
Quantification of Arsenic in Soil Samples Collected in an Industrial Area of Brindisi (Apulia, Italy): Speciation Analysis and Availability


Abstract
:1. Introduction
2. Results and Discussion
2.1. Sample Collection and Total As Determination
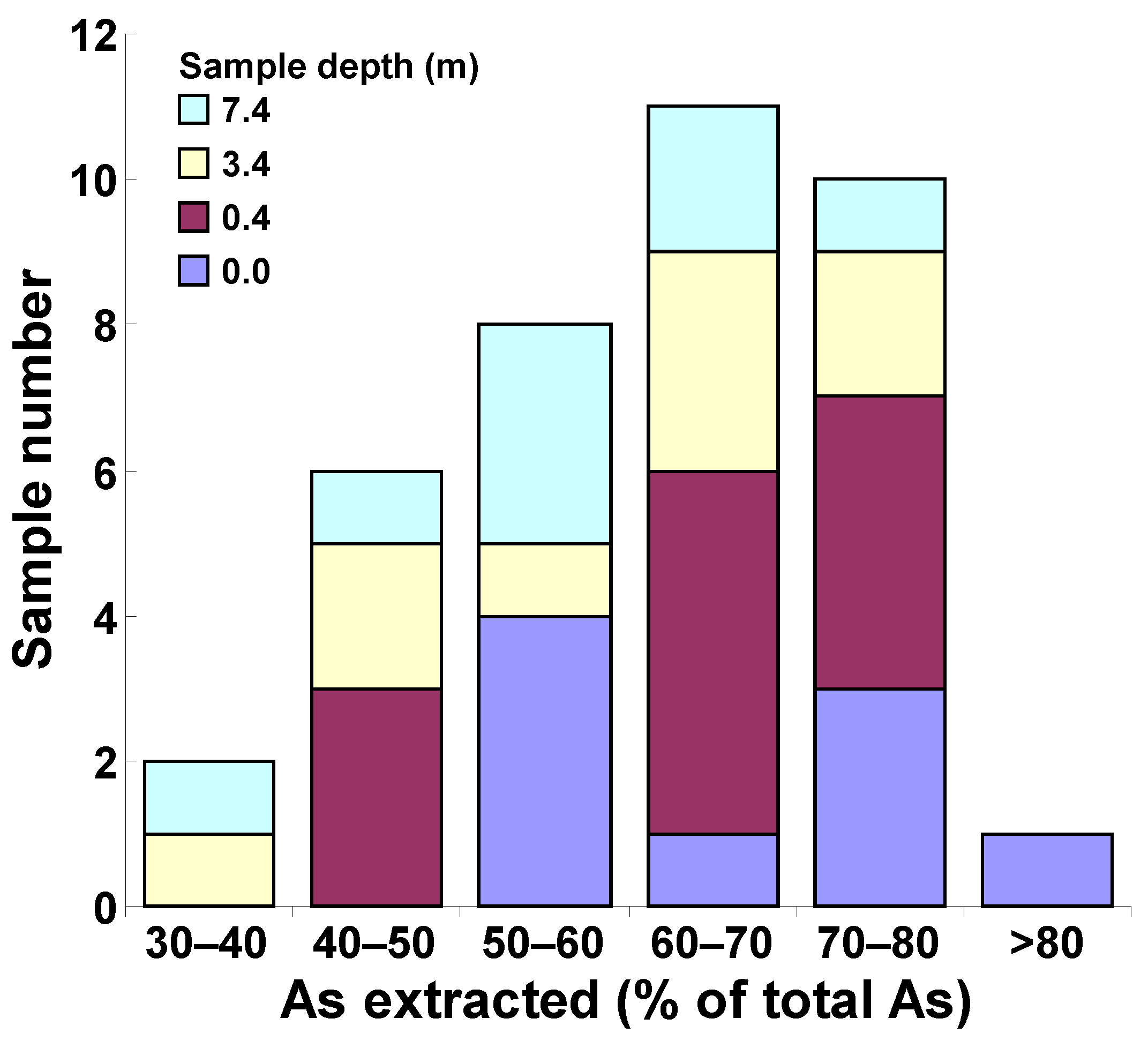
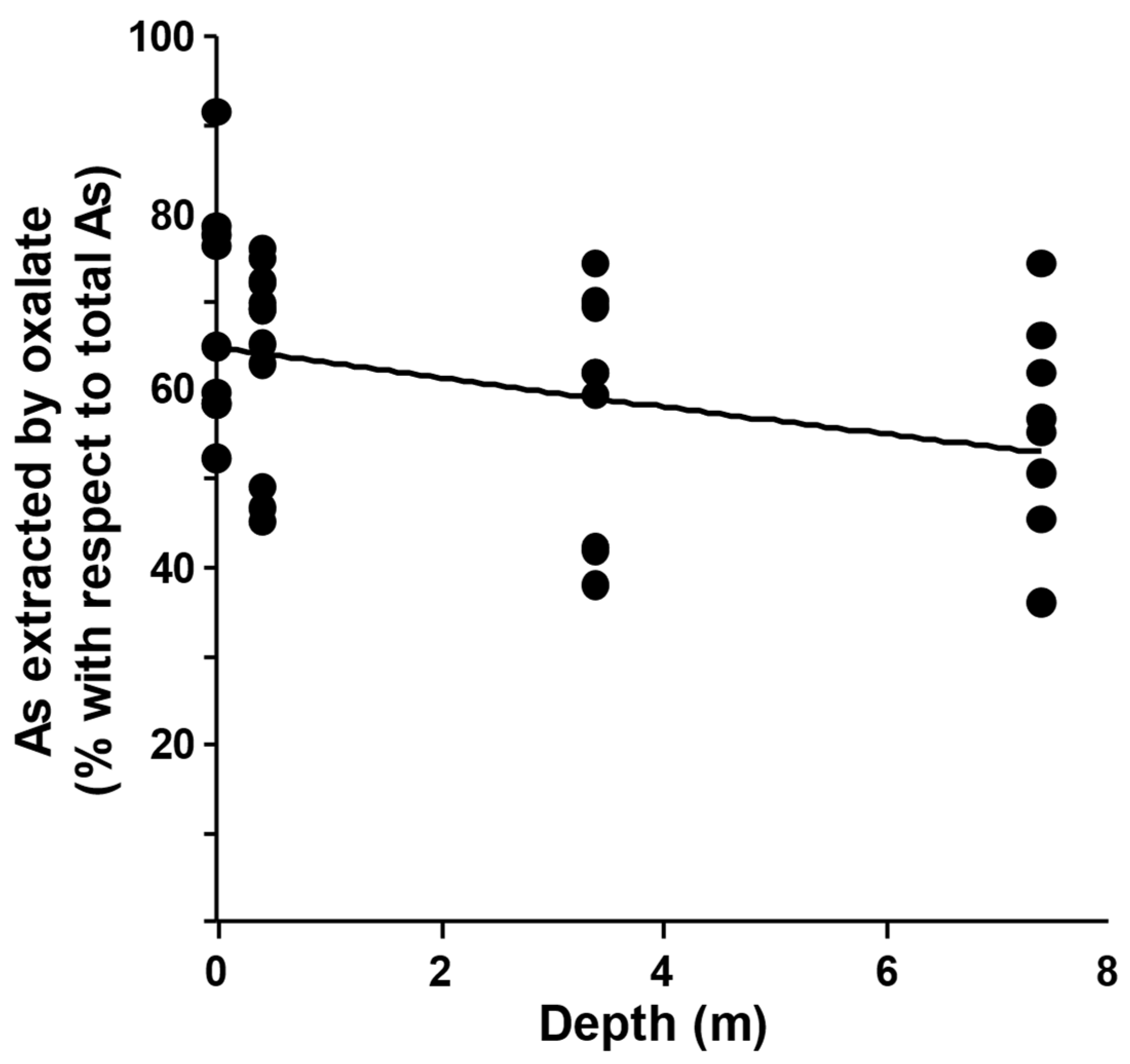
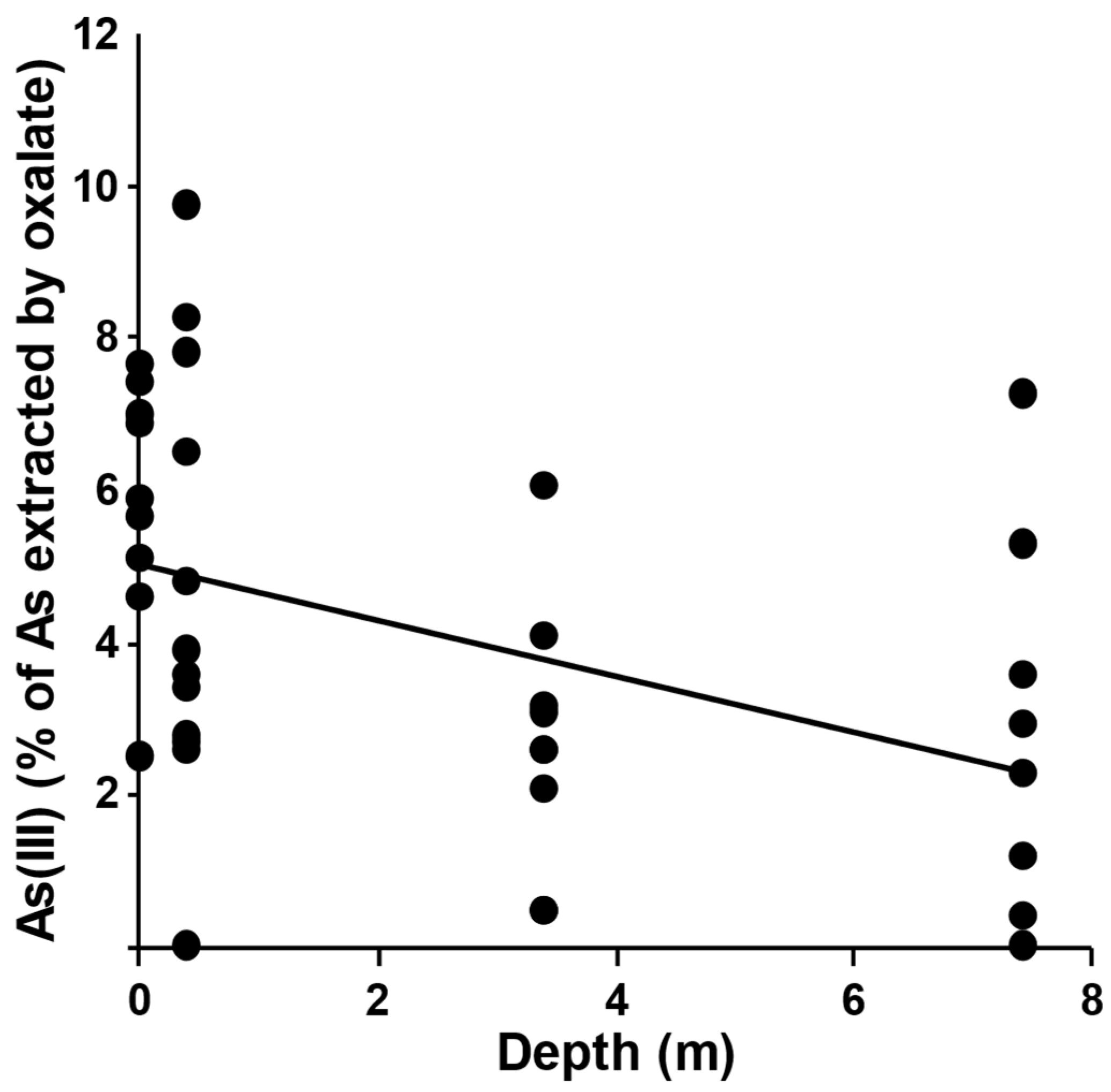
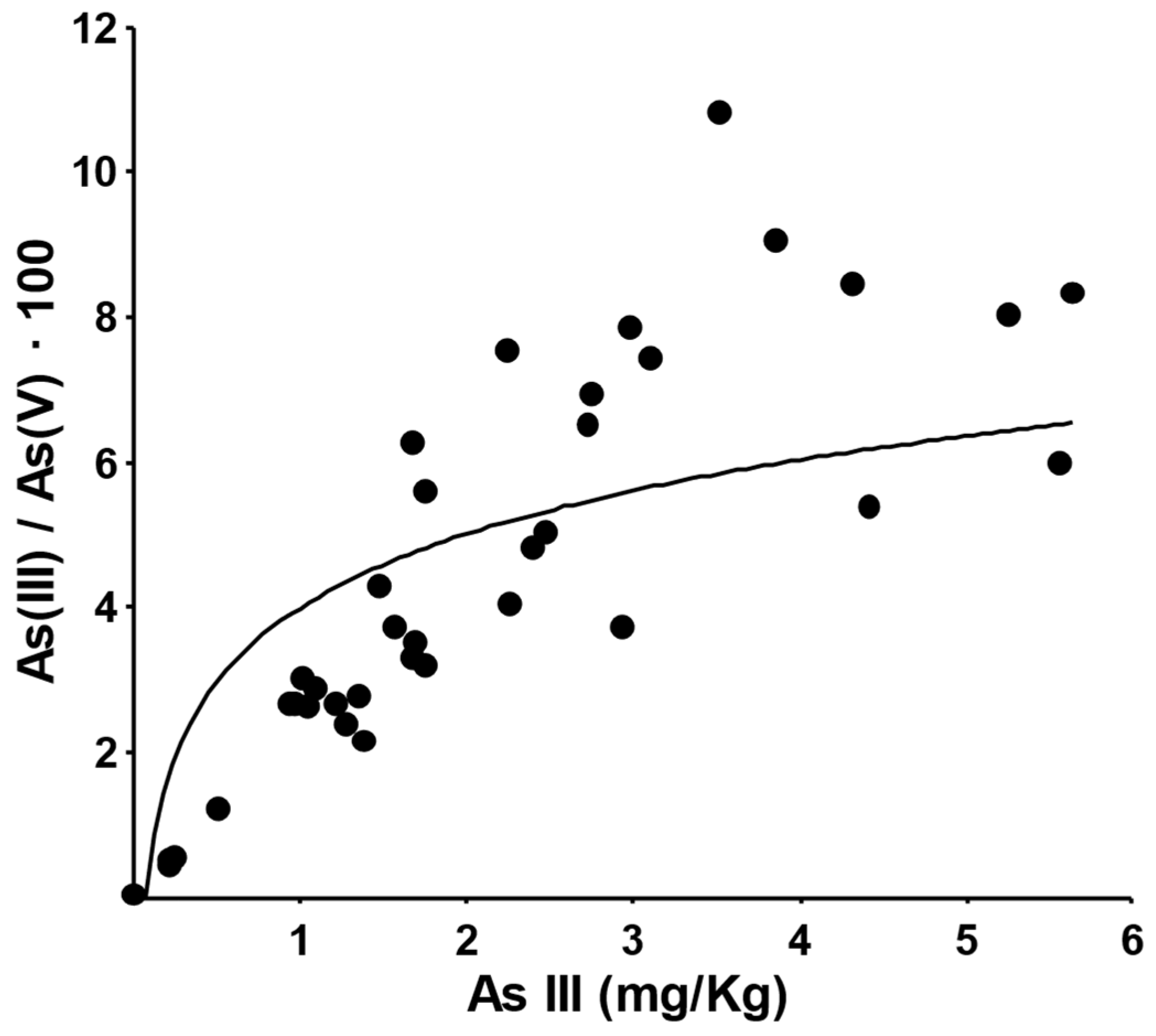
2.2. Ammonium Oxalate Extraction and As Speciation
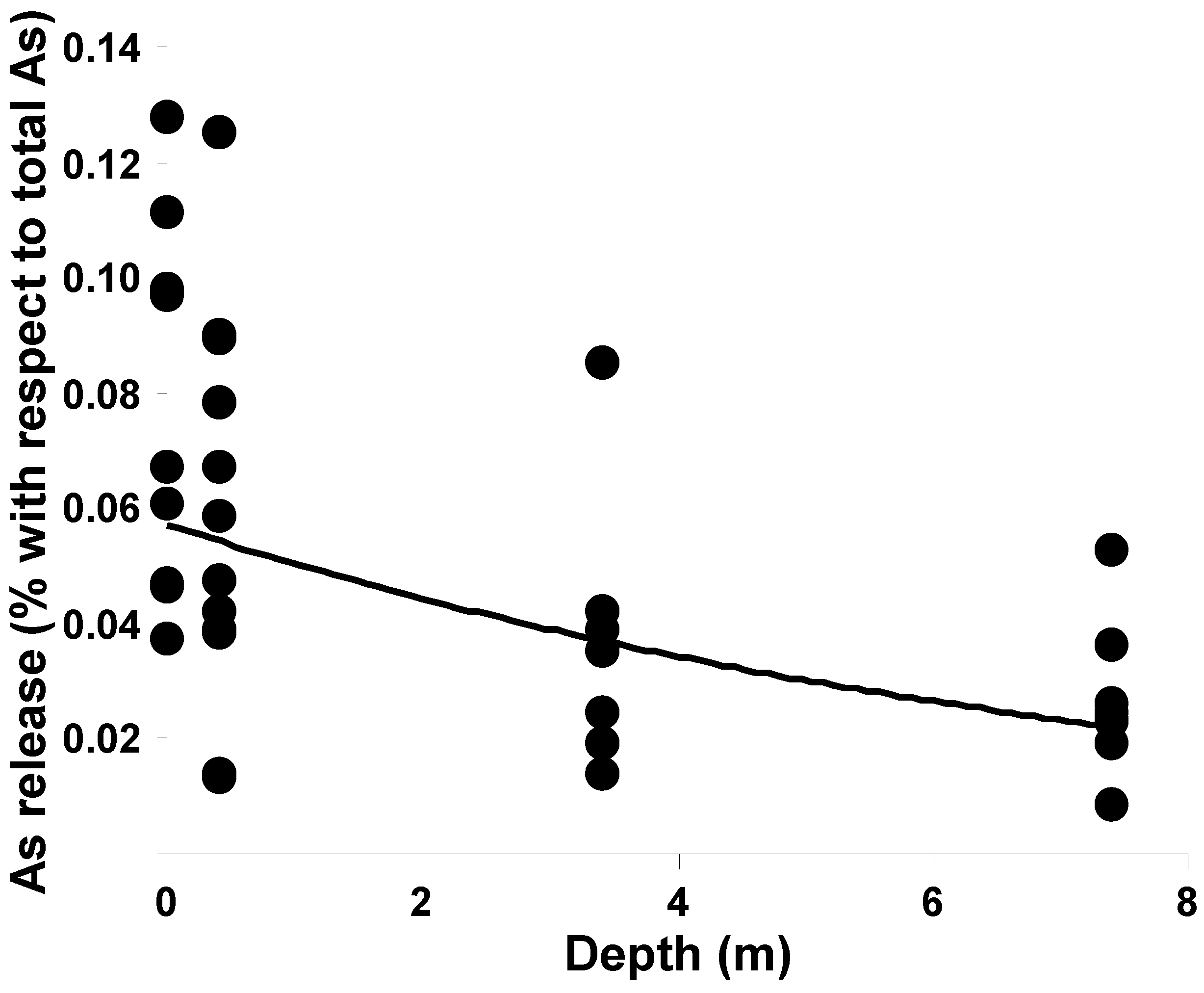
2.3. Cession Test
2.4. Comparison of Sites
3. Materials and Methods
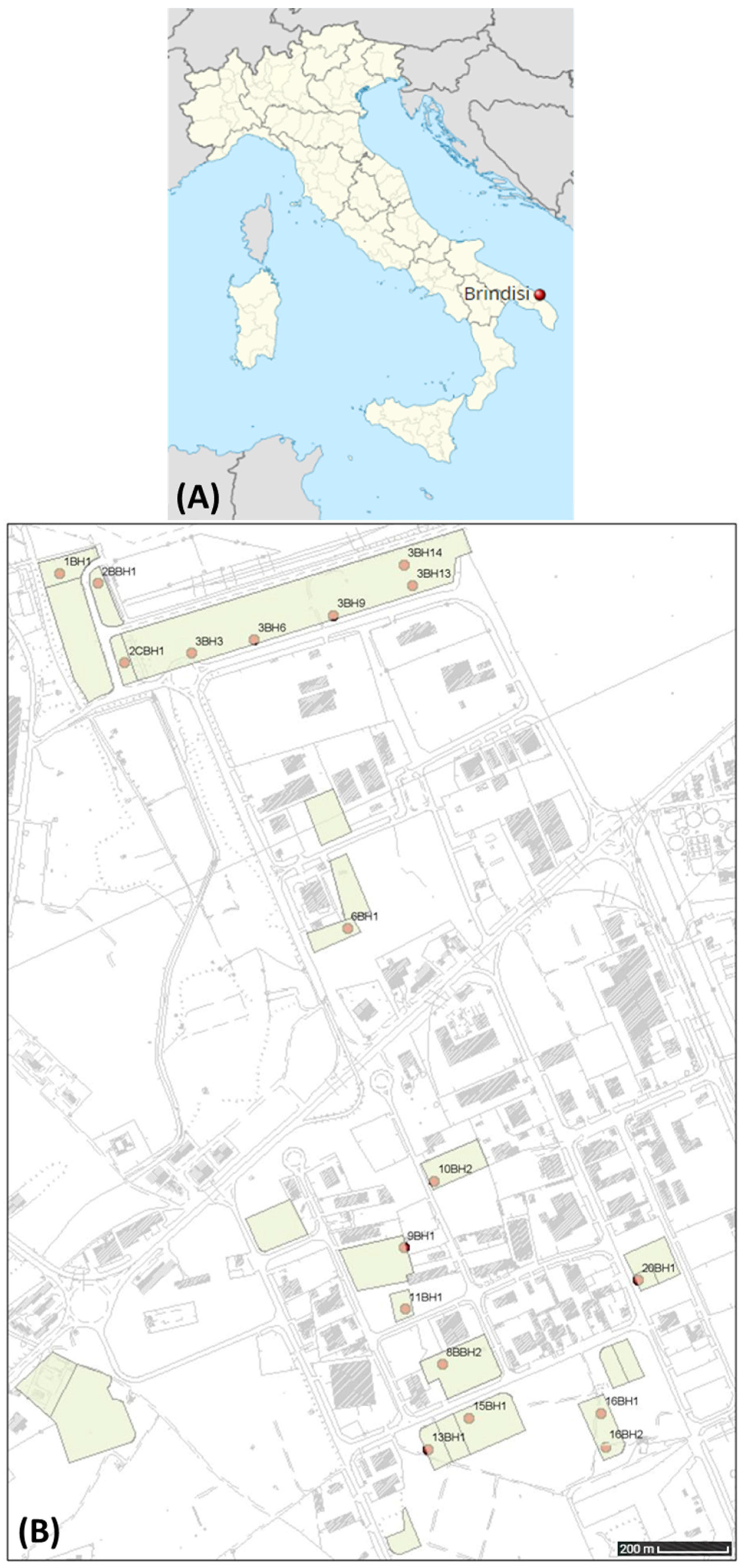
3.1. Sample Collection
3.2. Total As Determination
3.3. Ammonium Oxalate Extraction and As Speciation Analysis
3.4. Cession Test
- -
- Elution ratio of 1:20 (i.e., 50 g of sample per 1000 g of water);
- -
- Contact time of 24 h with moderate continuous stirring;
- -
- Analysis of the filtered solution (0.45 µm) carried out using atomic absorption spectrophotometry with a hydride generation system (HG-AAS).
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- National Research Council (US) Committee on Medical and Biological Effects of Environmental Pollutants. Arsenic: Medical and Biological Effects of Environmental Pollutants; The National Academies Press: Washington, DC, USA, 1977. [Google Scholar]
- Cullen, W.R.; Reimer, K.J. Arsenic Speciation in the Environment. Chem. Rev. 1989, 89, 713–764. [Google Scholar] [CrossRef]
- Cullen, W.R.; Liu, Q.; Lu, X.; McKnight-Whitford, A.; Peng, H.; Popowich, A.; Yan, X.; Zhang, Q.; Fricke, M.; Sun, H.; et al. Methylated and Thiolated Arsenic Species for Environmental and Health Research—A Review on Synthesis and Characterization. J. Environ. Sci. 2016, 49, 7–27. [Google Scholar] [CrossRef] [PubMed]
- McBride, M.B. Environmental Chemistry of Soils; Oxford University Press: Oxford, UK, 1994; ISBN 978-0-19-507011-8. [Google Scholar]
- Garelick, H.; Jones, H.; Dybowska, A.; Valsami-Jones, E. Arsenic Pollution Sources. In Reviews of Environmental Contamination Volume 197: International Perspectives on Arsenic Pollution and Remediation; Springer: New York, NY, USA, 2008; pp. 17–60. ISBN 978-0-387-79284-2. [Google Scholar]
- Mandal, B.K.; Suzuki, K.T. Arsenic Round the World: A Review. Talanta 2002, 58, 201–235. [Google Scholar] [CrossRef] [PubMed]
- Golfinopoulos, S.K.; Varnavas, S.P.; Alexakis, D.E. The Status of Arsenic Pollution in the Greek and Cyprus Environment: An Overview. Water 2021, 13, 224. [Google Scholar] [CrossRef]
- Nickson, R.T.; McArthur, J.M.; Ravenscroft, P.; Burgess, W.G.; Ahmed, K.M. Mechanism of Arsenic Release to Groundwater, Bangladesh and West Bengal. Appl. Geochem. 2000, 15, 403–413. [Google Scholar] [CrossRef]
- Shrivastava, A.; Ghosh, D.; Dash, A.; Bose, S. Arsenic Contamination in Soil and Sediment in India: Sources, Effects, and Remediation. Curr. Pollut. Rep. 2015, 1, 35–46. [Google Scholar] [CrossRef]
- Teixeira, M.C.; Santos, A.C.; Fernandes, C.S.; Ng, J.C. Arsenic Contamination Assessment in Brazil—Past, Present and Future Concerns: A Historical and Critical Review. Sci. Total Environ. 2020, 730, 138217. [Google Scholar] [CrossRef]
- Moore, J.W.; Ramamoorthy, S. Impact of Heavy Metals in Natural Waters. In Heavy Metals in Natural Waters; Springer: New York, NY, USA, 1984; pp. 205–233. [Google Scholar] [CrossRef]
- Smith, E.; Naidu, R.; Alston, A.M. Arsenic in the Soil Environment: A Review. In Advances in Agronomy; Sparks, D.L., Ed.; Academic Press: Cambridge, MA, USA, 1998; Volume 64, pp. 149–195. ISBN 0065-2113. [Google Scholar]
- Gleyzes, C.; Tellier, S.; Sabrier, R.; Astruc, M. Arsenic Characterisation in Industrial Soils by Chemical Extractions. Environ. Technol. 2001, 22, 27–38. [Google Scholar] [CrossRef]
- Zhao, C.; Yang, J.; Zheng, Y.; Yang, J.; Guo, G.; Wang, J.; Chen, T. Effects of Environmental Governance in Mining Areas: The Trend of Arsenic Concentration in the Environmental Media of a Typical Mining Area in 25 Years. Chemosphere 2019, 235, 849–857. [Google Scholar] [CrossRef]
- Wang, S.; Mulligan, C.N. Effect of Natural Organic Matter on Arsenic Release from Soilsand Sediments into Groundwater. Environ. Geochem. Health 2006, 28, 197–214. [Google Scholar] [CrossRef]
- Nriagu, J.O. Arsenic in the Environment. In Part I: Cycling and Characterization; John Wiley & Sons: New York, NY, USA, 1994. [Google Scholar]
- Pongratz, R. Arsenic Speciation in Environmental Samples of Contaminated Soil. Sci. Total Environ. 1998, 224, 133–141. [Google Scholar] [CrossRef]
- Moreda–Piñeiro, J.; Alonso-Rodríguez, E.; Romarís-Hortas, V.; Moreda-Piñeiro, A.; López-Mahía, P.; Muniategui-Lorenzo, S.; Prada-Rodríguez, D.; Bermejo-Barrera, P. Assessment of the Bioavailability of Toxic and Non-Toxic Arsenic Species in Seafood Samples. Food Chem. 2012, 130, 552–560. [Google Scholar] [CrossRef]
- Van Herreweghe, S.; Swennen, R.; Vandecasteele, C.; Cappuyns, V. Solid Phase Speciation of Arsenic by Sequential Extraction in Standard Reference Materials and Industrially Contaminated Soil Samples. Environ. Pollut. 2003, 122, 323–342. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.S.; Hoy, K.S.; Schofield, J.R.M.; Uppal, J.S.; Lin, Y.; Lu, X.; Peng, H.; Le, X.C. Arsenic Speciation Analysis: A Review with an Emphasis on Chromatographic Separations. TrAC Trends Anal. Chem. 2020, 123, 115770. [Google Scholar] [CrossRef]
- Rochette, E.A.; Li, G.C.; Fendorf, S.E. Stability of Arsenate Minerals in Soil under Biotically Generated Reducing Conditions. Soil Sci. Soc. Am. J. 1998, 62, 1530–1537. [Google Scholar] [CrossRef]
- Migoni, D.; Papadia, P.; Cannito, F.; Fanizzi, F.P. Sequential Extraction Analysis of Arsenic in Soil Samples Collected in an Agricultural Area of Brindisi, Apulia (Italy), in the Proximity of a Coal-Burning Power Plant. Appl. Sci. 2021, 11, 2115. [Google Scholar] [CrossRef]
- Gleyzes, C.; Tellier, S.; Astruc, M. Fractionation Studies of Trace Elements in Contaminated Soils and Sediments: A Review of Sequential Extraction Procedures. TrAC Trends Anal. Chem. 2002, 21, 451–467. [Google Scholar] [CrossRef]
- Gómez-Ariza, J.L.; Sánchez-Rodas, D.; Giráldez, I. Selective Extraction of Iron Oxide Associated Arsenic Species from Sediments for Speciation with Coupled HPLC-HG-AAS. J. Anal. At. Spectrom. 1998, 13, 1375–1379. [Google Scholar] [CrossRef]
- Styblo, M.; Del Razo, L.M.; Vega, L.; Germolec, D.R.; LeCluyse, E.L.; Hamilton, G.A.; Reed, W.; Wang, C.; Cullen, W.R.; Thomas, D.J. Comparative Toxicity of Trivalent and Pentavalent Inorganic and Methylated Arsenicals in Rat and Human Cells. Arch. Toxicol. 2000, 74, 289–299. [Google Scholar] [CrossRef]
- Ardini, F.; Dan, G.; Grotti, M. Arsenic Speciation Analysis of Environmental Samples. J. Anal. At. Spectrom. 2020, 35, 215–237. [Google Scholar] [CrossRef]
- Szpunar, J. Bio-Inorganic Speciation Analysis by Hyphenated Techniques. Analyst 2000, 125, 963–988. [Google Scholar] [CrossRef] [PubMed]
- Akter, K.F.; Owens, G.; Davey, D.E.; Naidu, R. Arsenic Speciation and Toxicity in Biological Systems. In Reviews of Environmental Contamination and Toxicology; Ware, G.W., Albert, L.A., Crosby, D.G., de Voogt, P., Hutzinger, O., Knaak, J.B., Mayer, F.L., Morgan, D.P., Park, D.L., Tjeerdema, R.S., et al., Eds.; Springer: New York, NY, USA, 2005; pp. 97–149. ISBN 978-0-387-27565-9. [Google Scholar]
- Patel, K.S.; Pandey, P.K.; Martín-Ramos, P.; Corns, W.T.; Varol, S.; Bhattacharya, P.; Zhu, Y. A Review on Arsenic in the Environment: Contamination, Mobility, Sources, and Exposure. RSC Adv. 2023, 13, 8803–8821. [Google Scholar] [CrossRef] [PubMed]
- Guerin, T.; Astruc, A.; Astruc, M. Speciation of Arsenic and Selenium Compounds by HPLC Hyphenated to Specific Detectors: A Review of the Main Separation Techniques. Talanta 1999, 50, 1–24. [Google Scholar] [CrossRef]
- Gong, Z.; Lu, X.; Cullen, W.R.; Chris Le, X. Unstable Trivalent Arsenic Metabolites, Monomethylarsonous Acid and Dimethylarsinous Acid. J. Anal. At. Spectrom. 2001, 16, 1409–1413. [Google Scholar] [CrossRef]
- National Research Council. Arsenic in Drinking Water; The National Academies Press: Washington, DC, USA, 1999; ISBN 978-0-309-06333-3. [Google Scholar]
- Guerin, T.; Molenat, N.; Astruc, A.; Pinel, R. Arsenic Speciation in Some Environmental Samples: A Comparative Study of HG-GC-QFAAS and HPLC-ICP-MS Methods. Appl. Organomet. Chem. 2000, 14, 401–410. [Google Scholar] [CrossRef]
- Montperrus, M.; Bohari, Y.; Bueno, M.; Astruc, A.; Astruc, M. Comparison of Extraction Procedures for Arsenic Speciation in Environmental Solid Reference Materials by High-Performance Liquid Chromatography–Hydride Generation-Atomic Fluorescence Spectroscopy. Appl. Organomet. Chem. 2002, 16, 347–354. [Google Scholar] [CrossRef]
- Ure, A.M.; Quevauviller, P.; Muntau, H.; Griepink, B. Speciation of Heavy Metals in Soils and Sediments. An Account of the Improvement and Harmonization of Extraction Techniques Undertaken Under the Auspices of the BCR of the Commission of the European Communities. Int. J. Environ. Anal. Chem. 1993, 51, 135–151. [Google Scholar] [CrossRef]
- Templeton, D.M.; Ariese, F.; Cornelis, R.; Danielsson, L.-G.; Muntau, H.; van Leeuwen, H.P.; Lobinski, R. Guidelines for Terms Related to Chemical Speciation and Fractionation of Elements. Definitions, Structural Aspects, and Methodological Approaches (IUPAC Recommendations 2000). Pure Appl. Chem. 2000, 72, 1453–1470. [Google Scholar] [CrossRef]
- Gong, Z.; Lu, X.; Ma, M.; Watt, C.; Le, X.C. Arsenic Speciation Analysis. Talanta 2002, 58, 77–96. [Google Scholar] [CrossRef]
- APAT—IRSA/CNR. Metodi Analitici per Le Acque; APAT—IRSA/CNR: Rome, Italy, 2003; Volume 29, ISBN 88-448-0083-7. [Google Scholar]
- Rodriguez, R.R.; Basta, N.T.; Casteel, S.W.; Armstrong, F.P.; Ward, D.C. Chemical Extraction Methods to Assess Bioavailable Arsenic in Soil and Solid Media. J. Environ. Qual. 2003, 32, 876–884. [Google Scholar] [CrossRef]
- Srithongkul, C.; Krongchai, C.; Santasup, C.; Kittiwachana, S. An Investigation of the Effect of Operational Conditions on a Sequential Extraction Procedure for Arsenic in Soil in Thailand. Chemosphere 2020, 242, 125230. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Blodau, C. Mobilization of Arsenic by Dissolved Organic Matter from Iron Oxides, Soils and Sediments. Sci. Total Environ. 2006, 354, 179–190. [Google Scholar] [CrossRef]
- Huang, J.-H.; Matzner, E. Mobile Arsenic Species in Unpolluted and Polluted Soils. Sci. Total Environ. 2007, 377, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Morosini, C.; Terzaghi, E.; Raspa, G.; Grotti, M.; Armiraglio, S.; Anelli, S.; Di Guardo, A. Arsenic Movement and Fractionation in Agricultural Soils Which Received Wastewater from an Adjacent Industrial Site for 50 Years. Sci. Total Environ. 2023, 898, 165422. [Google Scholar] [CrossRef] [PubMed]
- Billmann, M.; Hulot, C.; Pauget, B.; Badreddine, R.; Papin, A.; Pelfrêne, A. Oral Bioaccessibility of PTEs in Soils: A Review of Data, Influencing Factors and Application in Human Health Risk Assessment. Sci. Total Environ. 2023, 896, 165263. [Google Scholar] [CrossRef]
- Mir, K.A.; Rutter, A.; Koch, I.; Smith, P.; Reimer, K.J.; Poland, J.S. Extraction and Speciation of Arsenic in Plants Grown on Arsenic Contaminated Soils. Talanta 2007, 72, 1507–1518. [Google Scholar] [CrossRef]
- Su, R.; Ou, Q.; Wang, H.; Dai, X.; Chen, Y.; Luo, Y.; Yao, H.; Ouyang, D.; Li, Z.; Wang, Z. Organic–Inorganic Composite Modifiers Enhance Restoration Potential of Nerium Oleander L. to Lead–Zinc Tailing: Application of Phytoremediation. Environ. Sci. Pollut. Res. 2023, 30, 56569–56579. [Google Scholar] [CrossRef]
- Cornara, D.; Cavalieri, V.; Dongiovanni, C.; Altamura, G.; Palmisano, F.; Bosco, D.; Porcelli, F.; Almeida, R.P.P.; Saponari, M. Transmission of Xylella Fastidiosa by Naturally Infected Philaenus Spumarius (Hemiptera, Aphrophoridae) to Different Host Plants. J. Appl. Entomol. 2017, 141, 80–87. [Google Scholar] [CrossRef]
- Papadia, P.; Barozzi, F.; Angilé, F.; Migoni, D.; Piro, G.; Fanizzi, F.P.; Di Sansebastiano, G.-P. Evaluation of Dittrichia Viscosa Performance in Substrates with Moderately Low Levels of As and Cd Contamination. Plant Biosyst. Int. J. Deal. All Asp. Plant Biol. 2020, 154, 983–989. [Google Scholar] [CrossRef]
- Barozzi, F.; Papadia, P.; Stefano, G.; Renna, L.; Brandizzi, F.; Migoni, D.; Fanizzi, F.P.; Piro, G.; Di Sansebastiano, G.-P. Variation in Membrane Trafficking Linked to SNARE AtSYP51 Interaction with Aquaporin NIP1;1. Front. Plant Sci. 2019, 9, 1949. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Depth (m) | As(III) (mg/kg) | As(V) (mg/kg) | MMAA (mg/kg) | DMAA (mg/kg) | Speciation Total As (mg/kg) | Total As (mg/Kg) | Cession Test As (μg/L) | Speciation Total As/Total As (%) | Cession Test As/Total As (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1BH1/0.0–0.1 | 0 | 2.24 | 29.76 | <0.11 | <0.15 | 32 | 53.5 | 1.26 | 60 | 0.0471 |
| 2BBH1/1 | 0.4 | 2.47 | 49.06 | <0.11 | <0.15 | 51.53 | 78.9 | 2.65 | 65 | 0.0672 |
| 2BBH1/4 | 3.4 | 0.26 | 50.68 | <0.11 | <0.15 | 50.94 | 72.6 | 1.53 | 70 | 0.0421 |
| 2BBH1/8 | 7.4 | 1.01 | 33.51 | <0.11 | <0.15 | 34.52 | 55.7 | 0.72 | 62 | 0.0259 |
| 2CBH1/1 | 0 | 5.57 | 93.3 | <0.11 | <0.15 | 98.87 | 169.6 | 8.31 | 58 | 0.0980 |
| 3BH3/1 | 0.4 | 4.32 | 51.05 | <0.11 | <0.15 | 55.37 | 118.3 | 2.29 | 47 | 0.0387 |
| 3BH3/4 | 3.4 | 2.73 | 42.05 | <0.11 | <0.15 | 44.78 | 106.9 | 0.74 | 42 | 0.0138 |
| 3BH3/8 | 7.4 | 0.21 | 49.98 | <0.11 | <0.15 | 50.19 | 99.1 | 1.13 | 51 | 0.0228 |
| 3BH6/8 | 7.4 | 1.57 | 42.02 | <0.11 | <0.15 | 43.59 | 65.9 | 0.81 | 66 | 0.0246 |
| 3BH9/1 | 0.4 | 0.01 | 38.44 | <0.11 | <0.15 | 38.45 | 61 | 3.81 | 63 | 0.1249 |
| 3BH13/1 | 0.4 | 3.87 | 42.74 | <0.11 | <0.15 | 46.61 | 62.2 | 2.79 | 75 | 0.0897 |
| 3BH13/4 | 3.4 | 1.48 | 34.58 | <0.11 | <0.15 | 36.06 | 58.2 | 2.48 | 62 | 0.0852 |
| 3BH14/1 | 0.4 | 2.26 | 55.71 | <0.11 | <0.15 | 57.97 | 128.5 | 5.74 | 45 | 0.0893 |
| 3BH14/4 | 3.4 | 1.76 | 55.01 | <0.11 | <0.15 | 56.77 | 134.2 | 2.35 | 42 | 0.0350 |
| 3BH14/8 | 7.4 | 0.51 | 42.08 | <0.11 | <0.15 | 42.59 | 118.7 | 2.14 | 36 | 0.0361 |
| 6BH1/0.0–0.1 | 0 | 5.25 | 65.44 | <0.11 | <0.15 | 70.69 | 77.1 | 3.73 | 92 | 0.0968 |
| 6BH1/1 | 0.4 | 1.35 | 48.57 | <0.11 | <0.15 | 49.92 | 65.7 | 1.26 | 76 | 0.0384 |
| 8BBH2/1 | 0 | 1.06 | 40.82 | <0.11 | <0.15 | 41.88 | 71.8 | 3.99 | 58 | 0.1111 |
| 8BBH2/4 | 3.4 | 0.21 | 41.21 | <0.11 | <0.15 | 41.42 | 66.7 | 0.64 | 62 | 0.0192 |
| 9BH1/1 | 0.4 | 1.09 | 38.03 | <0.11 | <0.15 | 39.12 | 54.2 | 1.28 | 72 | 0.0472 |
| 10BH2/0.0–0.1 | 0 | 3.11 | 41.92 | <0.11 | <0.15 | 45.03 | 57.3 | 1.07 | 79 | 0.0373 |
| 10BH2/1 | 0.4 | 2.75 | 39.64 | <0.11 | <0.15 | 42.39 | 61.2 | 0.41 | 69 | 0.0134 |
| 11BH1/0.0–0.1 | 0 | 1.67 | 26.64 | <0.11 | <0.15 | 28.31 | 54.1 | 1.64 | 52 | 0.0606 |
| 11BH1/1 | 0.4 | 1.22 | 45.74 | <0.11 | <0.15 | 46.96 | 96 | 2.82 | 49 | 0.0588 |
| 11BH1/4 | 3.4 | 0.97 | 36.19 | <0.11 | <0.15 | 37.16 | 98.1 | 1.73 | 38 | 0.0353 |
| 11BH1/8 | 7.4 | 1.76 | 31.43 | <0.11 | <0.15 | 33.19 | 72.8 | 1.92 | 46 | 0.0527 |
| 13BH1/1 | 0.4 | 3.53 | 32.59 | <0.11 | <0.15 | 36.12 | 51.8 | 0.36 | 70 | 0.0139 |
| 15BH1/0.0–0.1 | 0 | 4.42 | 82.01 | <0.11 | <0.15 | 86.43 | 113.4 | 2.62 | 76 | 0.0462 |
| 15BH1/1 | 0.4 | 2.93 | 78.44 | <0.11 | <0.15 | 81.37 | 117.4 | 4.58 | 69 | 0.0780 |
| 15BH1/4 | 3.4 | 1.39 | 65.18 | <0.11 | <0.15 | 66.57 | 112.3 | 1.36 | 59 | 0.0242 |
| 15BH1/8 | 7.4 | 0.01 | 39.76 | <0.11 | <0.15 | 39.77 | 71.9 | 0.69 | 55 | 0.0192 |
| 16BH1/4 | 3.4 | 0.94 | 35.21 | <0.11 | <0.15 | 36.15 | 52.1 | 0.92 | 69 | 0.0353 |
| 16BH1/8 | 7.4 | 2.99 | 37.99 | <0.11 | <0.15 | 40.98 | 55.2 | 0.64 | 74 | 0.0232 |
| 16BH2/0.0–0.1 | 0 | 5.64 | 67.78 | <0.11 | <0.15 | 73.42 | 94.8 | 3.19 | 77 | 0.0673 |
| 16BH2/1 | 0.4 | 1.69 | 48.12 | <0.11 | <0.15 | 49.81 | 68.9 | 1.44 | 72 | 0.0418 |
| 16BH2/4 | 3.4 | 1.68 | 51.16 | <0.11 | <0.15 | 52.84 | 71.1 | 1.38 | 74 | 0.0388 |
| 16BH2/8 | 7.4 | 1.29 | 54.95 | <0.11 | <0.15 | 56.24 | 99 | 0.43 | 57 | 0.0087 |
| 20BH1/1 | 0 | 2.4 | 49.78 | <0.11 | <0.15 | 52.18 | 80.3 | 5.12 | 65 | 0.1275 |
| HPLC: Ion Exchange Column Hamilton PRP-X100 (250 × 4.6 mm) with Pre-Column | |
|---|---|
| Mobile phase | Sol. A: ammonium phosphate (NH4H2PO4/(NH4)2HPO4) 5 mM, pH 8.0 Sol. B: ammonium phosphate (NH4H2PO4/(NH4)2HPO4) 30 mM, pH 8.0 |
| Injected volume | 200 µL |
| Flow rate | 1 mL min−1 |
| Elution gradient | from 0 to 4 min, Sol A from 4 to 10 min, Sol B from 10 to 20 min, Sol A |
| Column temperature | Room temperature (~25 °C) |
| Hydride Generation | |
| Argon flow | 40 mL min−1 |
| Reducing agent | NaBH4 0.16 M |
| Reducing agent flow | 1 mL min−1 |
| Acid | HCl 6 M |
| Acid flow | 1 mL min−1 |
| Atomic Adsorption Spectrometer | |
| Lamp | hollow cathode lamp |
| Wavelength | 193.7 nm |
| Slit width | 0.5 nm |
| Cell temperature | 925 °C (electrothermal temperature controller) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Migoni, D.; Papadia, P.; Fanizzi, F.P. Quantification of Arsenic in Soil Samples Collected in an Industrial Area of Brindisi (Apulia, Italy): Speciation Analysis and Availability. Sustainability 2023, 15, 14666. https://doi.org/10.3390/su152014666
Migoni D, Papadia P, Fanizzi FP. Quantification of Arsenic in Soil Samples Collected in an Industrial Area of Brindisi (Apulia, Italy): Speciation Analysis and Availability. Sustainability. 2023; 15(20):14666. https://doi.org/10.3390/su152014666
Chicago/Turabian StyleMigoni, Danilo, Paride Papadia, and Francesco Paolo Fanizzi. 2023. "Quantification of Arsenic in Soil Samples Collected in an Industrial Area of Brindisi (Apulia, Italy): Speciation Analysis and Availability" Sustainability 15, no. 20: 14666. https://doi.org/10.3390/su152014666