Soybean Oil-Derived Poly-Unsaturated Fatty Acids Enhance Liver Damage in NAFLD Induced by Dietary Cholesterol

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
3. Results
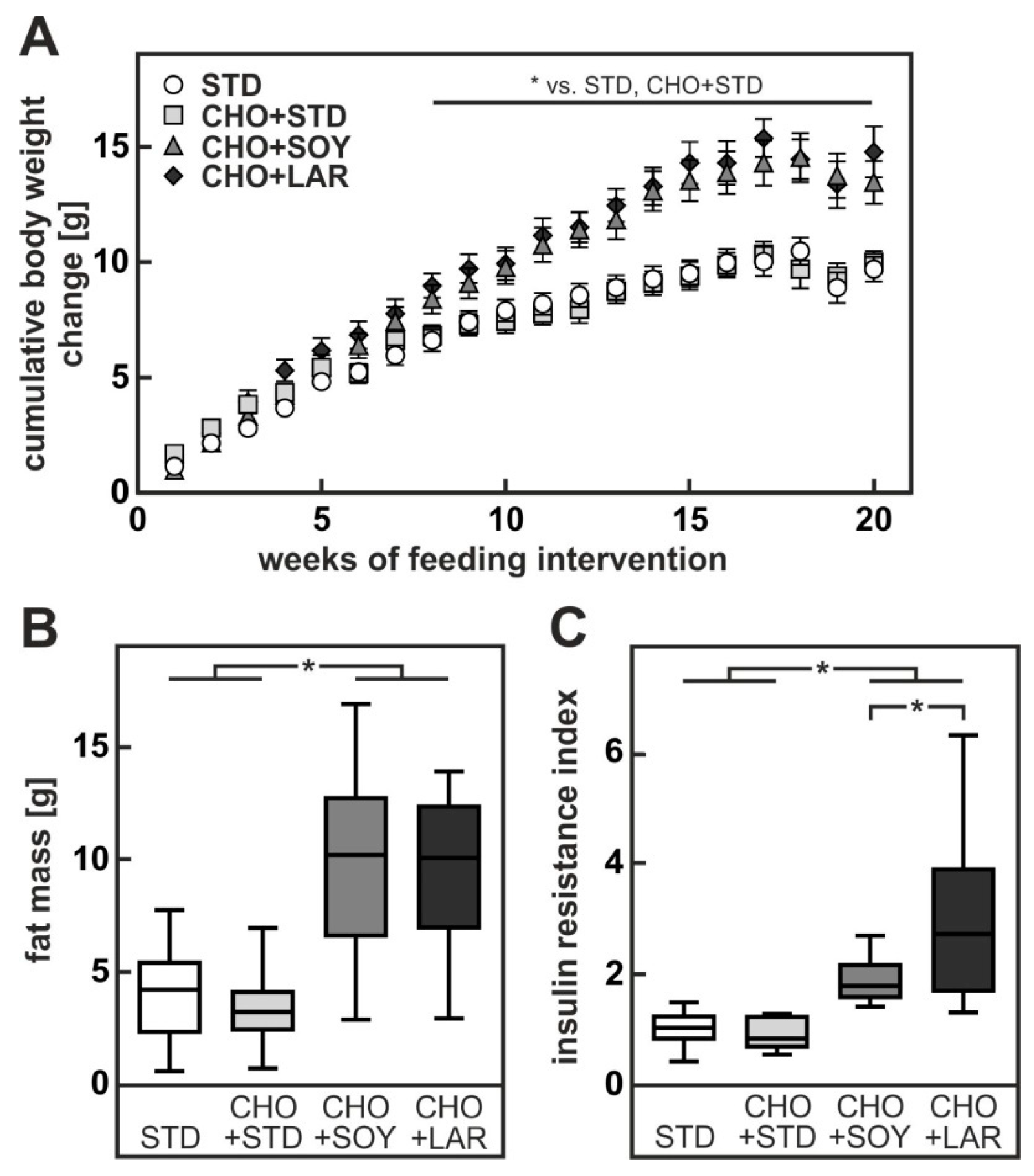
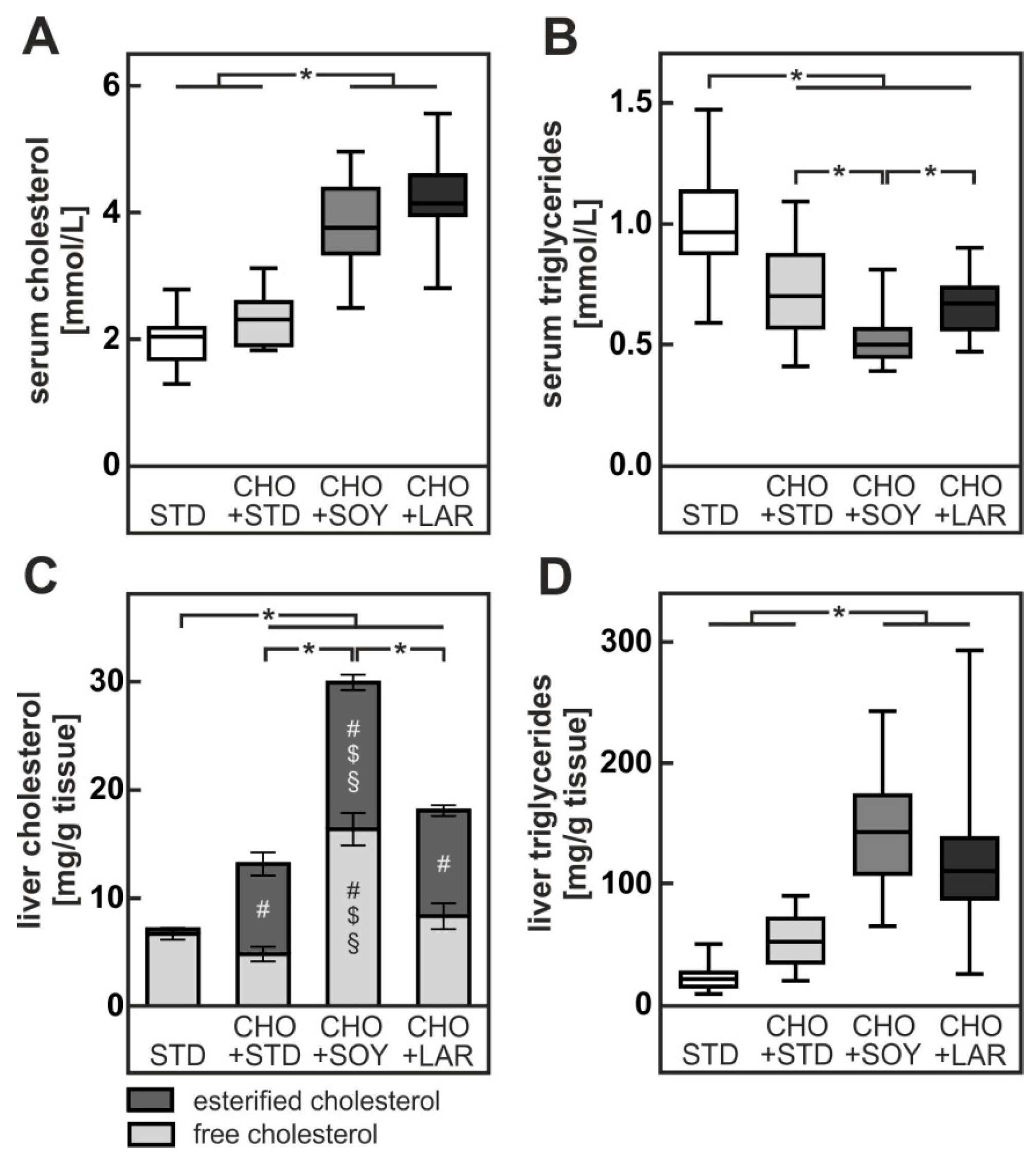
3.1. Diet-Induced Weight Gain, Insulin Resistance and NAFLD
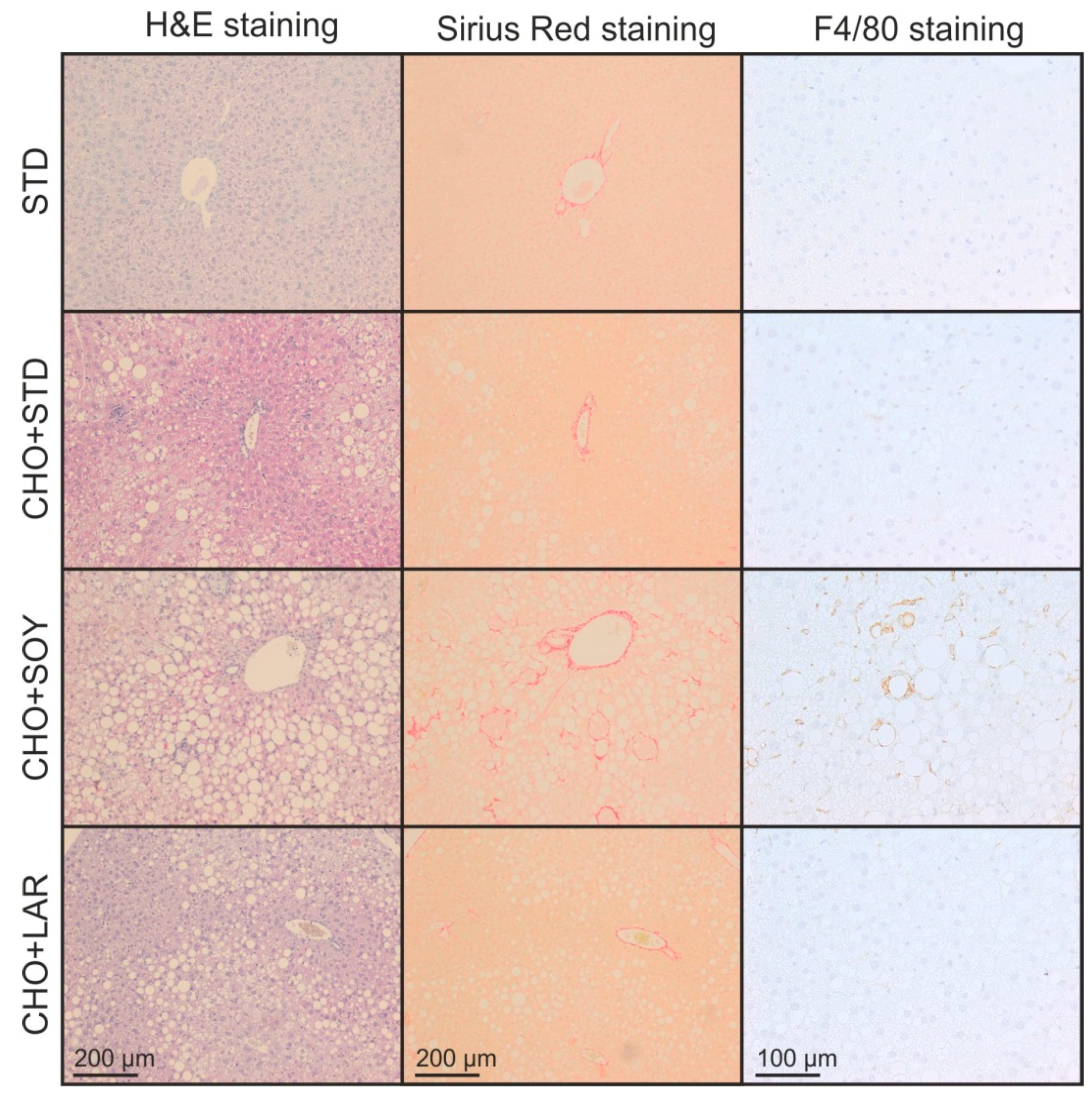
3.2. Diet-Induced Inflammation and Fibrosis
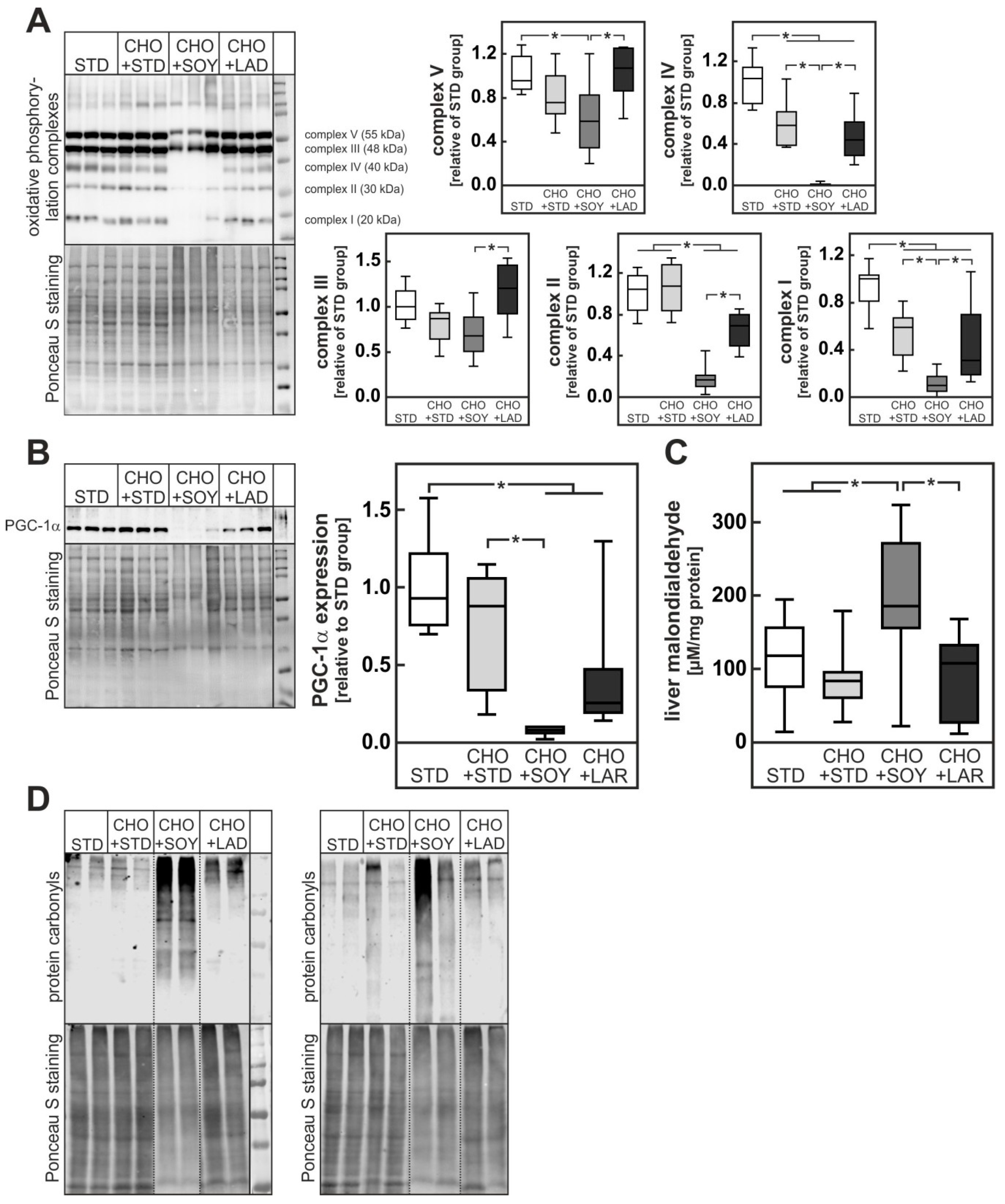
3.3. Diet-Induced Mitochondrial Damage and Oxidative Stress
3.4. Oxidative Stress Preceding Inflammation
4. Discussion
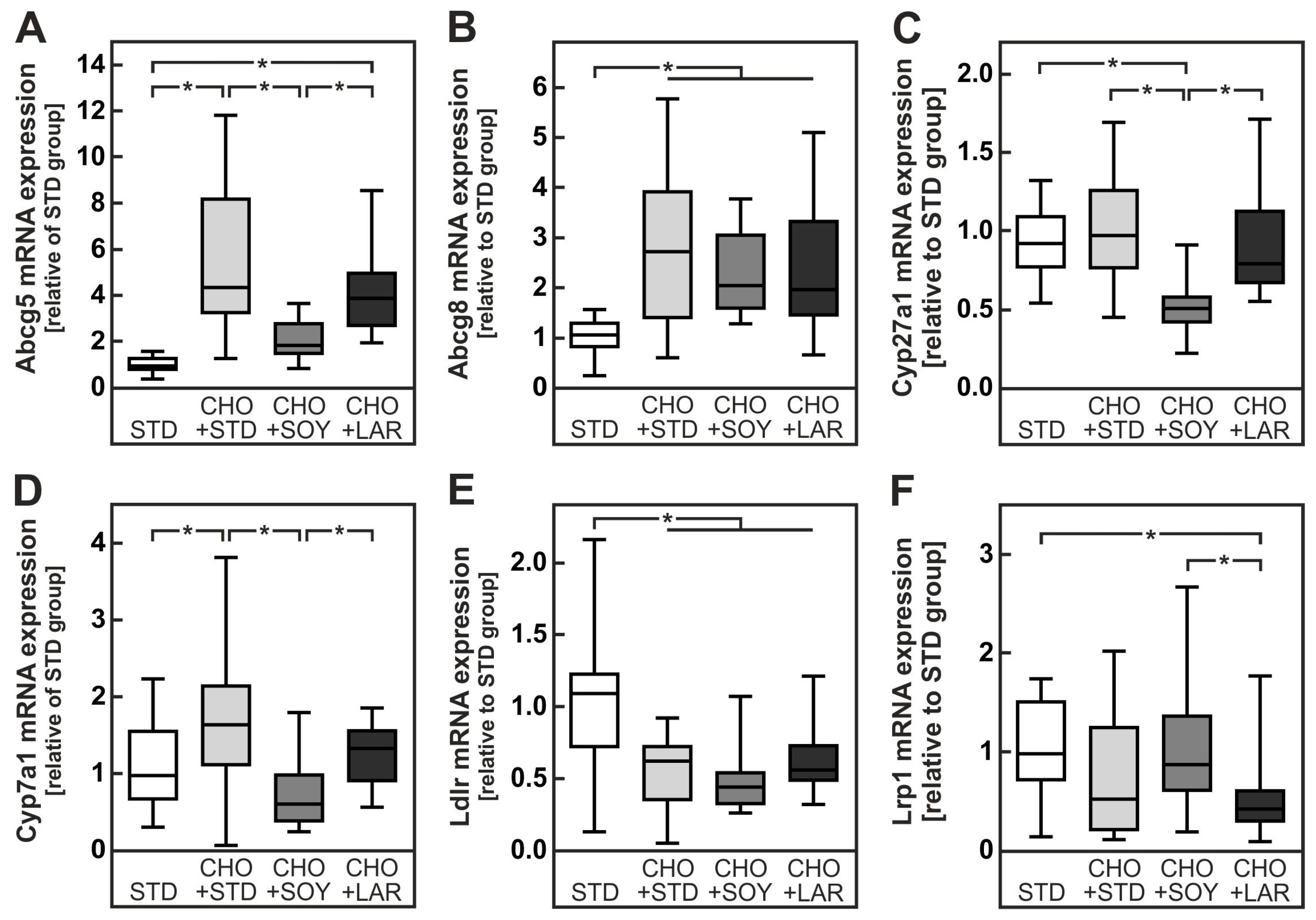
4.1. PUFA-Dependent Increase in Cholesterol Accumulation
4.2. PUFA-Dependent Enhancement of Oxidative Stress
4.3. Possible Clinical Impact
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, Y.T.; Laxton, V.; Lin, H.Y.; Chan, Y.W.F.; Fitzgerald-Smith, S.; To, T.L.O.; Yan, B.P.; Liu, T.; Tse, G. Animal models of atherosclerosis. Biomed. Rep. 2017, 6, 259–266. [Google Scholar] [CrossRef] [PubMed]
- McNamara, D.J. Dietary cholesterol, heart disease risk and cognitive dissonance. Pro. Nut. Soc. 2014, 73, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.; Raman, G.; Vishwanathan, R.; Jacques, P.F.; Johnson, E.J. Dietary cholesterol and cardiovascular disease: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2015, 102, 276–294. [Google Scholar] [CrossRef] [PubMed]
- Brownawell, A.M.; Falk, M.C. Cholesterol: Where science and public health policy intersect. Nutr. Rev. 2010, 68, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Vinué, Á.; Herrero-Cervera, A.; González-Navarro, H. Understanding the impact of dietary cholesterol on chronic metabolic diseases through studies in rodent models. Nutrients 2018, 10, 939. [Google Scholar] [CrossRef] [PubMed]
- Jahn, D.; Kircher, S.; Hermanns, H.M.; Geier, A. Animal models of NAFLD from a hepatologist’s point of view. BBA-Mol. Basis. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Andronescu, C.I.; Purcarea, M.R.; Babes, P.A. Nonalcoholic fatty liver disease: Epidemiology, pathogenesis and therapeutic implications. J. Med. Life 2018, 11, 20–23. [Google Scholar] [PubMed]
- Brunt, E.M.; Wong, V.W.S.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Nonalcoholic fatty liver disease. Nat. Rev. Dis. Primers 2015, 1, 15080. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Goodspeed, L.; Wang, S.; Kim, J.; Zeng, L.; Ioannou, G.N.; Haigh, W.G.; Yeh, M.M.; Kowdley, K.V.; O’Brien, K.D.; et al. Dietary cholesterol exacerbates hepatic steatosis and inflammation in obese LDL receptor-deficient mice. J. Lipid Res. 2011, 52, 1626–1635. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.; Coleman, C.D.; Schraplau, A.; Jӧhrens, K.; Weber, D.; Castro, J.P.; Hugo, M.; Schulz, T.J.; Krämer, S.; Schürmann, A.; et al. Induction of steatohepatitis (NASH) with insulin resistance in wildtype B6 mice by a western-type diet containing soybean oil and cholesterol. Mol. Med. 2017, 23, 70. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N.; Subramanian, S.; Chait, A.; Haigh, W.G.; Yeh, M.M.; Farrell, G.C.; Lee, S.P.; Savard, C. Cholesterol crystallization within hepatocyte lipid droplets and its role in murine NASH. J. Lipid Res. 2017, 58, 1067–1079. [Google Scholar] [CrossRef] [PubMed]
- Buettner, R.; Ascher, M.; Gäbele, E.; Hellerbrand, C.; Kob, R.; Bertsch, T.; Bollheimer, L.C. Olive oil attenuates the cholesterol-induced development of nonalcoholic steatohepatitis despite increased insulin resistance in a rodent model. Horm. Metab. Res. 2013, 45, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.H.; Feigh, M.; Veidal, S.S.; Rigbolt, K.T.; Vrang, N.; Fosgerau, K. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov. Today 2017, 22, 1707–1718. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ren, Q.; Wu, T.; Guo, Y.; Liang, Y.; Liu, S. Ezetimibe prevents the development of non-alcoholic fatty liver disease induced by high-fat diet in C57BL/6J mice. Mol. Med. Rep. 2014, 10, 2917–2923. [Google Scholar] [CrossRef] [PubMed]
- Nakade, Y.; Murotani, K.; Inoue, T.; Kobayashi, Y.; Yamamoto, T.; Ishii, N.; Ohashi, T.; Ito, K.; Fukuzawa, Y.; Yoneda, M. Ezetimibe for the treatment of non-alcoholic fatty liver disease: A meta-analysis. Hepatol. Res. 2017, 47, 1417–1428. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, O. Hypoxic signaling and cholesterol lipotoxicity in fatty liver disease progression. Oxid. Med. Cell. Longev. 2018, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Siedel, J.; Rollinger, W.; Röschlau, P.; Ziegenhorn, J.; Bergmeyer, H.U. Total cholesterol, end-point and kinetic method. In Methods of Enzymatic Analysis; Verlag Chemie GmbH: Weinheim, Germany, 1984; pp. 139–148. [Google Scholar]
- Kleiner, D.E.; Brunt, E.M.; van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.-C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Menke, A.L.; Driessen, A.; Koek, G.H.; Lindeman, J.H.; Stoop, R.; Havekes, L.M.; Kleemann, R.; van den Hoek, A.M. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS ONE 2014, 9, e115922. [Google Scholar] [CrossRef] [PubMed]
- Manowsky, J.; Camargo, R.G.; Kipp, A.P.; Henkel, J.; Püschel, G.P. Insulin-induced cytokine production in macrophages causes insulin resistance in hepatocytes. Am. J. Physiol. Endocrin. Met. 2016, 310, E938–E946. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.; Neuschäfer-Rube, F.; Pathe-Neuschäfer-Rube, A.; Püschel, G.P. Aggravation by prostaglandin E2 of interleukin-6-dependent insulin resistance in hepatocytes. Hepatology 2009, 50, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.J.; Fan, J.G.; Ding, X.D.; Qiao, L.; Wang, G.L. Characterization of high-fat, diet-induced, non-alcoholic steatohepatitis with fibrosis in rats. Digest. Dis. Sci. 2010, 55, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, N.; Takamura, T.; Kurita, S.; Misu, H.; Ota, T.; Ando, H.; Yokoyama, M.; Honda, M.; Zen, Y.; Nakanuma, Y.; et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology 2007, 46, 1392–1403. [Google Scholar] [CrossRef] [PubMed]
- Wiggers, K.D.; Richard, M.J.; Stewart, J.W.; Jacobson, N.L.; Berger, P.J. Type and amount of dietary fat affect relative concentration of cholesterol in blood and other tissues of rats. Atherosclerosis 1977, 27, 27–34. [Google Scholar] [CrossRef]
- Sheril, A.; Jeyakumar, S.M.; Jayashree, T.; Giridharan, N.V.; Vajreswari, A. Impact of feeding polyunsaturated fatty acids on cholesterol metabolism of dyslipidemic obese rats of WNIN/GR-Ob strain. Atherosclerosis 2009, 204, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.; Benndorf, R.A. ABC transport proteins in cardiovascular disease—A brief summary. Molecules 2017, 22, 589. [Google Scholar] [CrossRef] [PubMed]
- Pizzini, A.; Lunger, L.; Demetz, E.; Hilbe, R.; Weiss, G.; Ebenbichler, C.; Tancevski, I. The role of omega-3 fatty acids in reverse cholesterol transport: A review. Nutrients 2017, 9, 1099. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C.; Lin, M.H.; Huang, P.C. A high cholesterol, (n-3) polyunsaturated fatty acid diet induces hypercholesterolemia more than a high cholesterol (n-6) polyunsaturated fatty acid diet in hamsters. J. Nutr. 1996, 126, 1759–1765. [Google Scholar] [PubMed]
- Wang, S.; Matthan, N.R.; Wu, D.; Reed, D.B.; Bapat, P.; Yin, X.; Grammas, P.; Shen, C.L.; Lichtenstein, A.H. Lipid content in hepatic and gonadal adipose tissue parallel aortic cholesterol accumulation in mice fed diets with different omega-6 PUFA to EPA plus DHA ratios. Clin. Nutr. 2014, 33, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Jump, D.B.; Depner, C.M.; Tripathy, S.; Lytle, K.A. Impact of dietary fat on the development of non-alcoholic fatty liver disease in Ldlr−/− mice. Proc. Nutr. Soc. 2016, 75, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shang, T.; Liu, L.; Zhou, J.; Zhang, M.; Hu, Q.; Fang, M.; Wu, Y.; Yao, P.; Gong, Z. Protective effects of various ratios of DHA/EPA supplementation on high-fat diet-induced liver damage in mice. Lipids Health Dis. 2017, 16, 65. [Google Scholar] [CrossRef] [PubMed]
- Pachikian, B.D.; Essaghir, A.; Demoulin, J.B.; Neyrinck, A.M.; Catry, E.; de Backer, F.C.; Dejeans, N.; Dewulf, E.M.; Sohet, F.M.; Portois, L.; et al. Hepatic n-3 polyunsaturated fatty acid depletion promotes steatosis and insulin resistance in mice: Genomic analysis of cellular targets. PLoS ONE 2011, 6, e23365. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.; Tomé, D.; Baars, A.; Hooiveld, G.J.; Müller, M. Dietary protein affects gene expression and prevents lipid accumulation in the liver in mice. PLoS ONE 2012, 7, e47303. [Google Scholar] [CrossRef] [PubMed]
- Legry, V.; van Rooyen, D.M.; Lambert, B.; Sempoux, C.; Poekes, L.; Español-Suñer, R.; Molendi-Coste, O.; Horsmans, Y.; Farrell, G.C.; Leclercq, I.A. Endoplasmic reticulum stress does not contribute to steatohepatitis in obese and insulin-resistant high-fat-diet-fed foz/foz mice. Clin. Sci. 2014, 127, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; García-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Schrauwen, P.; Schrauwen-Hinderling, V.; Hoeks, J.; Hesselink, M.K.C. Mitochondrial dysfunction and lipotoxicity. BBA-Mol. Cell Biol. Lipids 2010, 1801, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Bellanti, F.; Mitarotonda, D.; Tamborra, R.; Blonda, M.; Iannelli, G.; Petrella, A.; Sanginario, V.; Iuliano, L.; Vendemiale, G.; Serviddio, G. Oxysterols induce mitochondrial impairment and hepatocellular toxicity in non-alcoholic fatty liver disease. Free Radic. Biol. Med. 2014, 75, S16–S17. [Google Scholar] [CrossRef] [PubMed]
- Vejux, A.; Guyot, S.; Montange, T.; Riedinger, J.M.; Kahn, E.; Lizard, G. Phospholipidosis and down-regulation of the PI3-K/PDK-1/Akt signalling pathway are vitamin E inhibitable events associated with 7-ketocholesterol-induced apoptosis. J. Nutr. Biochem. 2009, 20, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, T.; Hyogo, H.; Honda, A.; Miyazaki, T.; Tokushige, K.; Hashimoto, E.; Inui, K.; Matsuzaki, Y.; Tazuma, S. Increased serum liver X receptor ligand oxysterols in patients with non-alcoholic fatty liver disease. J. Gastroenterol. 2012, 47, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Serviddio, G.; Blonda, M.; Bellanti, F.; Villani, R.; Iuliano, L.; Vendemiale, G. Oxysterols and redox signaling in the pathogenesis of non-alcoholic fatty liver disease. Free Radic. Res. 2013, 47, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N.; Haigh, W.G.; Thorning, D.; Savard, C. Hepatic cholesterol crystals and crown-like structures distinguish NASH from simple steatosis. J. Lipid Res. 2013, 54, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepat. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Ferré, N.; Martínez-Clemente, M.; López-Parra, M.; González-Périz, A.; Horrillo, R.; Planagumà, A.; Camps, J.; Joven, J.; Tres, A.; Guardiola, F.; et al. Increased susceptibility to exacerbated liver injury in hypercholesterolemic ApoE-deficient mice: Potential involvement of oxysterols. Am. J. Physiol. Gastr. Liver Physiol. 2009, 296, G553–G562. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N.; Morrow, O.B.; Connole, M.L.; Lee, S.P. Association between dietary nutrient composition and the incidence of cirrhosis or liver cancer in the United States population. Hepatology 2009, 50, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; de Michieli, F.; Cassader, M.; Rizzetto, M.; Durazzo, M.; Fagà, E.; Silli, B.; Pagano, G. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology 2003, 37, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Yasutake, K.; Nakamuta, M.; Shima, Y.; Ohyama, A.; Masuda, K.; Haruta, N.; Fujino, T.; Aoyagi, Y.; Fukuizumi, K.; Yoshimoto, T.; et al. Nutritional investigation of non-obese patients with non-alcoholic fatty liver disease: The significance of dietary cholesterol. Scand. J. Gastroenterol. 2009, 44, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Jun, D.W.; Kim, H.J.; Oh, H.; Saeed, W.K.; Ahn, H.; Cheung, R.C.; Nguyen, M.H. Ezetimibe decreased nonalcoholic fatty liver disease activity score but not hepatic steatosis. Korean J. Int. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- He, X.X.; Wu, X.L.; Chen, R.P.; Chen, C.; Liu, X.G.; Wu, B.J.; Huang, Z.M. Effectiveness of omega-3 polyunsaturated fatty acids in non-alcoholic fatty liver disease: A meta-analysis of randomized controlled trials. PLoS ONE 2016, 11, e0162368. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| STD | CHO + STD | CHO + SOY | CHO + LAR | |
|---|---|---|---|---|
| Metabolizing energy (kcal/g) | 3.06 | 3.06 | 4.64 | 4.73 |
| Energy from carbohydrates (%) | 65 | 65 | 35 | 35 |
| Energy from protein (%) | 25 | 25 | 16 | 20 |
| Energy from fat (%) | 10 | 10 | 49 | 45 |
| Cholesterol (%) | 0.00 | 0.75 | 0.75 | 0.75 |
| Fatty acid composition | ||||
| Saturated fatty acids (g/100g) | 0.55 | 0.55 | 4.00 | 7.26 |
| Mono-unsaturated fatty acids (g/100g) | 0.64 | 0.64 | 5.75 | 8.69 |
| Poly-unsaturated fatty acids (g/100g) | 2.01 | 2.01 | 14.50 | 6.36 |
| Scoring Parameter | STD | CHO + STD | CHO + SOY | CHO + LAR |
|---|---|---|---|---|
| Steatosis | 0.00 ± 0.00 | 1.53 ± 0.37 (#) | 3.77 ± 0.08 (#,$,§) | 2.41 ± 0.33 (#) |
| Hepatocyte hypertrophy | 0.00 ± 0.00 | 0.35 ± 0.15 | 1.73 ± 0.10 (#,$,§) | 0.76 ± 0.18 (#) |
| Inflammation | 0.10 ± 0.05 | 0.53 ± 0.15 | 1.50 ± 0.13 (#,$,§) | 0.41 ± 0.12 |
| Fibrosis | 0.26 ± 0.08 | 0.59 ± 0.12 | 0.80 ± 0.07 (#) | 0.47 ± 0.12 |
| NASH activity score (NAS) | 0.36 ± 0.10 | 3.00 ± 0.59 (#) | 7.80 ± 0.20 (#,$,§) | 4.06 ± 0.49 (#) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Henkel, J.; Alfine, E.; Saín, J.; Jöhrens, K.; Weber, D.; Castro, J.P.; König, J.; Stuhlmann, C.; Vahrenbrink, M.; Jonas, W.; et al. Soybean Oil-Derived Poly-Unsaturated Fatty Acids Enhance Liver Damage in NAFLD Induced by Dietary Cholesterol. Nutrients 2018, 10, 1326. https://doi.org/10.3390/nu10091326
Henkel J, Alfine E, Saín J, Jöhrens K, Weber D, Castro JP, König J, Stuhlmann C, Vahrenbrink M, Jonas W, et al. Soybean Oil-Derived Poly-Unsaturated Fatty Acids Enhance Liver Damage in NAFLD Induced by Dietary Cholesterol. Nutrients. 2018; 10(9):1326. https://doi.org/10.3390/nu10091326
Chicago/Turabian StyleHenkel, Janin, Eugenia Alfine, Juliana Saín, Korinna Jöhrens, Daniela Weber, José P. Castro, Jeannette König, Christin Stuhlmann, Madita Vahrenbrink, Wenke Jonas, and et al. 2018. "Soybean Oil-Derived Poly-Unsaturated Fatty Acids Enhance Liver Damage in NAFLD Induced by Dietary Cholesterol" Nutrients 10, no. 9: 1326. https://doi.org/10.3390/nu10091326
APA StyleHenkel, J., Alfine, E., Saín, J., Jöhrens, K., Weber, D., Castro, J. P., König, J., Stuhlmann, C., Vahrenbrink, M., Jonas, W., Kleinridders, A., & Püschel, G. P. (2018). Soybean Oil-Derived Poly-Unsaturated Fatty Acids Enhance Liver Damage in NAFLD Induced by Dietary Cholesterol. Nutrients, 10(9), 1326. https://doi.org/10.3390/nu10091326