Physicochemical Properties
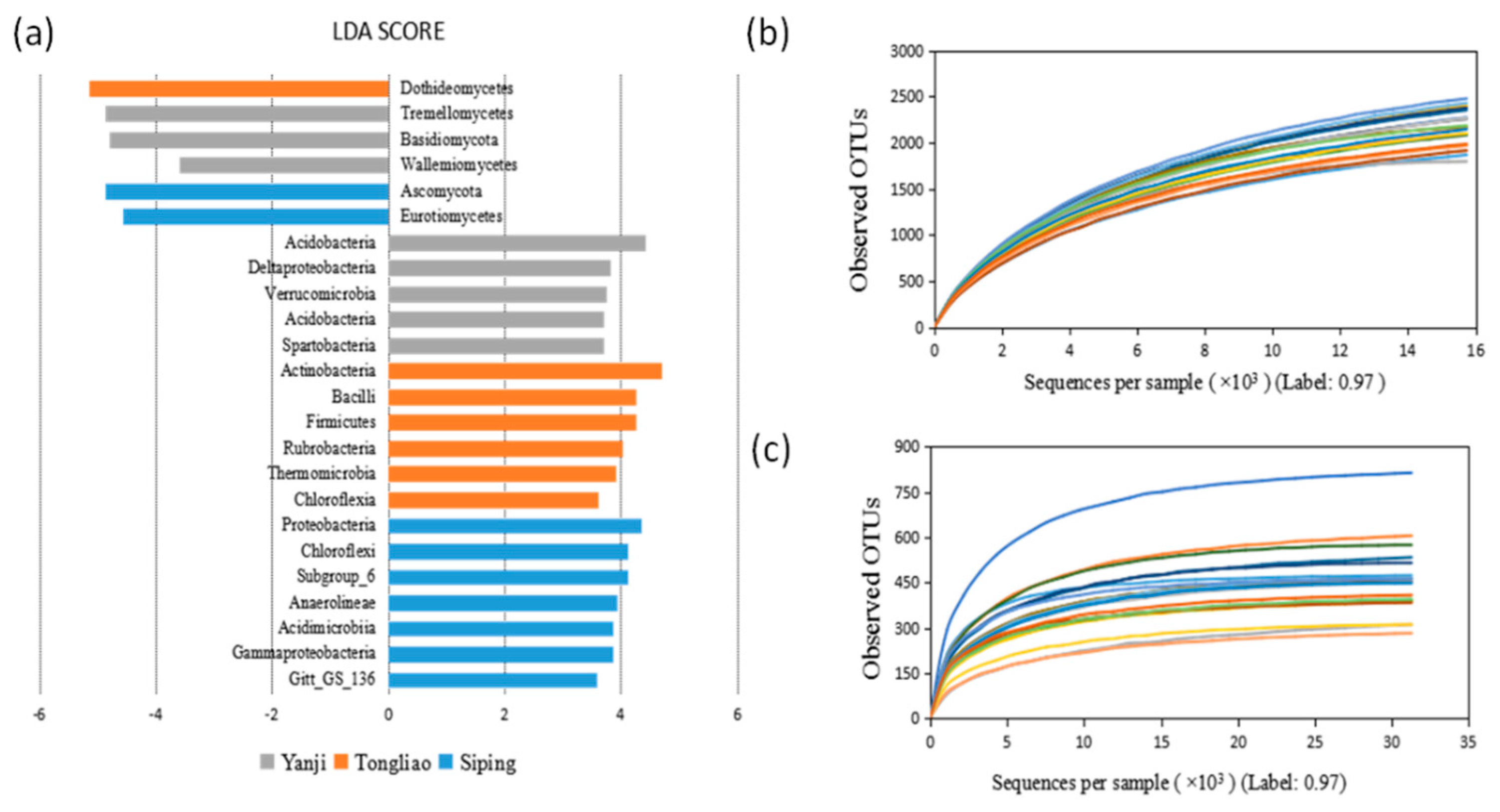
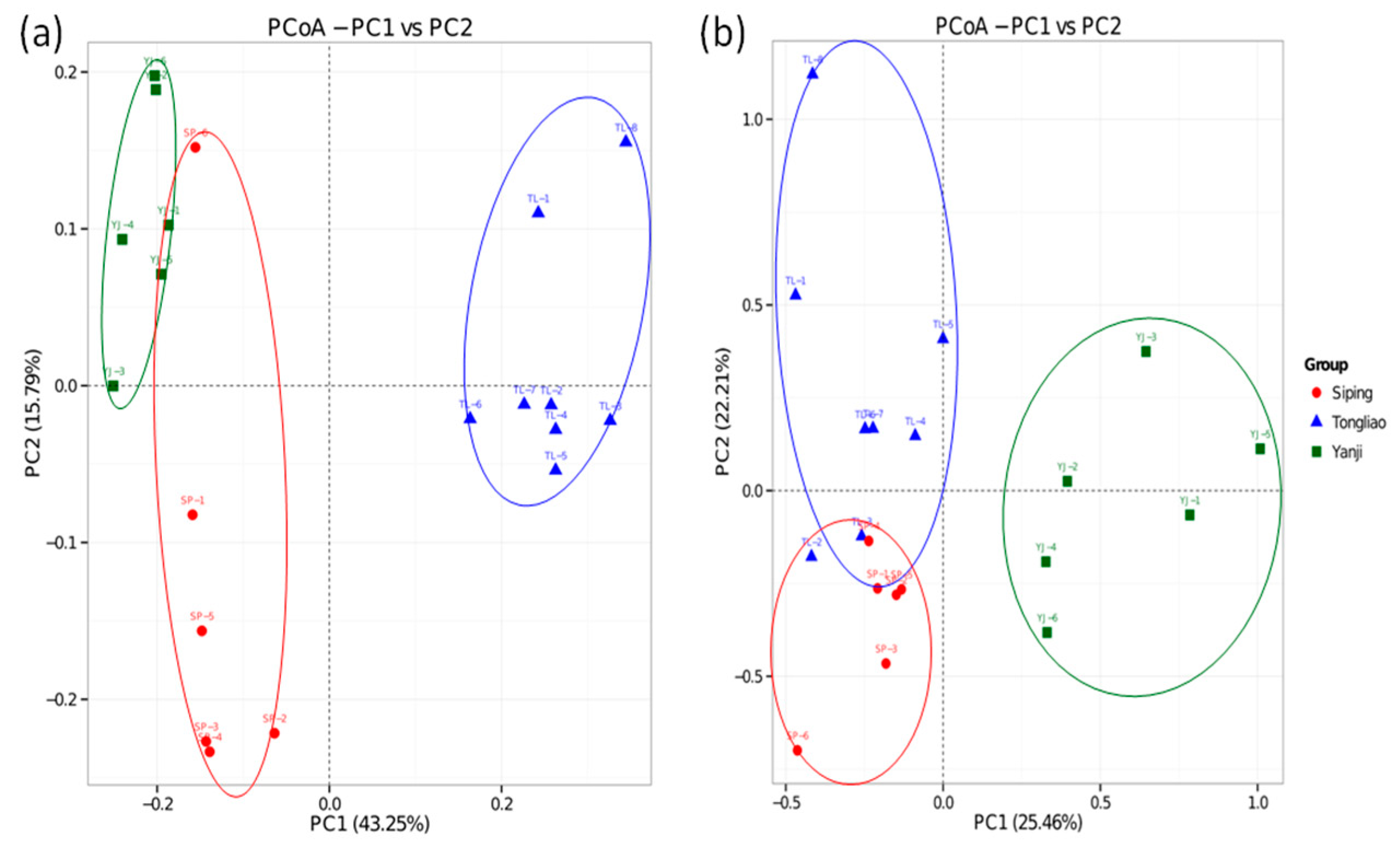
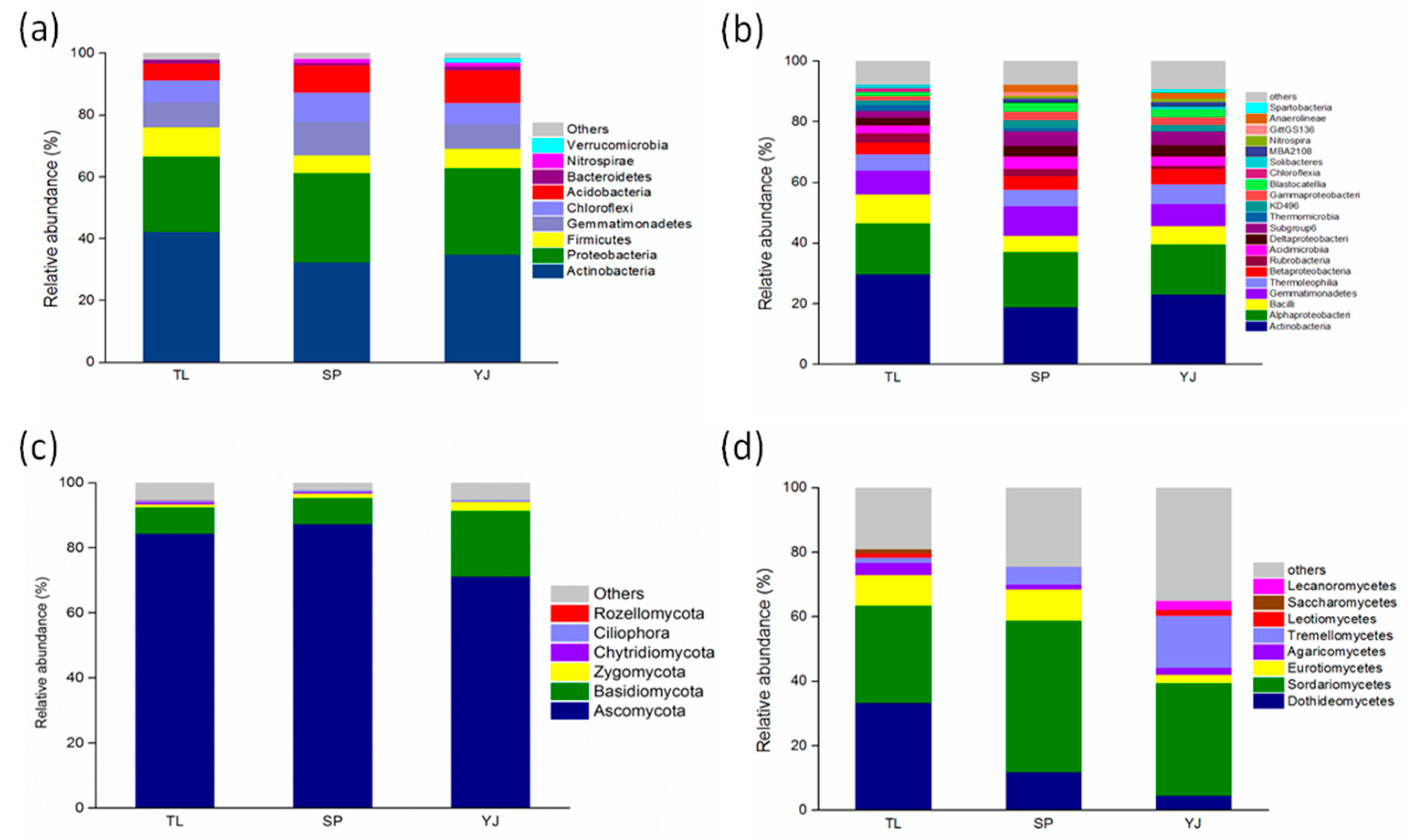
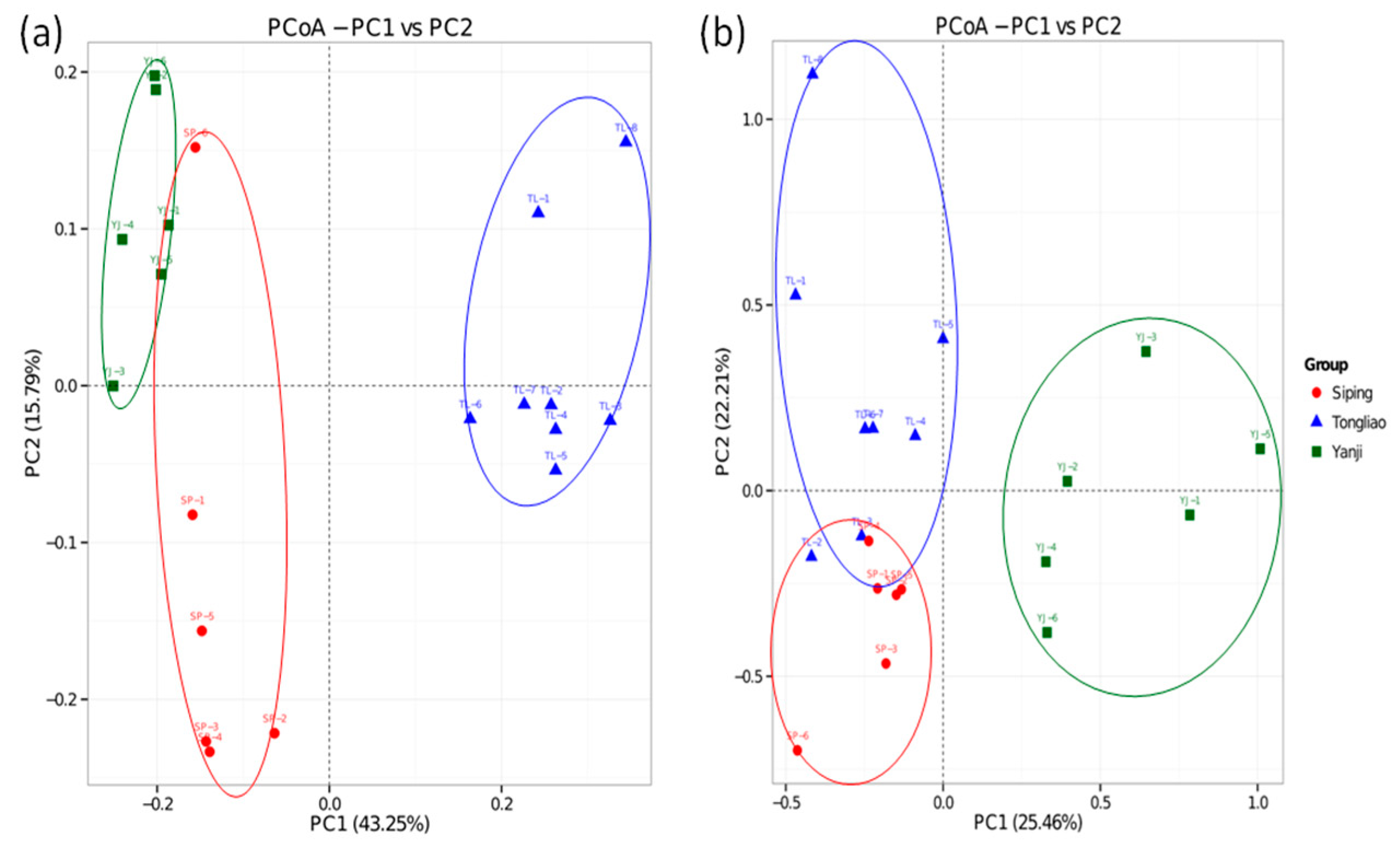
3.2. Differences in the Microbial Community in Three Sampling Sites
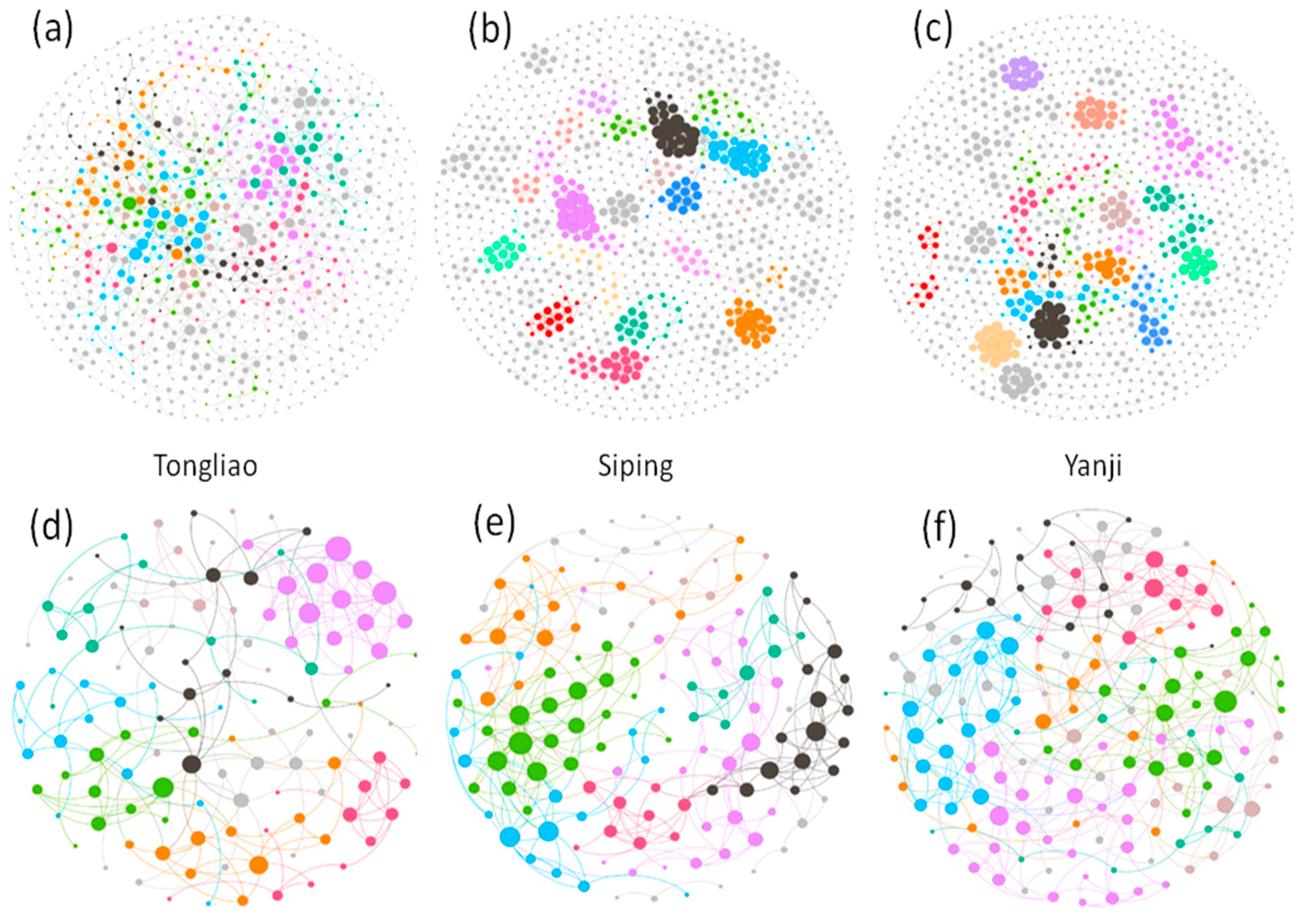
3.3. Comparison of Soil Microbial Co-Occurrence Networks
4. Discussion
4.1. Comparison of Soil Characterizations Among Three Land Use Types
4.2. Comparison of Bacterial and Fungal Composition and Abundance
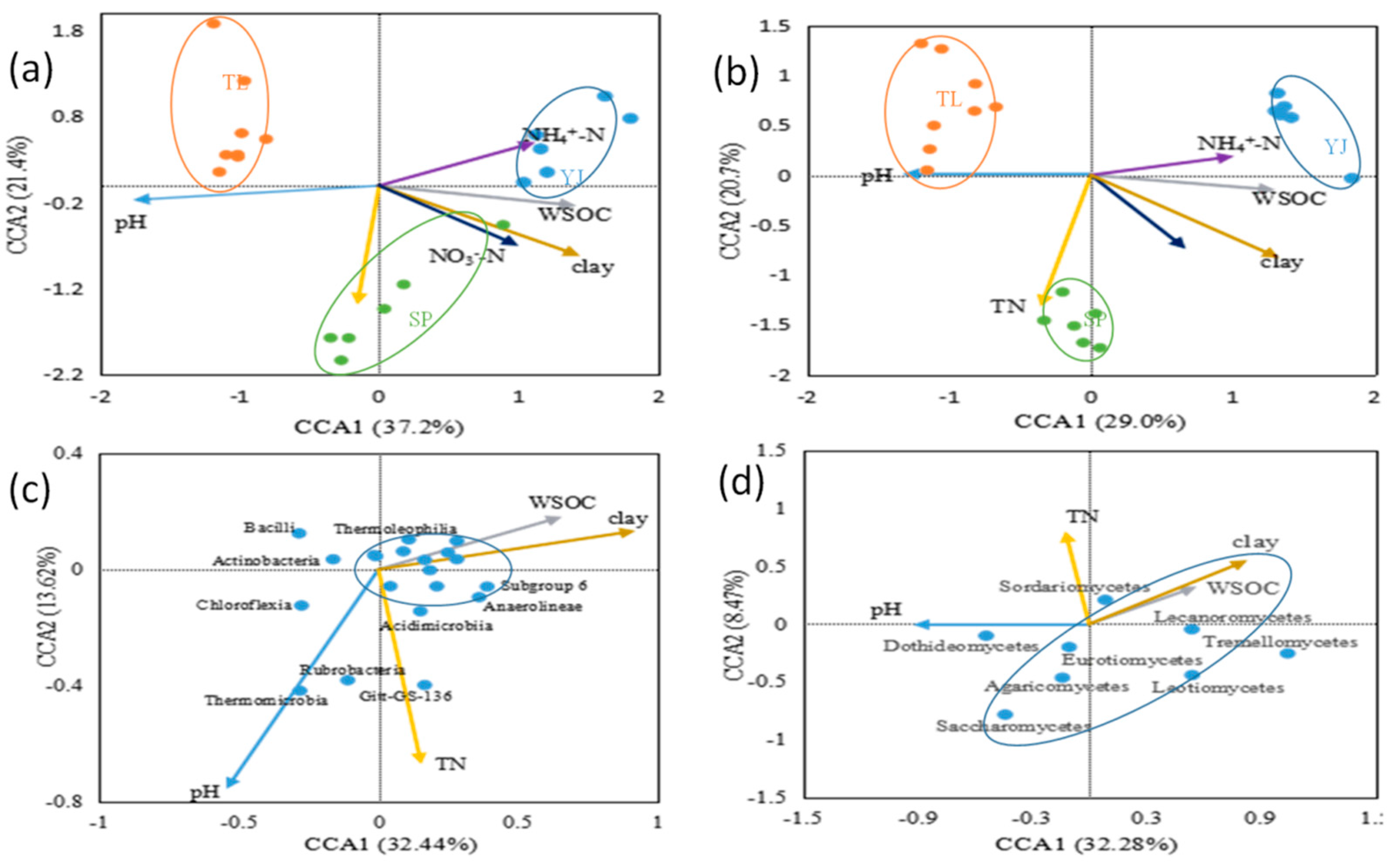
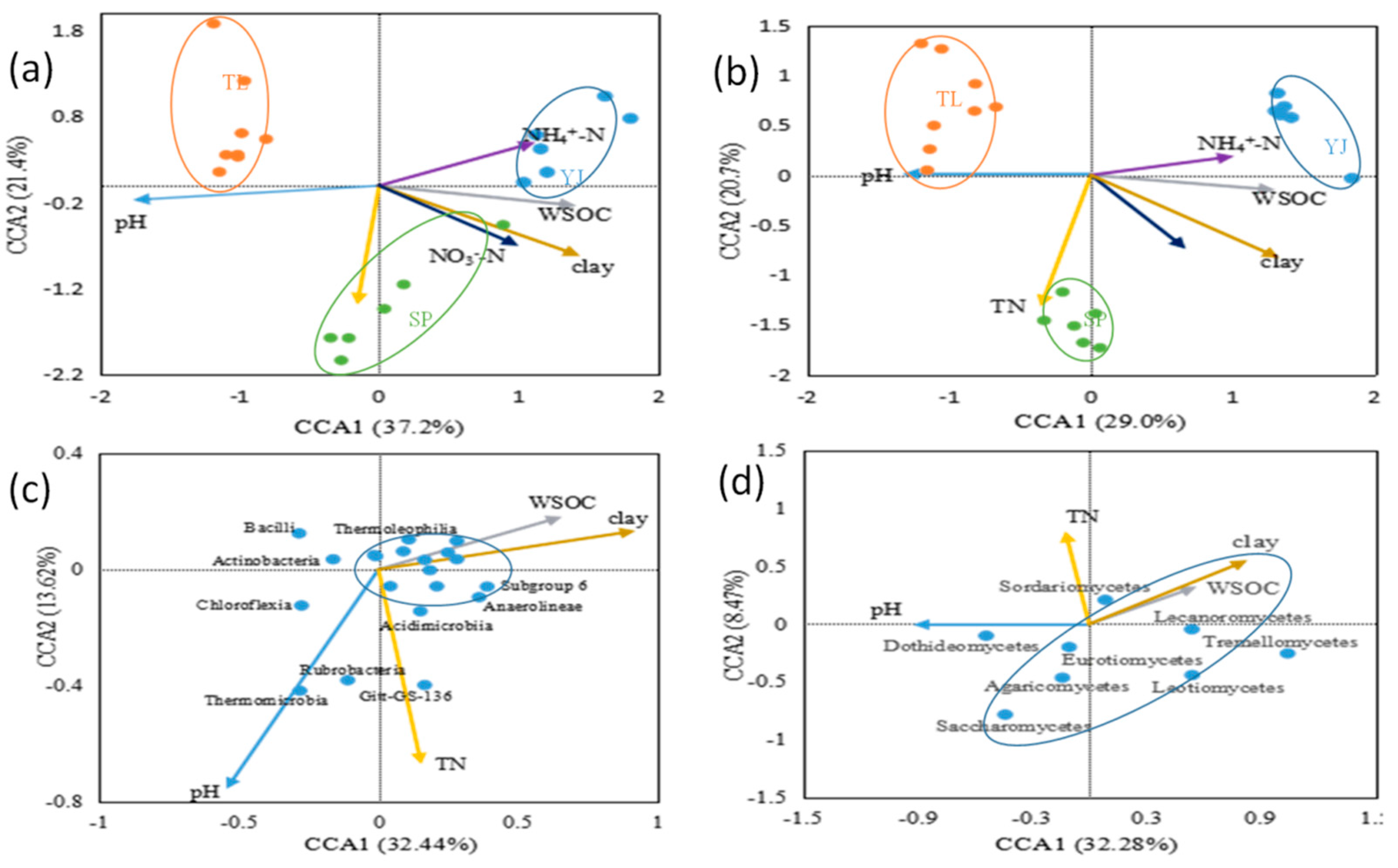
4.3. The Influence of Soil Properties on Bacterial and Fungal Community
4.4. Topological Features of Microbial Structures
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Basilio, A.; Gonzalez, I.; Vicente, M.F.; Gorrochategui, J.; Cabello, A.; Gonzalez, A.; Genilloud, O. Patterns of Antimicrobial Activities from Soil Actinomycetes Isolated under Different Conditions of Ph and Salinity. J. Appl. Microbiol. 2003, 95, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Widmer, F. Community Structure Analyses Are More Sensitive to Differences in Soil Bacterial Communities Than Anonymous Diversity Indices. Appl. Environ. Microbiol. 2006, 72, 7804–7812. [Google Scholar] [CrossRef] [PubMed]
- Baath, E.; Anderson, T.H. Comparison of Soil Fungal/Bacterial Ratios in a Ph Gradient Using Physiological and Plfa-Based Techniques. Soil Biol. Biochem. 2003, 35, 955–963. [Google Scholar] [CrossRef]
- Nacke, H.; Thürmer, A.; Wollherr, A.; Will, C.; Hodac, L.; Herold, N.; Schöning, I.; Schrumpf, M.; Daniel, R. Pyrosequencing-Based Assessment of Bacterial Community Structure Along Different Management Types in German Forest and Grassland Soils. PLoS ONE 2011, 6, e17000. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Xiong, J.; Zhang, H.; Feng, Y.; Lin, X.; Li, X.; Liang, W.; Chu, H. Soil Ph Drives the Spatial Distribution of Bacterial Communities Along Elevation on Changbai Mountain. Soil Biol. Biochem. 2013, 57, 204–211. [Google Scholar] [CrossRef]
- Liu, J.; Sui, Y.; Yu, Z.; Shi, Y.; Chu, H.; Jin, J.; Liu, X.; Wang, G. High Throughput Sequencing Analysis of Biogeographical Distribution of Bacterial Communities in the Black Soils of Northeast China. Soil Biol. Biochem. 2014, 70, 113–122. [Google Scholar] [CrossRef]
- Marschner, P.; Kandeler, E.; Marschner, B. Structure and Function of the Soil Microbial Community in a Long-Term Fertilizer Experiment. Soil Biol. Biochem. 2003, 35, 453–461. [Google Scholar] [CrossRef]
- Stark, C.; Condron, L.M.; Stewart, A.; Di, H.J.; O’Callaghan, M. Influence of Organic and Mineral Amendments on Microbial Soil Properties and Processes. Appl. Soil Ecol. 2007, 35, 79–93. [Google Scholar] [CrossRef]
- Dong, W.Y.; Zhang, X.Y.; Dai, X.Q.; Fu, X.L.; Yang, F.T.; Liu, X.Y.; Sun, X.M.; Wen, X.F.; Schaeffer, S. Changes in Soil Microbial Community Composition in Response to Fertilization of Paddy Soils in Subtropical China. Appl. Soil Ecol. 2014, 84, 140–147. [Google Scholar] [CrossRef]
- Bahram, M.F.; Hildebrand, S.K.; Forslund, J.L.; Anderson, N.A.; Soudzilovskaia, P.M.; Bodegom, J.; Bengtsson-Palme, S.; Anslan, L.P.; Coelho, H.; Harend, J.; et al. Structure and Function of the Global Topsoil Microbiome. Nature 2018, 560, 233–237. [Google Scholar] [CrossRef]
- Lauber, C.L.; Strickland, M.S.; Bradford, M.A.; Fierer, N. The Influence of Soil Properties on the Structure of Bacterial and Fungal Communities across Land-Use Types. Soil Biol. Biochem. 2008, 40, 2407–2415. [Google Scholar] [CrossRef]
- Rath, K.M.; Fierer, N.; Murphy, D.V.; Rousk, J. Linking Bacterial Community Composition to Soil Salinity Along Environmental Gradients. ISME J. 2019, 13, 836. [Google Scholar] [CrossRef] [PubMed]
- Rousk, J.; Bååth, E.; Brookes, P.C.; Lauber, C.L.; Lozupone, C.; Caporaso, J.G.; Knight, R.; Fierer, N. Soil Bacterial and Fungal Communities across a Ph Gradient in an Arable Soil. ISME J. 2010, 4, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sui, Y.; Yu, Z.; Shi, Y.; Chu, H.; Jin, J.; Liu, X.; Wang, G. Soil Carbon Content Drives the Biogeographical Distribution of Fungal Communities in the Black Soil Zone of Northeast China. Soil Biol. Biochem. 2015, 83, 29–39. [Google Scholar] [CrossRef]
- de Boer, W.; Verheggen, P.; Gunnewiek, P.J.K.; Kowalchuk, G.A.; van Veen, J.A. Microbial Community Composition Affects Soil Fungistasis. Appl. Environ. Microbiol. 2003, 69, 835–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegen, J.C.; Lin, X.; Konopka, A.E.; Fredrickson, J.K. Stochastic and Deterministic Assembly Processes in Subsurface Microbial Communities. ISME J. 2012, 6, 1653–1664. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Deng, Y.; Luo, F.; He, Z.; Yang, Y. Phylogenetic Molecular Ecological Network of Soil Microbial Communities in Response to Elevated Co2. MBio 2011, 2, e00122-11. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Nuccio, E.E.; Shi, Z.J.; He, Z.; Zhou, J.; Firestone, M.K. The Interconnected Rhizosphere: High Network Complexity Dominates Rhizosphere Assemblages. Ecol. Lett. 2016, 19, 926–936. [Google Scholar] [CrossRef]
- Deng, Y.; Zhang, P.; Qin, Y.; Tu, Q.; Yang, Y.; He, Z.; Schadt, C.W.; Zhou, J. Network Succession Reveals the Importance of Competition in Response to Emulsified Vegetable Oil Amendment for Uranium Bioremediation. Environ. Microbiol. 2016, 18, 205–218. [Google Scholar] [CrossRef]
- Kozdroj, J.; van Elsas, J.D. Structural Diversity of Microbial Communities in Arable Soils of a Heavily Industrialised Area Determined by Pcr-Dgge Fingerprinting and Fame Profiling. Appl. Soil Ecol. 2001, 17, 31–42. [Google Scholar] [CrossRef]
- Rousk, J.; Brookes, P.C.; Bååth, E. Investigating the Mechanisms for the Opposing Ph Relationships of Fungal and Bacterial Growth in Soil. Soil Biol. Biochem. 2010, 42, 926–934. [Google Scholar] [CrossRef]
- Schmidt, S.K.; Nemergut, D.R.; Darcy, J.L.; Lynch, R. Do Bacterial and Fungal Communities Assemble Differently During Primary Succession? Mol. Ecol. 2014, 23, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Vance, E.D.; Brookes, P.C.; Jenkinson, D.S. An Extraction Method for Measuring Soil Microbial Biomass C. Soil Biol. Biochem. 1987, 19, 703–707. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Keller, K.; Brodie, E.L.; Larsen, N.; Piceno, Y.M.; Phan, R.; Andersen, G.L. Nast: A Multiple Sequence Alignment Server for Comparative Analysis of 16s Rrna Genes. Nucleic Acids Res. 2006, 34, W394–W399. [Google Scholar] [CrossRef] [PubMed]
- Dunfield, P.F.; Yuryev, A.; Senin, P.; Smirnova, A.V.; Stott, M.B.; Hou, S.; Ly, B.; Saw, J.H.; Zhou, Z.; Ren, Y.; et al. Methane Oxidation by an Extremely Acidophilic Bacterium of the Phylum Verrucomicrobia. Nature 2007, 450, 879–882. [Google Scholar] [CrossRef]
- Niţă, A.; Buttler, A.; Rozylowicz, L.; Pătru-Stupariu, I. Perception and Use of Landscape Concepts in the Procedure of Environmental Impact Assessment: Case Study—Switzerland and Romania. Land Use Policy 2015, 44, 145–152. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic Biomarker Discovery and Explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Deng, Y.; Jiang, Y.H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular Ecological Network Analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef]
- Manolache, S.; Nita, A.; Ciocanea, C.M.; Popescu, V.D.; Rozylowicz, L. Power, Influence and Structure in Natura 2000 Governance Networks. A Comparative Analysis of Two Protected Areas in Romania. J. Environ. Manag. 2018, 212, 54–64. [Google Scholar] [CrossRef]
- Faust, K.; Raes, J. Microbial Interactions: From Networks to Models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. In Proceedings of the Third international AAAI Conference on Weblogs and Social Media, San Jose, CA, USA, 17–20 May 2009. [Google Scholar]
- Peacock, A.G.; Mullen, M.D.; Ringelberg, D.B.; Tyler, D.D.; Hedrick, D.B.; Gale, P.M.; White, D.C. Soil Microbial Community Responses to Dairy Manure or Ammonium Nitrate Applications. Soil Biol. Biochem. 2001, 33, 1011–1019. [Google Scholar] [CrossRef]
- Cookson, W.R.; Abaye, D.A.; Marschner, P.; Murphy, D.V.; Stockdale, E.A.; Goulding, K.W. The Contribution of Soil Organic Matter Fractions to Carbon and Nitrogen Mineralization and Microbial Community Size and Structure. Soil Biol. Biochem. 2005, 37, 1726–1737. [Google Scholar] [CrossRef]
- Elfstrand, S.; Hedlund, K.; Mårtensson, A. Soil Enzyme Activities, Microbial Community Composition and Function after 47 Years of Continuous Green Manuring. Appl. Soil Ecol. 2007, 35, 610–621. [Google Scholar] [CrossRef]
- Galvez, A.; Sinicco, T.; Cayuela, M.L.; Mingorance, M.D.; Fornasier, F.; Mondini, C. Short Term Effects of Bioenergy by-Products on Soil C and N Dynamics, Nutrient Availability and Biochemical Properties. Agric. Ecosyst. Environ. 2012, 160, 3–14. [Google Scholar] [CrossRef]
- Banerjee, S.; Schlaeppi, K.; Van Der Heijden, M.G. Keystone Taxa as Drivers of Microbiome Structure and Functioning. Nat. Rev. Microbiol. 2018, 16, 1. [Google Scholar] [CrossRef]
- Wang, X.B.; Lü, X.T.; Yao, J.; Wang, Z.W.; Deng, Y.; Cheng, W.X.; Zhou, J.Z.; Han, X.G. Habitat-Specific Patterns and Drivers of Bacterial Beta-Diversity in China’s Drylands. ISME J. 2017, 11, 1345–1358. [Google Scholar] [CrossRef] [PubMed]
- Clegg, C.D.; Lovell, R.D.; Hobbs, P.J. The Impact of Grassland Management Regime on the Community Structure of Selected Bacterial Groups in Soils. FEMS Microbiol. Ecol. 2003, 43, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an Ecological Classification of Soil Bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef]
- Jones, R.T.; Robeson, M.S.; Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. A Comprehensive Survey of Soil Acidobacterial Diversity Using Pyrosequencing and Clone Library Analyses. ISME J. 2009, 3, 442–453. [Google Scholar] [CrossRef]
- Eichorst, S.A.; Breznak, J.A.; Schmidt, T.M. Isolation and Characterization of Soil Bacteria That Define Terriglobus Gen. Nov., in the Phylum Acidobacteria. Appl. Environ. Microbiol. 2007, 73, 2708–2717. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sui, Y.; Yu, Z.; Yao, Q.; Shi, Y.; Chu, H.; Jin, J.; Liu, X.; Wang, G. Diversity and Distribution Patterns of Acidobacterial Communities in the Black Soil Zone of Northeast China. Soil Biol. Biochem. 2016, 95, 212–222. [Google Scholar] [CrossRef]
- de Castro, V.H.L.; Schroeder, L.F.; Quirino, B.F.; Kruger, R.H.; Barreto, C.C. Acidobacteria from Oligotrophic Soil from the Cerrado Can Grow in a Wide Range of Carbon Source Concentrations. Can. J. Microbiol. 2013, 59, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Bardgett, R.D.; McAlister, E. The Measurement of Soil Fungal: Bacterial Biomass Ratios as an Indicator of Ecosystem Self-Regulation in Temperate Meadow Grasslands. Biol. Fertil. Soils 1999, 29, 282–290. [Google Scholar] [CrossRef]
- Lovell, R.D.; Jarvis, S.C.; Bardgett, R.D. Soil Microbial Biomass and Activity in Long-Term Grassland: Effects of Management Changes. Soil Biol. Biochem. 1995, 27, 969–975. [Google Scholar] [CrossRef]
- De Vries, F.T.; Hoffland, E.; van Eekeren, N.; Brussaard, L.; Bloem, J. Fungal/Bacterial Ratios in Grasslands with Contrasting Nitrogen Management. Soil Biol. Biochem. 2006, 38, 2092–2103. [Google Scholar] [CrossRef]
- Maestre, F.T.; Delgado-Baquerizo, M.; Jeffries, T.C.; Eldridge, D.J.; Ochoa, V.; Gozalo, B.; Quero, J.L.; Garcia-Gomez, M.; Gallardo, A.; Ulrich, W.; et al. Increasing Aridity Reduces Soil Microbial Diversity and Abundance in Global Drylands. Proc. Natl. Acad. Sci. USA 2015, 112, 15684–15689. [Google Scholar] [CrossRef]
- Wagner, M.; Horn, M. The Planctomycetes, Verrucomicrobia, Chlamydiae and Sister Phyla Comprise a Superphylum with Biotechnological and Medical Relevance. Curr. Opin. Biotechnol. 2006, 17, 241–249. [Google Scholar] [CrossRef]
- Newton, R.J.; Jones, S.E.; Eiler, A.; McMahon, K.D.; Bertilsson, S. A Guide to the Natural History of Freshwater Lake Bacteria. Microbiol. Mol. Biol. Rev. 2011, 75, 14–49. [Google Scholar] [CrossRef] [Green Version]
- Hanson, R.S.; Hanson, T.E. Methanotrophic Bacteria. Microbiol. Rev. 1996, 60, 439–471. [Google Scholar]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-Based Assessment of Soil Ph as a Predictor of Soil Bacterial Community Structure at the Continental Scale. Appl. Environ. Microbiol. 2009, 75, 5111–5120. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Fierer, N.; Lauber, C.L.; Caporaso, J.G.; Knight, R.; Grogan, P. Soil Bacterial Diversity in the Arctic Is Not Fundamentally Different from That Found in Other Biomes. Environ. Microbiol. 2010, 12, 2998–3006. [Google Scholar] [CrossRef] [PubMed]
- Tedersoo, L.; Bahram, M.; Põlme, S.; Kõljalg, U.; Yorou, N.S.; Wijesundera, R.; Ruiz, L.V.; Vasco-Palacios, A.M.; Thu, P.Q.; Suija, A.; et al. Global Diversity and Geography of Soil Fungi. Science 2014, 346, 1256688. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, S.N.; Waite, I.S.; Blackburn, A.; Husband, R.; Rushton, S.P.; Manning, D.C.; O’Donnell, A.G. Actinobacterial Community Dynamics in Long Term Managed Grasslands. Antonie Van Leeuwenhoek 2009, 95, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Deng, M.; Yuan, A.; Wang, J.; Li, H.; Ma, J. Vertical Variation of a Black Soil’s Properties in Response to Freeze-Thaw Cycles and Its Links to Shift of Microbial Community Structure. Sci. Total Environ. 2018, 625, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Nevarez, L.; Vasseur, V.; Le Madec, A.; Le Bras, M.A.; Coroller, L.; Leguérinel, I.; Barbier, G. Physiological Traits of Penicillium Glabrum Strain Lcp 08.5568, a Filamentous Fungus Isolated from Bottled Aromatized Mineral Water. Int. J. Food Microbiol. 2009, 130, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Rousk, J.; Demoling, L.A.; Bahr, A.; Baath, E. Examining the Fungal and Bacterial Niche Overlap Using Selective Inhibitors in Soil. FEMS Microbiol. Ecol. 2008, 63, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Ranjard, L.; Richaume, A. Quantitative and Qualitative Microscale Distribution of Bacteria in Soil. Res. Microbiol. 2001, 152, 707–716. [Google Scholar] [CrossRef]
- Ma, J.; Ibekwe, A.M.; Yang, C.H.; Crowley, D.E. Bacterial Diversity and Composition in Major Fresh Produce Growing Soils Affected by Physiochemical Properties and Geographic Locations. Sci. Total Environ. 2016, 563, 199–209. [Google Scholar] [CrossRef]
- Klein, D.A.; McLendon, T.; Paschke, M.W.; Redente, E.F. Nitrogen Availability and Fungal-Bacterial Responses in Successional Semiarid Steppe Soils. Arid Soil Res. Rehabil. 1996, 10, 321–332. [Google Scholar] [CrossRef]
- Denef, K.; Six, J.; Bossuyt, H.; Frey, S.D.; Elliott, E.T.; Merckx, R.; Paustian, K. Influence of Dry-Wet Cycles on the Interrelationship between Aggregate, Particulate Organic Matter, and Microbial Community Dynamics. Soil Biol. Biochem. 2001, 33, 1599–1611. [Google Scholar] [CrossRef]
- Zhou, J.; Xia, B.; Treves, D.S.; Wu, L.Y.; Marsh, T.L.; O’Neill, R.V.; Palumbo, A.V.; Tiedje, J.M. Spatial and Resource Factors Influencing High Microbial Diversity in Soil. Appl. Environ. Microbiol. 2002, 68, 326–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mummey, D.; Holben, W.; Six, J.; Stahl, P. Spatial Stratification of Soil Bacterial Populations in Aggregates of Diverse Soils. Microb. Ecol. 2006, 51, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Bohme, L.; Langer, U.; Bohme, F. Microbial Biomass, Enzyme Activities and Microbial Community Structure in Two European Long-Term Field Experiments. Agric. Ecosyst. Environ. 2005, 109, 141–152. [Google Scholar] [CrossRef]
- Li, H.; Parent, L.E.; Karam, A.; Tremblay, C. Potential of Sphagnum Peat for Improving Soil Organic Matter, Water Holding Capacity, Bulk Density and Potato Yield in a Sandy Soil. Plant and Soil 2004, 265, 355–365. [Google Scholar] [CrossRef]
- Pereira e Silva, M.C.; Dias, A.C.F.; van Elsas, J.D.; Salles, J.F. Spatial and Temporal Variation of Archaeal, Bacterial and Fungal Communities in Agricultural Soils. PLoS ONE 2012, 7, e51554. [Google Scholar] [CrossRef] [PubMed]
- de Vries, F.T.; Griffiths, R.I.; Bailey, M.; Craig, H.; Girlanda, M.; Gweon, H.S.; Hallin, S.; Kaisermann, A.; Keith, A.M.; Kretzschmar, M.; et al. Soil Bacterial Networks Are Less Stable under Drought Than Fungal Networks. Nat. Commun. 2018, 9, 3033. [Google Scholar] [CrossRef] [PubMed]
- Zinger, L.; Lejon, D.P.; Baptist, F.; Bouasria, A.; Aubert, S.; Geremia, R.A.; Choler, P. Contrasting Diversity Patterns of Crenarchaeal, Bacterial and Fungal Soil Communities in an Alpine Landscape. PLoS ONE 2011, 6, e19950. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Tongliao | Siping | Yanji | MDA b |
|---|---|---|---|---|
| Clay | 16.72 ** | 10.35 ** | 7.93 ** | 16.22 ** |
| pH | 8.44 * | 0.21 | 7.86 * | 9.31 * |
| WSOC | 7.48 ** | 8.08 * | 14.88 ** | 14.74 ** |
| TN | 2.95 | 10.70 * | 6.78 * | 10.44 * |
| NO3−-N | 8.16 ** | 6.79 * | −0.18 | 8.02 ** |
| NH4+-N | 4.19 | 1.86 | 7.77 * | 6.92 * |
| Bacteria | Fungi | |||||
|---|---|---|---|---|---|---|
| δ | F | R | δ | F | R | |
| Total | 0.427 *** | 7.899 *** | 0.897 *** | 0.660 *** | 5.192 *** | 0.874 *** |
| TL vs. SP | 0.434 ** | 6.710 *** | 0.831 *** | 0.715 *** | 3.457 ** | 0.761 *** |
| TL vs. YJ | 0.420 ** | 11.273 *** | 1.000 *** | 0.651 *** | 6.875 ** | 0.959 *** |
| SP vs. YJ | 0.427 ** | 5.614 ** | 0.802 ** | 0.606 ** | 5.688 ** | 0.926 ** |
| Bacteria | Fungi | |||
|---|---|---|---|---|
| r | p | r | p | |
| Clay | 0.431 | 0.001 | 0.329 | 0.003 |
| pH | 0.511 | 0.001 | 0.352 | 0.002 |
| WSOC | 0.284 | 0.006 | 0.251 | 0.005 |
| TN | 0.175 | 0.034 | 0.217 | 0.017 |
| NO3−-N | −0.372 | 0.999 | −0.240 | 0.999 |
| NH4+-N | −0.091 | 0.833 | −0.235 | 0.998 |
| Network Parameters | Bacterial | Fungal | ||||
|---|---|---|---|---|---|---|
| Tongliao | Siping | Yanji | Tongliao | Siping | Yanji | |
| nodes | 901 | 1093 | 1099 | 124 | 142 | 170 |
| links | 1137 | 2336 | 1790 | 217 | 307 | 384 |
| average degree | 1.262 | 2.137 | 1.629 | 1.750 | 2.162 | 2.276 |
| network diameter | 8 | 6 | 6 | 5 | 7 | 5 |
| modularity | 0.893 | 0.967 | 0.977 | 0.764 | 0.742 | 0.751 |
| average clustering coefficient | 0.084 | 0.303 | 0.262 | 0.158 | 0.220 | 0.223 |
| average path length | 2.093 | 1.510 | 1.406 | 1.659 | 2.082 | 1.914 |
| Bacteria | Fungi | |||
|---|---|---|---|---|
| r | p | r | p | |
| clay | 0.115 | 0.002 | 0.013 | 0.337 |
| pH | 0.245 | 0.001 | 0.197 | 0.002 |
| WSOC | 0.171 | 0.001 | 0.232 | 0.002 |
| TN | −0.108 | 0.999 | −0.059 | 0.871 |
| NO3−-N | −0.068 | 0.973 | −0.115 | 0.981 |
| NH4+-N | 0.130 | 0.009 | −0.106 | 0.977 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, J.; Nergui, S.; Han, Z.; Huang, G.; Li, H.; Zhang, R.; Zhu, L.; Liao, J. The Variation of the Soil Bacterial and Fungal Community Is Linked to Land Use Types in Northeast China. Sustainability 2019, 11, 3286. https://doi.org/10.3390/su11123286
Ma J, Nergui S, Han Z, Huang G, Li H, Zhang R, Zhu L, Liao J. The Variation of the Soil Bacterial and Fungal Community Is Linked to Land Use Types in Northeast China. Sustainability. 2019; 11(12):3286. https://doi.org/10.3390/su11123286
Chicago/Turabian StyleMa, Jincai, Sumiya Nergui, Ziming Han, Guannan Huang, Huiru Li, Rui Zhang, Liyue Zhu, and Jiafen Liao. 2019. "The Variation of the Soil Bacterial and Fungal Community Is Linked to Land Use Types in Northeast China" Sustainability 11, no. 12: 3286. https://doi.org/10.3390/su11123286