Slowing Down Ageing: The Role of Nutrients and Microbiota in Modulation of the Epigenome


Nencki Institute of Experimental Biology, Polish Academy of Sciences, 3 Pasteur St., 02-093 Warsaw, Poland
*
Author to whom correspondence should be addressed.
Nutrients 2019, 11(6), 1251; https://doi.org/10.3390/nu11061251
Submission received: 5 April 2019
/
Revised: 27 May 2019
/
Accepted: 28 May 2019
/
Published: 1 June 2019
(This article belongs to the Special Issue Nutrition and Age-Related Disorders)
{kind=link}
{kind=link}
{kind=link}
Abstract
:The human population is getting ageing. Both ageing and age-related diseases are correlated with an increased number of senescent cells in the organism. Senescent cells do not divide but are metabolically active and influence their environment by secreting many proteins due to a phenomenon known as senescence associated secretory phenotype (SASP). Senescent cells differ from young cells by several features. They possess more damaged DNA, more impaired mitochondria and an increased level of free radicals that cause the oxidation of macromolecules. However, not only biochemical and structural changes are related to senescence. Senescent cells have an altered chromatin structure, and in consequence, altered gene expression. With age, the level of heterochromatin decreases, and less condensed chromatin is more prone to DNA damage. On the one hand, some gene promoters are easily available for the transcriptional machinery; on the other hand, some genes are more protected (locally increased level of heterochromatin). The structure of chromatin is precisely regulated by the epigenetic modification of DNA and posttranslational modification of histones. The methylation of DNA inhibits transcription, histone methylation mostly leads to a more condensed chromatin structure (with some exceptions) and acetylation plays an opposing role. The modification of both DNA and histones is regulated by factors present in the diet. This means that compounds contained in daily food can alter gene expression and protect cells from senescence, and therefore protect the organism from ageing. An opinion prevailed for some time that compounds from the diet do not act through direct regulation of the processes in the organism but through modification of the physiology of the microbiome. In this review we try to explain the role of some food compounds, which by acting on the epigenetic level might protect the organism from age-related diseases and slow down ageing. We also try to shed some light on the role of microbiome in this process.
1. Introduction
Ageing is defined as a continuous accumulation of detrimental changes in the whole organism due to the impaired functioning of many tissues and organs. A constant loss of function of the muscle and skeletal systems, memory disabilities and a higher vulnerability to, for example, infections, are observed with age [1]. Age is the main risk factor for certain diseases, called age-related diseases (ARD). Ageing is associated with chronic low grade inflammation, which is probably both the cause and the result of increased age [2]. Because the population of people over 65 years old is increasing very quickly, it is becoming necessary to develop some new approaches to improve their quality of life. It is generally accepted that it is much more reasonable to protect from ageing than to treat a particular disease, especially given that age-related diseases usually arise jointly, which entails the need to take many different medicines that very often act antagonistically [3]. To slow down ageing and to reduce or postpone ARD, well-defined and efficient strategies must be developed. These strategies should preferably be compatible with our lifestyle and habits. Such an attitude should not be complicated and inscribed in a normal everyday functioning. Currently, nobody needs to be persuaded that a daily diet is one of the most important elements of our health and wellbeing. The existing data clearly show that what we eat and how much we consume determines our healthspan. One of the most convincing examples is the population of Okinawa Island, on which the number of centenarians (people over 100 years of age) is the highest in the world. The diagnosed reason of such a long lifespan is a lower calorie consumption [4,5].
The observations of some beneficial effects exerted by a diet are supported by a great deal of evidence from scientific studies. Certain mechanisms of such an impact have been already defined and certain molecular factors have been recognized. It has been proven that the ageing process is malleable, and that by appropriate proceedings, we can influence the pace and the course of ageing and ARD [6]. However, currently, the most appreciated anti-ageing intervention is caloric restriction or certain approaches mimicking it [7]. To such activities belong: a strongly defined lifestyle consisting of a proper healthy diet, mild physical activity, and the avoidance, in the broad sense, of stress. Since it is already well recognized that ageing can be modified, our aware attitude should help to elongate our healthspan and possibly also the lifespan.
It has been documented that one of the most recognized causes of ageing is cellular senescence. Firstly, it has been observed that senescent cells accumulate with ageing in many tissues [8,9,10] and later on, it has been proved that elimination of such cells improves the functioning of the organism and slightly elongates the lifespan of model animals [11,12,13]. A reduction of the number of senescent cells was achieved mostly in genetically modified animals, which is not possible to perform in humans. The non-genetic eradication of senescent cells is not easy but recent studies have shown that it is possible. A new approach, named senotherapy, is being intensively developed and some promising results, which can be transferred to humans, were obtained [14].
One of the most spectacular and significant changes observed in senescent cells is modification of the chromatin structure [15]. The role of the genotype in ageing and longevity is an important issue, however, a growing evidence has shown that the role of epigenetics cannot be neglected and can be equally or, in some situations, even more important than the genetic profile. Epigenetic changes can be influenced by a variety of environmental factors [16,17,18,19]. Functional foods and nutraceuticals seem to be among those most important [19] as they can promote health and longevity and prevent ARD [20]. Lower global chromatin compaction and locally increased condensation lead to an elevated vulnerability of the cell nucleus to destruction, and may also modify gene expression. This is due to epigenetic and post-translational modifications of DNA and histones, respectively. Acquired epigenetic marks are inherited by offspring, which clearly proves their significance. Moreover, manipulation of epigenetic modifications seems to be less complicated and does not raise any ethical doubts as does genetic intervention. Nutrients can modify epigenetic marks; however, the impact of diet on chromatin may not always be direct. There are some suggestions that the influence can be mediated by the microbiome [17]. The composition of the gut bacteria depends on the age and it varies in health and disease [21]. The elderly population very often suffers from detrimental changes of several biological functions, which are associated with intestinal microbiota rearrangement. It has been proven that nutrients are involved in the shaping of the microbiota composition, which gives a promise that a well-composed diet can alleviate some age-related disabilities. The role of the microbiome in ageing is intensively studied and it is believed that the composition and the quality of the microbiome can determine healthspan and longevity [22]. The links between the diet, the microbiome and chromatin structure will be discussed in this review.
2. Ageing, Cellular Senescence and Anti-Ageing Interventions
Ageing is associated with some features, which are characteristic for all elderly; however, age-related diseases do not affect all of them equally. To common age-related health problems belong: impaired visions and hearing, sarcopenia, osteoporosis, increased vulnerability to infections, impaired wound healing and decreased skin elasticity. With age increases the risk of appearance of ARD, which include: neurodegenerative diseases (e.g., Alzheimer’s and Parkinson’s diseases, AD and PD, respectively), cardiovascular diseases (CVD, hypertension and atherosclerosis), type II diabetes, lung diseases (chronic obstructive pulmonary disease, COPD and idiopathic pulmonary fibrosis, IPF), osteoarthritis, cataract, glaucoma and certain types of tumors (e.g., breast, colon, lung cancer) [1]. It has been documented that there is increased number of senescent cells in tissues and organs affected by all age-related diseases [1,23,24,25,26,27,28]. For this reason, the role of cellular senescence in ageing and ARD cannot be neglected. Therefore, cellular senescence is intensively studied and there are examples showing that an improvement of organismal functioning is associated with the alleviation of the senescent phenotype [29] and vice versa, the eradication of senescent cells is followed by a reduction of certain age-related disabilities [11,12].
Cellular senescence is not always detrimental, as it may appear based on its engagement in ageing and ARD. The function of this basic cellular process is complex and the relevance of senescence depends on the age of the organism [30]. Senescence is essential for proper body shaping during embryonic development [31,32]. In a young organism it serves a beneficial function in tissue regeneration, in protection from fibrosis [33,34] and as a barrier from cancer (cessation of proliferation of damaged cells) [30]. However, in an old organism the number of senescent cells increases. They are involved in the generation of a chronic low-grade inflammatory state via the excessive secretion of certain proteins (senescence-associated secretory phenotype, SASP), and an enhanced production of reactive oxygen species (ROS). Such properties of senescent cells lead to microenvironmental changes supporting tumor progression, lowering of the regenerative potential and the renewal of tissues, and to senescence of neighboring non-senescent cells.
Cellular senescence can occur as a result of telomere shortening, and is then called replicative senescence, or as a response to stress and is then termed stress-induced premature senescence (SIPS) [35,36]. Senescence can be induced by intrinsic (ROS production, metabolic malfunctions, oncogene overexpression, DNA damage, ER-stress, and by genetic and epigenetic changes e.g., chromatin structure dysfunction) or extrinsic (exposition to physical or chemical factors) stimuli [37,38].
Cellular senescence is listed as one of the nine cellular hallmarks which contribute to ageing: genomic instability, telomere attrition, epigenetic alterations, stem cells exhaustion, altered intercellular communication, cellular senescence, mitochondrial dysfunction, loss of proteostasis, deregulated nutrient sensing [39]. However, all of the mentioned features are strongly related to cellular senescence. Either they can lead to cell senescence (genomic instability, telomere attrition, epigenetic alterations, mitochondrial dysfunction and loss of proteostasis) or are the result of senescence (stem cell exhaustion, altered intercellular communication, deregulated nutrient sensing).
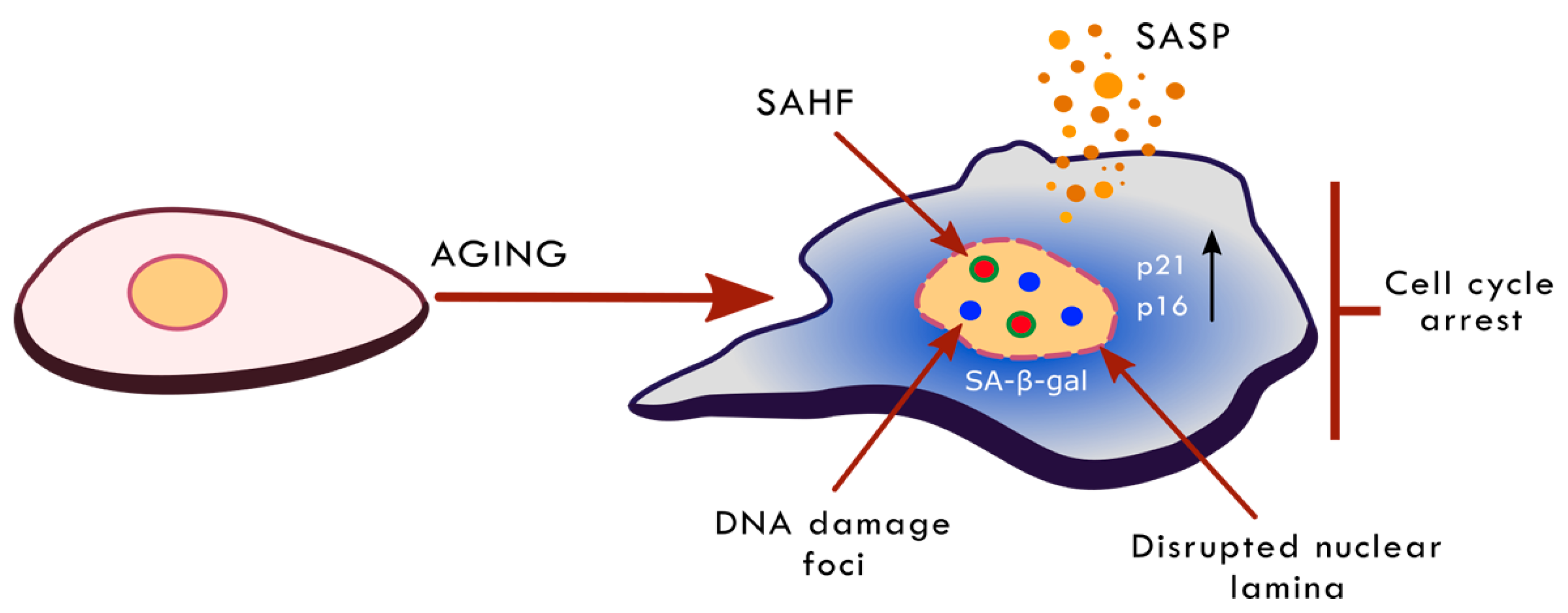
Senescent cells are characterized by multiple alterations in cell morphology, physiology and biochemistry (summarized in Figure 1). The first most important and essential feature of senescence is the permanent cessation of proliferation. However, senescence concerns not only the proliferation-competent cell but also post-mitotic cells, such as neurons, skeletal muscle cells and cardiac myocytes [40]. The most recognized senescence markers include: increased activity of a lysosomal enzyme, senescence-associated β-galactosidase (SA-β-gal), increased level of cell-cycle inhibitors (p21, p16), increased frequency of DNA damage and activation of the DNA damage response (DDR) pathway, increased the production and secretion of SASP components (a bystander effect is senescence induction in neighboring cells) and changes in chromatin structure and gene expression [33,41]. Until now, there was no universal marker of cell senescence. The most widely accepted reason of cellular senescence is DNA damage [38,42,43]. However, there are some examples showing that cells undergo senescence without DNA breaks [38,40,44]. Currently, there is an assumption that senescence is a response to the widely understood stress conditions. There are some examples to show that cell senescence may be the result of reticular stress, nucleolar stress, chromatin changes, the activation of the DDR pathway without DNA damage, and osmotic stress [38].
Studies on the mechanism of ageing and senescence have led to the development of certain anti-ageing approaches, which have proved their worth on animal models. Some of the most promising approaches include caloric/diet restriction (CR/DR), which reduces the calorie intake without causing malnutrition, and some activities mimicking it, such as certain micro-nutrients or mild physical activity [45,46]. Dietary restriction has been shown to be effective in several species (yeast, fruit flies, nematodes, rats, dogs, primates) [7,47,48,49]; however, it is difficult to apply it to humans. Nonetheless, a similar approach, namely intermittent fasting (IF), gave comparable results [50,51]. The effects of CR are related not only to lifespan extension but, first of all, to healthspan improvement, that is alleviation of neurodegeneration, sarcopenia, cardiovascular and metabolic diseases, and decreased incidence of cancer [52,53,54]. In humans, short-term CR increased multiple markers of metabolic and cardiovascular health [55]. There are some suggestions concerning the beneficial effects of the mechanism involved in CR. The role in CR is ascribed to a group of enzymes involved in the regulation of many cellular processes indispensable for maintaining homeostasis, namely sirtuins, reviewed in [7]. Sirtuin 1 and 6 (SIRT1 and 6) are strongly involved in the modulation of chromatin structure by histone deacetylation and have an impact on DNA methylation. The level of almost all sirtuins is elevated due to CR [56]. On the other hand, the expression and activity of these enzymes decrease with age [7,44,57]; moreover, SIRT1 and 3 were suggested as markers of frailty, one of the most characteristic features associated with ageing [58].
The main signalling pathways involved in longevity, and thus proposed to be a target for ageing intervention, include mTOR–S6K signalling and the GH/IGF-1 axis, which require AMPK and specific sirtuins [54]. They are mutually related and regulated. Inhibition of mTOR and IGF-1, or its receptor, is a recognized approach to elongate the lifespan, reviewed in [7]. The mTOR–S6K pathway is inhibited by AMPK, which regulates, and is regulated by sirtuins. SIRT1, in turn, is involved in the p53 pathway, which antagonizes IGF-1 signalling [59].
3. Epigenetics and Nuclear Landscape; Ageing-Associated Changes of Chromatin Structure
The genetic material of all Eukaryotes is confined to a highly organized nucleus measuring only a few microns. To fit in such a restrained volume, 146 kb long segments of double stranded DNA are wound on histone octamers forming the basic structure of a nucleosome. The nucleosomes are then further condensed by linker histones and scaffold proteins into higher order chromatin. The chromatin is enclosed by a double nuclear membrane spiked with complexes of transmembrane proteins—nucleoporins. The interior membrane is lined with a mesh of intermediate filaments, namely lamins, that constantly interact with chromatin domains [60,61].
Epigenetics is defined as the study of heritable changes in gene function that do not entail any alteration in DNA sequence [62]. The epigenome acts as a molecular link between the genome and the environment. The environmental impact is related to the lifestyle, including diet, physical activity, stressors, chemical exposition and addictions such as smoking and drug abuse. All these factors are able to alter the epigenetic landscape and favor ageing-related disease phenotype. The epigenetic mechanisms include: DNA methylation, histone modifications and post transcriptional regulation of gene expression by non-coding RNA (microRNA, miRNA) [63]. These processes are able to regulate synergistically and cooperatively gene expression by changing chromatin organization and DNA accessibility. Although epigenetic marks are to a great extent inherited, the epigenetic landscape is not given once and for all. It is constantly prone to perpetual fluctuations throughout the lifespan and is profoundly altered in ageing organisms. The resulting gene expression profile, however, can be relatively easily modified by pharmaceuticals [64], physical exercise [65], diet [66] and even gut microbiota [67].
3.1. DNA Methylation
One of the prominent examples of epigenetic modification is DNA methylation, which is characterized by covalent transfer of methyl group from S-adenosylmethionine (SAM) to the fifth position of cytosine ring (5mC) within cytosine-phosphate-guanine (CpG) dinucleotide. DNA methylation in vertebrates is mainly restricted to CpG sites (CpGs) and 60–80% of CpGs in the human genome are methylated. Approximately 7% of CpGs are located in CpG islands (CGIs), which are regions of high CG density [68]. Depending on the number of methylated cytosines within the site, DNA can be either hypo- or hypermethylated. Hypermethylation of CpG islands is associated with transcriptional repression and gene silencing whereas hypomethylation with transcriptional activation [69]. This often applies CpG islands (CGIs) in promoter regions of particular genes. Approximately 60–70% of genes have a CpG island within their promoters [70]. Although these regions are considered mostly unmethylated [71], this state changes drastically in pathological conditions like cancer, or most importantly, during ageing. As the organism is getting older, hypermethylation of tissue specific genes or genes encoding transcription factors is observed [72,73] in contrast to global loss of methylation [74,75], leading to drastic changes in gene expression profile as compared to young controls. This phenomenon has found its application in several recently established ‘epigenetic clocks’, where biological and chronological age can be estimated on the basis of DNA methylation level [76,77].
In humans, four DNA methyltransferases (DNMT) genes, namely DNMT1, DNMT2, DNMT3A, and DNMT3B have been identified [78]. Three of them are classic methyltransferases (DNMT1, DNMT3A, DNMT3B) and the function of DNMT2 is a little controversial. Moreover, DNMT-like regulatory enzyme DNMT3L (DNA methyltransferase 3 like) is worth to be mentioned. DNMT1 was the first methyltransferase identified and is responsible for replicating the methylation pattern on newly synthesized DNA strands based on the hemi-methylated template strand, hence it plays a maintenance role. On the other hand, DNMT3A and DNMT3B are believed to methylate DNA de novo. Nevertheless, recent studies have suggested that these three enzymes can play complementary roles in mammals [79]. As far as DNMT3L is considered, it cannot be classified as a typical methyltransferase since it is catalytically inactive. Instead, it works as a cofactor in the regulation of methyltransferase activity by forming a complex with histone H3 that is unmethylated on lysine 4, to recruit DNMT3A and perform de novo DNA methylation [80]. Last but not least, DNMT2 shares a strong homology with other DNA methyltransferases although its methyltransferase activity is thought to be marginal [81]. It is mainly considered to play a leading role in tRNA methylation [82]. However, recent studies suggest a putative contribution of DNMT2 to DNA methylation. Khalil et al. show that the activity of DNMT2 in aged mouse macrophages is considerably increased, which leads to hypermethylation in promoter regions of autophagy genes Atg5 and LC3B. This results in reduced autophagy, which is one of the causes of chronic inflammation, a characteristic feature of aged organisms [73]. Moreover, DNMT2 is shown to be upregulated in replicatively senescent human fibroblasts, which suggests its role in longevity regulation. Interestingly, silencing of DNMT2 results in changes in proliferation-related and tumor suppressor miRNAs level and leads to proliferation inhibition and induction of cellular senescence mediated by oxidative stress [83]. DNMT2 silencing in mouse fibroblasts leads to, inter alia, telomere shortening, elevation of cell cycle inhibitors and DNA damage, resulting cell senescence [84]. It is believed that DNA demethylation is not only a passive process occurring as a result of the lack of DNMT1 but can be achieved by active demethylation [19]. The methylated cytosine is oxidized to 5-hydroxymethylcytosine (5hmC) by the ten-eleven translocation (TET) enzymes consisting of three family members, i.e., TET1, TET2 and TET3 [85]. These proteins can catalyze further 5hmC oxidation to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC), that usually ends up with the removal of the modified base by base excision repair or decarboxylation [86]. How the process of DNA demethylation proceeds in vivo, however, is still under extensive investigation. Nevertheless, different tissues seem to accumulate 5hmC at varying levels [87,88], and the enrichment is usually observed at promoters of specific genes [89]. This indicates, that 5hmC does not only serve as an intermediate in the active DNA demethylation but can also stand as an epigenetic regulatory mark controlling gene expression. The 5hmC is most abundant in embryonic stem cells, adult somatic stem cells and brain tissue [88,89] although localization of the 5hmC-enriched regions depends on the type of cell and developmental stage. Profound changes are found in ageing mouse brains; a study revealed a global increase in hippocampal 5hmc content, which was unrelated to oxidative stress [90]. The same trend was noted in substantia nigra, where the increase of 5hmC was observed in contrast to striatum which has stable DNA methylation status across ageing [91].
Moreover, chromatin accessibility is regulated via a crosstalk between DNA methylation and histone modifications. Methylated DNA recruits histone deacetylases and histone methyltransferases e.g., SuV39H1 which, by methylating H3K9 (histone H3 lysine 9), tightens the chromatin structure [19]. Moreover, HP-1 (heterochromatin protein 1) is responsible for recruitment of DNA methyltransferases, DNMTs [92].
3.2. Posttranslational Modification of Histones
The next level of nuclear organization and gene expression control concerns chromatin structure predominantly controlled by posttranslational modifications (PTMs) of histones. Histones are highly conserved DNA-binding proteins, that form a nucleosome core. Each nucleosome consists of two copies of canonical histones H2A, H2B, H3 and H4. The double stranded DNA is wound on the established histone octamer and the whole complex is stabilized by linker histone H1 [93]. Each core histone (except for H4) has variants which differ in amino acid sequence and can be synthesized independently of DNA replication both in mitotic and post-mitotic cells [94]. The incorporation of histone variants certainly adds up to the complexity of the nucleosome organization and gene expression control.
Histones can be post-translationally modified on the N-terminal tails protruding from the globular histone core. Depending on the type of modification the impact on chromatin accessibility and stability differs considerably. Among the plethora of PTMs the most prevalent are methylation, acetylation, phosphorylation and ubiquitination [95]. These modifications are precisely controlled by highly specialized “writers”, “erasers” and “readers”, that incorporate, remove and recognize PTMs in histones. Writers include histone acetyltransferases (HATs), that transfer an acetyl group from acetyl-CoA on specific lysine residues in the histone tail [96], and histone methyltransferases (HMTs) that can mono-, di- and tri-methylate lysine residues [96]. Acetylation of histones H3 and H4 leads to an open and active chromatin structure. On the other hand, histone methylation can both activate and repress gene expression. Trimethylation of K4, K36 and K79 in histone H3 results in active transcription, while trimethylation of K9, K27 and K20 is associated with silenced chromatin [97]. The trimethylation of histone H3 at lysine 9 (H3K9me3) is catalyzed by the Suv39H1 methyltransferase. This HMT can directly interact with HP-1 and regulates chromatin compaction [98]. Erasers, such as histone deacetylases (HDACs and Sirtuins) and histone demethylases (HDMs), counteract the action of HATs and HMTs and modify chromatin condensation and gene expression. Readers, on the other hand, are proteins that possess domains with high affinity for the modified histone sites to direct a particular transcriptional outcome [99]. Domains that recognize methylated histones include chromo, PHD, WD40, Tudor, MBT domains and more, while histone acetylation is recognized by bromo domains and tandem PHD domains. Interestingly, readers can target multiple PTMs within a histone, nucleosome and even multiple nucleosomes and their action can be controlled by non-coding RNAs. Moreover, readers can recruit factors that are involved in transcription, replication or DNA damage repair [100].
With the help of histone modifiers chromatin can adopt two dynamic conformations: that of transcriptionally inactive heterochromatin, condensed during the interphase and replicated in the late S phase, and that of euchromatin accessible to transcription, decondensed during the interphase and replicated in the early S phase [101]. Heterochromatin can be further divided into constitutive heterochromatin (telomeres, pericentromeric regions) rich in H3K9me3, protein HP1 (which stabilizes chromatin compaction) and H4K20me3, and facultative heterochromatin, characterized by high H3K27me3 level, which arises temporarily in euchromatic regions and silences individual genes. On the other hand, euchromatin is usually abundant in active chromatin markers, for example H3K4me3, H3K36me3 and acetylated histones H3 and H4 [102].
3.3. Sirtuins
Sirtuins, through histone deacetylation, take part in the formation of constitutive and facultative heterochromatin. The most studied sirtuins are SIRT1 and 6. SIRT1 is involved in deacetylation of lysine K9 in histone 3 (H3K9), which leads to its trimethylation (acetylation of K9 protects it from trimethylation) and facilitates the interaction with heterochromatin protein 1 α (HP1α). Such interaction keeps the chromatin in a closed state resulting in gene silencing [103,104]. SIRT1 preferentially deacetylates H4K16, H3K9, H3K56 H3K14, H1K26 and H1K9 [104,105]. Moreover, this enzyme influences chromatin condensation by regulating histone expression and the level and activity of some histone modifying enzymes, e.g., it inhibits Suv39h1 methyltransferase degradation and enhances its activity. It can also modulate the activity of the p300 histone acetyltransferase [106,107,108]. In turn, SIRT6 deacetylates mainly H3K9 in the promotor regions of genes involved in metabolism [109]. It is also suggested that by deacetylating H3K9 in telomeric regions it can protect the cells from telomere dysfunction [110].
3.4. Non-Coding RNA
MicroRNAs are small (21–25 nucleotides) RNAs, which bind to complementary nascent mRNAs and trigger their degradation leading to inhibition of expression of specific genes. It has been estimated that 1%–4% genes in the animal genome encode miRNAs [111] and computational analysis predicts that individual miRNA can target approximately 200 transcripts [112]. It is known that miRNAs play a role in lifespan regulation [113].
3.5. Nuclear Architecture
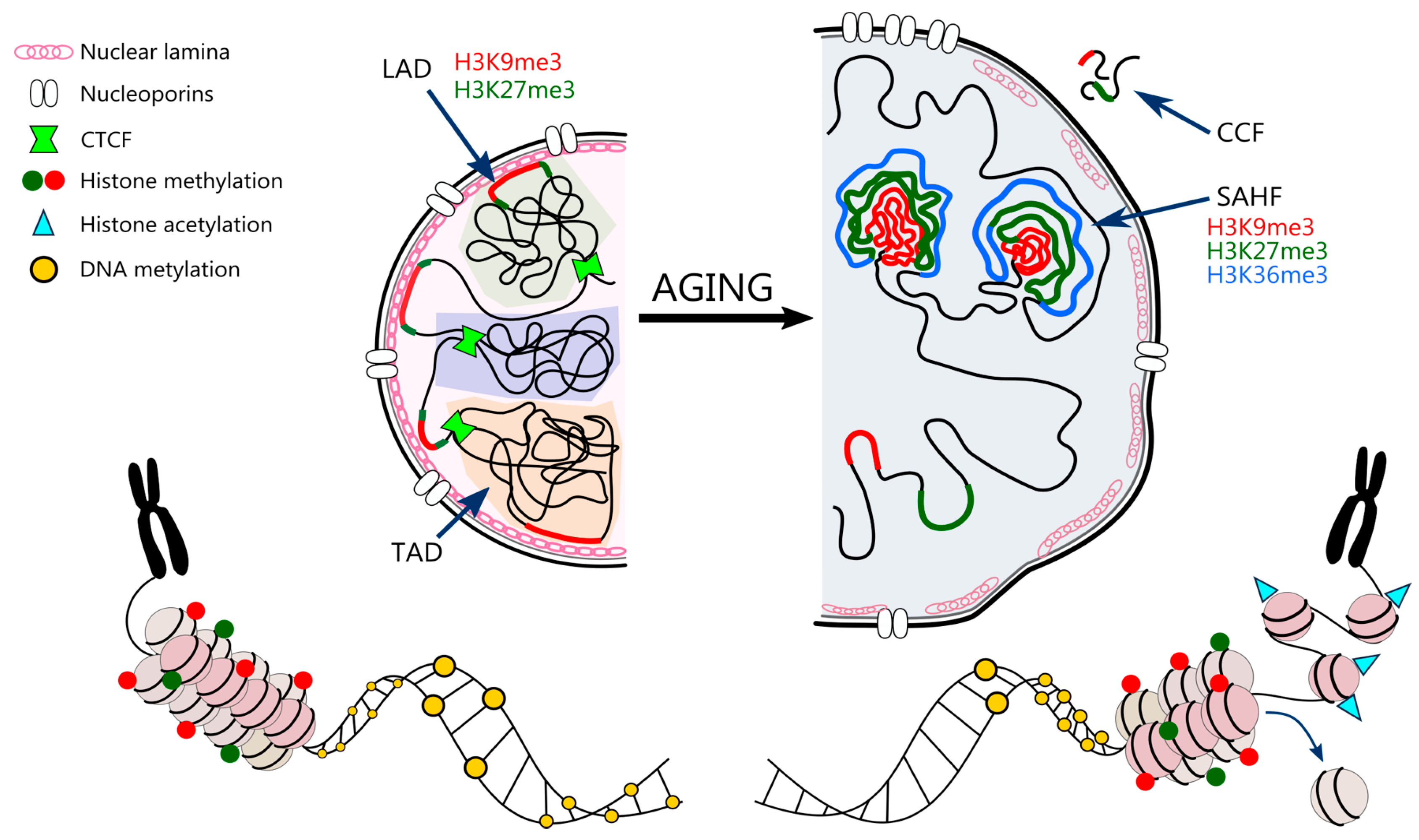
Higher order 3D chromatin architecture involves containment of certain chromatin regions inside particular domains, called topologically associated domains (TADs). These provide favorable environment for gene expression regulation via enhancer-promoter interactions. TADs are enclosed by CTCF and cohesin that insulate them from the neighboring domains [114]. Chromatin can be also assigned either to so called compartment A that consists of transcriptionally active, gene-rich DNA regions or compartment B that overlaps with lamina associated domains (LADs) and is represented mostly by heterochromatin [115]. LADs are regions of heterochromatin enriched with H3K9me3 and H3K27me3 that tightly interact with nuclear lamina to organize chromosomes and stabilize the nuclear architecture [116].
Mechanisms responsible for establishing and maintaining the chromatin structure are summarized in Figure 2.
3.6. Nuclear Landscape in Cell Senescence and Ageing
In ageing, the DNA methylation pattern is altered. The most clear alteration is related to a global decrease in DNA methylation observed in different species such as mouse, rat, cow, hamster and humans [117,118,119]. It has been demonstrated that the expression of DNMT1 (responsible for global DNA methylation) and DMNT3a decrease during ageing entailing a decline of DNA methylation [120]. However, gene-specific methylation of promoters is elevated, as it has been observed e.g., estrogen receptor and insulin-like growth factor-2 (IGF-2) [72,101,121].
Senescent cells are characterized by global loss of canonical histones both nucleosome forming (a “loss” of nucleosomes) and linker histones [122], which may occur due to histone proteins’ downregulation caused by cell-cycle arrest. A comparative quantitative analysis of global histone protein expression revealed a reduction in histone H3 and H4 content of about 30% and a 40% reduction in the turnover of H3 and H4 in senescent cells [123]. The loss of histones may also be a result of the induced autophagy of cytoplasmic chromatin fragments (CCFs) [124]. Senescent human fibroblasts form CCFs that contain DNA, γH2AX and repressive histone marks (mainly H3K9me3 and H3K27me3) [125], which are then digested in the lysosomes [126]. This leads to increased genome instability in ageing and senescence. On the other hand, histone variants are observed to accumulate with senescence [127]. In replicatively senescent and DNA damage-induced senescent mouse fibroblasts there is a significant increase in histone variant H2A.J as compared to proliferating cells [128]. Accumulation of H2A.J may promote expression of inflammatory genes contributing to SASP. Moreover, due to DNA damage, also γH2AX accumulates in senescent cells, as a part of DNA damage response machinery (DDR) [129]. In turn, prolonged DDR may lead to degradation of G9a and GLP methyltransferases causing reduction in H3K9me2, which is a part of constitutive heterochromatin [130]. Reduction in H3K9me3 in promoters of IL-8 and IL-6 encoding genes can be then compensated by acetylation of H3K9 and H4K16 leading to SASP. As shown by Hayakawa et al., senescent cells exhibit a gradual increase in the acetylation of H3K9 and H4K16 with the simultaneous downregulation of the histone deacetylase SIRT1 [131].
Generally, senescent cells exhibit global loss of HDACs and sirtuins [7,132] although certain genomic regions become enriched in acetylated histones. For example, senescent human fibroblasts show relatively elevated levels of H4K16ac in gene promoters. This impedes higher order chromatin packaging and results in an open chromatin structure in senescent cells that has been suggested to promote nucleosome exchange [133,134]. Moreover, studies have shown that H4K16ac is a prerequisite for the induction of H3K79 dimethylation while it acts as an inhibitor for H4K20me2 [135]. Contrarily, acetylation of K56 on H3 (H3K56ac) is reduced in replicatively senescent cells [135], probably due to SIRT6 activity [136]. It was shown, that SIRT6 also deacetylates H3K9ac at telomeric chromatin and maintains chromosomal stability [136]. The level/activity of majority of sirtuins decreases with age. It has been observed in many tissues and in many cell types in vitro (liver, arteries and vascular smooth muscle cells) [57,137,138]. One of the probable reasons of diminished sirtuins’ activity could be a reduction in NAD+ level.
On the other hand, during ageing, a global loss or redistribution of heterochromatin is observed, leading to increased genome instability and altered gene expression [15]. The loss of compaction is mostly due to both reduced methylation and usually increased acetylation of histones. Senescent cells are characterized by a drastic decrease in H3K9me3, and SUV39H1 methyltransferase, HP1α and H3K27me3 along with EZH2 methyltransferase. A similar trend of H3K9me3 and H3K27me3 loss is also observed in normal human ageing and premature ageing diseases, such as Hutchinson-Gilford progeria or Werner syndrome, suggesting that global loss of heterochromatin may be a common feature of ageing [139,140]. Oncogene-induced senescent (OIS) cells, in contrast, tend to have increased levels of repressive histone marks (H3K9me3 and H3K27me3) in comparison to non-senescent proliferating controls [141]. Moreover, the perinuclear heterochromatin associated with LAD reorganizes from the pre-existing heterochromatin into internal senescence-associated heterochromatin foci (SAHF) [142]. SAHFs are enriched in chromatin-associated proteins, namely HP1, HMGA and macro Histone 2A (mH2A). Additionally, SAHFs have a distinct layered structure: the core area is enriched in H3K9me3 [143], the periphery is abundant in H3K27me3 [144], while active chromatin marks can usually be found on the outside (e.g., H3K36me3) [145]. What is interesting, Chandra et al. revealed, that despite depletion of H3K9me3 and H3K27me3, OIS cells are still able to form SAHFs [144]. The downregulation of Lamin B1 and Lamin B Receptor (LBR) in OIS cells disrupts nuclear envelope structure and, as a result, disables heterochromatin anchorage. Moreover, recent studies show that the increased nuclear pore density in OIS contributes to SAHF formation [146]. The space beneath the nucleoporin complex is naturally depleted of heterochromatin; hence it shifts from the perinuclear to the internal space [147].
It is believed that an impact on chromatin compaction (forced induction of local chromatin condensation) could be sufficient to induce cell senescence due to its influence on the activation of the DDR pathway (ataxia telangiectasia mutated (ATM)- and ATR-dependent), which is not associated with DNA damage [148]. On the other hand, chromatin condensation is an integral part of the normal course of the DDR pathway and failure of this step results in impaired activation of this pathway. Moreover, downregulation of p300 histone acetyltransferase activity was sufficient to induce cell senescence [149].
MicroRNAs and their shuttles (extracellular vesicles in particular) regulate a large number of cellular functions and physiological processes and are currently recognized as regulators of healthy and unhealthy ageing [150]. In particular, the role of miRNAs is documented for brain functions in ageing [151,152,153]. For example, miRNAs control the level of two proteins, amyloid precursor protein (APP) and membrane-bound β-site APP-cleaving enzyme 1 (BACE1), which contribute to the formation of amyloid plaques in AD [154]. In a variety of ageing-related pathologies the miRNAs, similarly to other epigenetic regulators, are dysregulated [155,156]. The function of miRNAs in the regulation of the lifespan can be both inhibitory and activatory. In experimental studies, miRNAs were able to extend or shorten the lifespan of model organisms, such as C. elegans or mice [113,157,158,159]. For example, the loss of function mutation in the C. elegans miRNA lin-4 dramatically reduced the lifespan of the nematode and overexpression of this miRNA extended it [160]. The loss of function mutation in miRNA lin-14, elongated lifespan, which is disturbed by the overexpression of this type of miRNA. Both these miRNAs, lin-4 and lin-14, regulate the insulin/IGF-1 pathway. Other miRNAs, such as miR-375, which regulates insulin secretion in murine pancreatic islet cell [161] or miR-122 in murine liver, are related to this signaling pathway [162]. Some miRNAs that target genes encoding proteins and enzymes belonging to the nutrient sensing pathways can act as novel therapeutics. They are involved in diabetes as well as ageing, by regulating inflammation [150] and, therefore, may play key role in the modulation of the ageing process.
Chromatin structure is also influenced by changes in the nuclear envelope. The inner surface of the nuclear envelope is lined with two types of lamins—A and B. Senescence is associated with the depletion of lamin B1 and mutations in the lamin A gene resulting in accumulation of defective prelamin A [163]. This leads to an abnormal nuclear morphology and chromosomal aberrations. Prelamin A was shown to reduce the import of DNA repair factor 53BP1 by mislocalizing nucleoprotein NUP153 [164], which results in persistent DNA damage. In HGPS, progerin (untruncated prelamin A) anchors to the inner nuclear membrane and reduces the attachment of H3K9me3 and H3K27me3 [165]. Heterochromatin shifts from nuclear lamina (a filamentous protein meshwork created by lamins and inner nuclear membrane proteins) to the center of nucleus (like in OIS).
All age/senescence-related changes in chromatin structure and nucleus architecture are summarized in Figure 3.
3.7. Epigenetics in Age-Related Diseases
An increasing amount of evidence derived from both clinical and experimental studies indicates that epigenetic deregulation, especially of DNA methylation, is frequently associated with ageing and may underlie the etiology of chronic diseases e.g., diabetic complication, CVD (atherosclerosis), cancer, metabolic disorder and neurodegeneration [166,167,168,169,170]. In atherosclerotic plaques isolated from human aorta a specific DNA methylation profile was observed. Compared to the healthy controls, several hypermethylated genes associated with endothelial and smooth muscle functions were found [166]. Moreover, DNA methylation may represent a useful biomarker for a disease risk, e.g., increased global DNA methylation, which was observed in peripheral blood leukocytes (PBL) of Singapore Chinese, is positively correlated with increased incidence of CVD, hypertension, diabetes and obesity [169]. There are much more examples linking epigenetic modifications with adiposity and diabetes. It has been shown that the methylation status of the promoters of genes encoding the retinoid X receptor alpha (RXRA) and endothelial nitric oxide synthase (eNOS) in umbilical cord tissue in healthy newborns, and peroxisome proliferator-activated receptor g coactivator-1a (Pgc-1a) in the blood of children is associated with adiposity in childhood [171,172]. Epigenetic analyses performed at different phases of the human life could be a predictor of the individual vulnerability to later obesity and metabolic disease. The ability to reverse the epigenetic mark would be invaluable for the treatment of certain diseases. Epigenetic alteration was demonstrated to be related to age-associated organ dysfunctions as in the case of renal ageing [173] and in ageing-related decline of immune response reviewed in [174]. The impact on immune response is achieved, on the one hand, via the alteration of epigenetic decoration of promoters and enhancers of regulators of fundamental immune cell functions. On the other hand, it is related to the epigenetic regulation of the inflammatory state. For example the induction of inflammatory cytokines (IL-8, MIP-1α- macrophage inflammatory protein 1-alpha) was caused by the loss of H3K9 methylation at the promoter regions [175]. Moreover, it has been shown that an increased age-related genomic instability was associated with the loss of heterochromatin marks (H3K9me3) and decrease in Suv39H1 expression [176].
However, the role of epigenetics in ageing is much more complex and is not limited only to one generation [177]. It has been demonstrated in a mouse model that advanced age increases the susceptibility for disease in offspring. The offspring of aged fathers had an exacerbation of age-associated phenotypes, reduced lifespan and were more prone to age-related pathologies than animals sired by young fathers. This was accompanied by numerous epigenetic alterations in the paternal germ line and offspring tissue, which manifested themselves by altered activation states of longevity-related cell signaling. Genome-wide epigenetic studies have revealed differences in gene promoter methylation, which was enriched in the case of genes involved in regulation of longevity pathways, in DNA from sperm of aged males and tissues from old father offspring (increased activity of mTORC1). This is an excellent example to demonstrate the role of epigenetics in the inheritance of ageing-related changes acquired during the lifetime. Furthermore, such results provide a very convincing reason to develop strategies in order to avoid adverse effects in the case of an advanced age of the parents.
4. Impact of the Diet on Chromatin Structure and Gene Expression
The consequences of a diet for health can be observed on a long-term basis, counted even in decades. Diet has an impact on the onset of ADR and longevity, but some effects can be effectively counteracted/eliminated by certain approaches [63]. Epigenetic mechanisms mediated by the nutritional effect, but also other environmental factors, establish a certain type of cellular memory. Such modifications can be copied by cells during cell division and define gene expression in new generation of cells, without changing their primary DNA sequence. As it was described in the previous paragraphs, the involvement of chromatin structure changes in senescence and ageing is significant; therefore, the protective approaches or chromatin rejuvenation approaches are considered as potential tools of anti-ageing interventions. Structural changes are, on the one hand, associated with a higher vulnerability to DNA damage and, on the other hand, with altered gene expression. The products present in the diet are able to modulate chromatin structure and, in this manner, regulate protein composition. There is a large number of data showing the impact of diet on DNA methylation on the expression of particular genes in animal models reviewed in [19]. Some data are present also for humans. One of the most spectacular results concerns differences in DNA methylation in individuals exposed peri-conceptually to famine during the Dutch Hunger Winter [178,179]. This caused a decreased methylation of e.g., IGF-2 gene. Moreover, peri-conceptual folic acid supplementation influenced methylation of IGF-2 promoter [180]. Dietary restriction, one of the currently known anti-ageing interventions to be used in humans, also acts by changing DNA methylation [181]. DR is generally strongly protective against age-related changes in DNA methylation [181]. This is associated with decreased p16 methylation and increased activity of DMNT1 (potential reversal of the global hypomethylation) [182,183,184]. There is also growing evidence that the epigenome is much more malleable at an early stage of development (pre- and postnatal development), however modifications are possible in each period of life. In adulthood the plasticity of epigenome also exists, reviewed in [19]. It is believed that epigenetic modifications can be potentially reversible and it makes this feature an attractive target for interventions [185].
Nutrients directly regulate both the transcription and translational processes and interfere with metabolism by affecting DNA methylation, histone modifications and post transcriptional gene regulation by non-coding RNAs. High fat, low protein or energy restricted diet can alter the epigenetics marks [182,186,187].
4.1. Impact of Nutrients on DNA Methylation
Nutrition may affect both the establishment and maintenance of the DNA methylation pattern, which can exert long-lasting health effects. Gene expression can be modulated by both nutrient and non-nutrient dietary compounds. Currently, the major interest is focused on the effects of specific micronutrients and non-nutritional dietary components. To this group belong natural compounds, e.g., polyphenols, the most abundant phytochemicals in fruits, vegetables and plant-derived beverages [16]. There is a growing evidence that polyphenols can control epigenetic changes and it is believed that they display the ability to reverse epigenetic modifications related to some pathologies, such as metabolic disorders, cardiovascular and neurodegenerative diseases and certain types of cancer. Therefore, such compounds as flavonoids, curcuminoids and stilbenes are promising in both disease prevention and intervention because they can modify gene expression with consequences for disease risk and health maintenance.
Nutritional and dietary factors have been postulated to affect DNA methylation by changing the availability of the methyl donors and altering the activity of the DNMT enzymes. To the first group belong micronutrients which are co-factors for enzymes involved in one-carbon metabolism, including folate, vitamin B6, vitamin B12, choline and methionine and those which can affect one-carbon metabolism indirectly [63]. The dietary selenium caused an imbalance in the methylation cycle by decreasing homocysteine concentration and, in consequence, reducing its availability for the methionine cycle. It led to reduced global DNA methylation in rat [188]. Modulators of DNMT activity include: selenium, genistein, quercetin, curcumin and green tea polyphenols, e.g., EGCG (epigallocatechin-gallate), whose effect has been documented mostly in cancer cells [189,190,191,192] and apigentin and resveratrol [193]. Flavonoids from tea and fruits are able to reverse hypermethylation and, in this manner, to reactivate tumor suppressor genes [194]. Selenite inhibited the binding of AP-1 transcription factor to DNA, which, in turn, reduced DMNT1 association with DNA (decreased methylation) [195,196] and selenium inhibited DNMT expression [197]. EGCG can inhibit DNMT1 directly by fitting to the binding pocket [194,198]. Quercetin is able to inhibit cancer cell proliferation due to the demethylation of the promoter of the p16 gene, which results in an enhanced expression of this tumor suppressor [199]. A very good example of the link between nutrition, epigenetics and phenotype is the supplementation of maternal diet of agouti mouse (a retrotransposon inserted upstream of the agouti gene causes ectopic expression of agouti protein, resulting in yellow fur, obesity and diabetes) with folic acid (methyl donor) and cofactors (vitamin B12, choline and betaine) [200] or genistein [201]. DNA methylation of the retrotransposon in the vicinity of the agouti gene restores normal expression of the agouti gene, the visible marker of which is darker hair color in offspring. Moreover, supplementation with methyl donors or genistein of agouti females protects the offspring from obesity (one of the risk factor for diabetes and CVD), which is characteristic for these mice [201,202].
Gene regulation can be mediated by secondary DNA structures, e.g., G-quadruplexes (G4) [203]. It is believed that these highly stable tetra-stranded structures, resolved by DNA helicases, are a causal factor in disorders following mutations in DNA helicase genes and are related to the phenotype of premature ageing. Such G4 structures are subjects of dynamic DNA modifications, which can affect genome stability and integrity, and alter gene expression. It is proposed that dietary nutrients, such as folate and antioxidants, can stabilize G4 structures [204].
In some situations, the application of phytochemicals, which alter DNA methylation, can lead to adverse epigenetic changes. Such observation has been made in the case of premenopausal women receiving isoflavones (to which genistein belongs) in every day diet [205]. This caused increased methylation of the promoters of tumour suppressor genes. On the other hand, genistein increased expression of cell cycle inhibitors, which can reduce the proliferation potential of cancer cells [206,207].
4.2. Impact of Nutrients on Histones Modifications
There are a lot of data showing that histones can be modified by nutrients and dietary compounds. It is related to the inhibitory potential of some dietary constituents interacting with enzymes involved in histones decoration. Histones marks can be altered by changed abundance and/or efficacy of the enzymes involved in their decoration or by impaired availability of the enzyme substrate. As it was mentioned before the chromatin status is controlled by HATs, HDACs, HMTs and HDMs. However, histones are not only methylated and acetylated. Histones tails can be modified by phosphorylation, ribosylation, ubiquitination, sumoylation and biotinylation [63], which visibly shows the complexity of the regulatory mechanism. To HDACs class III belong sirtuins, which are named “the enzymes of youth”, reviewed in [7]. The level of sirtuin decreases with age and the restoration of their level leads to rejuvenation.
Numerous inhibitors of HDACs are recruited from polyphenolic compounds. A lot of data are related to sirtuins, especially 1 and 6 [193]. However, HDACs inhibitors appear to be nonselective. Such inhibitors have shown beneficial effects in neurodegeneration, cancer, and inflammatory disorders. HDACs can be inhibited by butyrate (a short-chain carboxylic acid produced in the colon by bacterial fermentation of carbohydrates) and some polyphenols present in garlic, soybeans (e.g., genistein), garcinol and cinnamon [63,193]. To HDAC1 inhibitors belong quercetin, green tea polyphenols e.g., EGCG, luteolin and genistein [190,208,209,210]. Curcumin, a natural polyphenol, has been reported to function as both HDAC and HAT inhibitor [211,212] and is recognized as a hypomethylating agent [192]. Moreover, curcumin, dependently on the concentration, can act as sirtuin inhibitor (cytostatic concentrations) or activator (concentration which do not impair proliferation potential) [44,57,213]. Similar function is ascribed to EGCG, which influences enzymatic activity and expression of HATs, HDACs (HDAC1, 2, 3), and sirtuins [193,214,215], and for quercetin [216,217]. In turn, genistein increased HAT and decreased HDAC activity, including SIRT 1, and in this manner alters DNA accessibility [193,206]. HATs can be inhibited by green tea polyphenols, including EGCG, quercetin and by copper [214,216,218], and HMTs by EGCG [219]. Reduced availability of methyl donors in the diet also contribute to reduced HMT activity [220]. Posttranslational histones modification can be also shaped by resveratrol and catechins, i.e., other dietary polyphenols [16].
The fact that sirtuins exert multiple beneficial effects in the organism, such as reduction of the symptoms of CVD, diabetes and neurodegeneration as well as are linked to longevity, [221], it is reasonable to look for factors, which can increase their expression or activity. The latter can be enhanced by certain natural and synthetic compounds. To the first group belong flavones, stilbenes, chalcones, and anthocyanidins, which directly activate particular sirtuins in vitro. Such activity was shown for quercetin, butein, curcumin, fisetin, kaempferol, catechins, resveratrol, cilostazol, paeonol, daidzein and piceatannol [7,193,222,223]. Activation of sirtuins was also observed following mild physical activity [224,225,226]. The role of sirtuins in ageing and disease has been discussed in details by us in Grabowska et al. [7].
HDAC inhibitors can be one of the most promising prevention strategies concerning epigenetic changes including reduction/reversal of the already existing detrimental alternations. The restoration of the transcription of genes involved in proper metabolism, which decreases with age, can delay the functional decline of the organism [227]. Indeed, some examples have shown that inhibitors of HDACs are potent in extending the lifespan. This has been shown for fruit fly e.g., after feeding with 4-phenylbutyrate (PBA) [228] or with other HDAC inhibitors [229,230]. A global increase in histone acetylation and altered gene expression (e.g., of SOD, superoxide dismutase, cytochrome P450, glutathione S-transferase) significantly increased the lifespan. HDAC inhibitors were proposed in treatment of type II diabetes, ageing-associated cognitive decline and neurodegenerative diseases, such as AD and PD [231,232,233,234,235].
Some limitations are imposed by the lack of specificity of HDAC inhibitors. This poses a real problem since inhibition of most HDACs is considered as beneficial, while sirtuins have to be activated to exert their anti-ageing/anti-ARD role. To this aim, careful studies to find out some unique compounds to be involved in the selective regulation of HDAC activity are necessary.
4.3. Impact of Nutrients on miRNA Regulation
Polyphenols, e.g., EGCG, curcumin, resveratrol and quercetin, are also regulators of miRNAs, as has been shown in cancer cells [236,237,238,239,240]. EGCG decreased the expression of oncogenic miRNAs and increased the expression of tumour-suppressor miRNAs, which indicates it precise regulatory function [236]. Quercetin increased expression of miRNAs recognized as negative regulators of inflammatory genes [237]. The anticancer potential of genistein is also related to miRNA deregulation [238].
5. Impact of the Exercise on Gene Expression
The remodeling of the epigenome can be carried out by environmental factors other than nutrients, for example, by exercise. A large body of data indicates that physical activity (PA) can modulate gene expression through epigenetic alterations. This could be a beneficial strategy in chronic diseases, such as metabolic syndrome, diabetes, cancer (breast, colorectal and gastric cancer), cardiovascular and neurodegenerative diseases, reviewed in [65]. Physical activity remodels the skeletal muscle and adipose tissue [241,242,243,244]. It has been shown that six months of exercise led to the hypermethylation of HDAC4 and NCOR2 genes, which had an impact on the adipose tissue metabolism [244]. HDAC4 activity is related to repression of GLUT4 transcription in adipocytes, and correlates with insulin resistance [245]. HDAC4 loss of function (export from the nucleus during exercise and loss of transcriptional repressive function) is also related to skeletal muscle adaptations to exercise; the latter effect has been interpreted as a result of activation of cellular GLUT4 expression [246]. In turn, NCOR2, a nuclear co-repressor, is involved in the recruitment of, among others, HDAC4 [247]. It has been shown that alteration in the methylation pattern due to physical activity reduced the level of inflammatory mediators and prevented the occurrence of diseases associated with low-grade chronic inflammation [248]. PA acts as modulator of DNA methylation, histone modifications (particularly H3 and H4) and of miRNAs [249]. In the case of miRNAs, exercises modulate certain miRNAs, especially these involved in cancer, metabolic diseases and CVD [250,251,252]. Interestingly, it has been shown that, at the epigenetic level, exercises not only protect patients from type 2 diabetes but also, through maternal transmission of epigenetic modifications of genes involved in important metabolic pathways, have a beneficial impact on the offspring [253]. MicroRNAs regulate myogenesis, atrophy and sarcopenia [254]. During exercise a remarkable reduction of some types of miRNA, such as miR-107, miR-208b, miR-221 and miR-499, in mouse soleus muscle tissue was observed. The skeletal muscle biopsy of old people (68–72 years old) has shown overexpression or lower expression of numerous miRNAs in comparison with young individuals reviewed in [254]. MicroRNAs regulate two most important signaling pathways involved in ageing and longevity, namely the IGF axis and the mTOR pathway. It is documented that exercise can modify gene expression and activate signaling pathways, and that some of these changes are related to the expression of various miRNAs [255]. In this manner PA can strengthen exercise adaptation [256].
6. Autophagy as a Target of Nutritionally-Altered Epigenetic Pattern
Autophagy is considered as an anti-ageing catabolic process and is linked with/ cell survival and longevity [257]. It is one of the basic processes involved in intracellular degradation of damaged macromolecules and organelles that leads to self-cleaning and recycling within the cell. The regulation of autophagy is multistep and occurs at several levels. One of the mechanisms that regulate autophagy gene expression is the post-translational modification of histones, which control, for example, the nutrient sensor TOR (target of rapamycin), a factor involved in longevity. Autophagy is very often elevated in long-lived model organisms. SIRT1 and miRNAs are involved in this process. SIRT1 has been demonstrated to stimulate autophagosome formation and autophagic degradation resulting in increased autophagic flux [258]. While the SIRT1-mediated deacetylation of cytoplasmic proteins is required for autophagy induction, the activity of SIRT1 in the nucleus limits autophagy [257]. SIRT1 regulates autophagy gene expression through histone deacetylation. By deacetylating H4K16, it represses expression of various genes related to the early and late steps of autophagy (Ulk1/ATG1, Ulk3/ATG1, Atg9A/ATG9, Lc3/ATG8, GabarapL2/ATG8, and vacuolar membrane protein Vmp1). Histone H3 hypoacetylation (K9, K14, K18), via spermidine inhibition of histone acetyltransferase activity, increased expression of genes involved in autophagy such as ATG7, ATG11, ATG15. Moreover, methyltransferase EHMT2/G9a, by targeting H3K9, repressed transcription of autophagy genes [259]. Conversely, the downregulation of histone acetylation by KAT8 (HAT) (deacetylation of H4K16) induced autophagy. In this manner, by autophagy induction, SIRT1 exerts a protective effect in certain neurodegenerative disease models (e.g., PD) [260]. Autophagy can be regulated by acetyl coenzyme A (acetyl-CoA), which is a donor for acetylation reactions [257]. Reduced acetyl-CoA levels extended the lifespan of Drosophila [261]. The cationic polyamines (e.g., spermine and spermidine), whose level decreases with age in several tissues, and which are involved in autophagy regulation, have also an impact on histone acetylation status by regulating the activity of HATs and HDACs [257,262]. Spermidine inhibits HAT activity and leads to hypoacetylation of H3 on K9, K14 and K18. It has been shown that repressive marks H3K9me3, which accumulate with age, and H3K4me3 are linked to alterations in autophagy gene expression, as reviewed in [257]. The second manner of the epigenetic modulation of autophagy are miRNAs. For example, miR-34 is linked with autophagy and longevity. A decreased level of this regulator was observed in mice subjected to DR and it was correlated with increased lifespan [263]. Upregulation of miR-34 during ageing inhibits expression of the autophagy genes (e.g., Atg9a) [264]. MIR34 also leads to the inhibition of the expression of the SIRT1 [265] and mTOR pathway components. Several other miRNAs such as MIR376A, MIR376B, MIR181, MIR30D, MIR101 are related to autophagy inhibition, reviewed in [257].
7. The Microbiota Changes in the Elderly
The human body makes an excellent environment for a highly dynamic and complex ecosystem of bacterial archaeal, fungal and viral species, which is referred to as human microbiota. These microorganisms constitute lifelong symbiotic companions that are present from birth until our final days. The exact starting point of microbial colonization, however, is currently a subject of heated debate [266]. It was suggested, that the whole process may already start in utero [267,268], although recent studies return to the hypothesis that placenta is rather sterile, hence the fetus cannot acquire any microflora at this stage [269,270]. Nevertheless, inhabitation can surely be estimated to start at labor [271] and the initial composition of microbiota largely depends on the mode of delivery [272]. During birth, the first microorganisms start to colonize our skin, lungs, oral cavity and gastrointestinal tract. This process is relatively stabilized by the age of three to five [273]. Further development partly relies on the host genome [274] and gender [275] but also on a plethora of external factors, which include the type of diet [276,277], physical activity [278,279], the use of medication [280,281], and the living environment in general [282,283]. However, it should be noted, that the microbial composition is highly individual, hence the aforementioned modulatory factors might affect each host differently. Another factor significantly influencing the bacterial composition is ageing and age-related diseases [284]. It is generally accepted that as we age, the overall number and diversity of bacterial species decreases, often resulting in an imbalance between commensal bacteria in favor of pathobionts. This leads to further colonization with pathogens, production of inhibitory substances or toxins, and development of chronic inflammation [285]. Progressing dysbiosis contributes to detrimental health and age-related conditions such as cardiovascular diseases, neurodegenerative diseases (Alzheimer’s and Parkinson’s diseases), type II diabetes, chronic obstructive pulmonary disease (COPD), osteoporosis and different types of cancers. Moreover, alterations in human microbiota are also thought to be connected with allergies [286], obesity [287] and even depression [288,289]. Microbiota can modulate age-related changes related to frailty, such as innate immunity, sarcopenia, and cognitive functions [22]. Very extensive studies have shown that the differences in the composition of the gut microbiota between older people and younger adults are related to the gradual changes with time and not to chronological age [22].
7.1. Gut Microbiota
The largest and most prominent microbial reservoir is residing in the lower gastrointestinal tract (GIT). Gut microbiota play significant role in human health and diseases [290,291]. The microbiome is required for the digestion of complex plant polysaccharides, synthesis of short chain fatty acids (SCFA), indispensable amino acids and vitamins. Moreover, it neutralizes drugs and carcinogens, protects the host against pathogenic infection and regulates oxidative stress, as reviewed in [292]. There is some kind of interdependency and mutual relationship between microbiota and the host [293,294]. Microbiota can modulate immune cell functions, metabolism and insulin sensitivity. Moreover, the microbiome can regulate host gene expression through multiple mediators, such as SCFA, antioxidants and mediators of inflammation. Nutrition plays a pivotal role in this process because all mentioned mediators synthesized by gut bacteria, are derived from the diet [295].
There are about 1000–1500 recognized bacterial species living in the gut and each person harbors about 150 different species, which together contain more genes than the human genome and constitute a sort of a second genome [294]. GIT is the most susceptible site of age-related changes in the bacterial composition, connected with gradual decrease in intestinal barrier function [296], impaired nutrient absorption [297], increased risk of developing cancers [298,299] and brain disorders [300,301]. The most visible alteration of gut microbiota is the reduction in their diversity, shifts in the dominant species, which is related to a decrease in beneficial microorganisms and increase in facultative anaerobic bacteria. This results in a lower availability of short chain fatty acids, because different compositions of microbiota have an impact on the production of bacterial metabolites [17,284]. Age-related changes in the gut microbiota are also associated with alteration in the physiology of gastrointestinal tract, such as a decrease in saliva production and gastric acid secretion, lower absorption of nutrients (iron and vitamin B12), slower gastrointestinal motor function and, finally, altered diet composition associated with decreased food intake and a decline in the functioning of the immune system. This makes elderly people more prone to infections and frailty [17,302,303,304,305].
GIT is composed of small and large intestines. Small intestine is responsible for nutrient absorption, facilitated by metabolic action of microbial cells that are rather low in abundancy but stable in composition [306]. During ageing however, the incidents of the microbial overgrowth, namely Small Intestinal Bacterial Overgrowth (SIBO) may occur. This leads to the malabsorption of micronutrients and, as a result, to malnutrition, abdominal distension, diarrhea and flatulence [307]. Large intestine, on the other hand, is the most densely colonized part of the human body. The average number of bacterial colonies ranges from 1011 to 1012, making their genome nearly 150 times larger than a human [294]. Bowels are a place where the final fermentation and absorption of key nutrients take place. The most abundant phyla in homeostatic conditions are Actinobacteria, Bacteroidetes, Firmicutes, Proteobacteria, and Verrucomicrobia. Along with mucosal secretions, the bacteria form a rather thick and resistant biofilm, that line the epithelium, working as a mediator between the lumen and host’s body [308]. These commensal bacteria produce short-chain fatty acids (SCFA), synthesize vitamins K and B and protect the host form the external pathogens (production of bacteriocins). All of these compounds exert an immunomodulatory effect by stimulating immunoglobulin A production, down-regulating proinflammatory cytokines and inducting regulatory T cells [309]. In ageing however, everything is disturbed as there is a decline in the number and diversity of commensals [310]. Bifidobacteria and Enterobacter decrease in elderly, while Bacteroidetes increase in comparison to young adults [311]. Moreover, beneficial bacteria are usually overtaken by pathobionts that disrupt the epithelial integrity [312] and lead to secretion of inflammatory markers, such as IL-6 and IL-8, resulting in elevated (and persistent) inflammation [22]. Intestinal inflammation is characterized by thinner or even degraded inner mucus layer, enabling bacteria to reach the epithelium. Pathogenic strains produce exotoxins [313] that induce changes to tight junctions, leading to increased gut permeability, also defined as “leaky gut”. This, in consequence, contributes to irritable bowel disease [314], the development of colon cancer and may also lead to a disrupted blood-brain axis detrimentally affecting neurodegenerative diseases [315,316,317]. Gut permeability, however, can be modified by beneficial bacterial strains, such as Lactobacillus plantarum, Escherichia coli, Nissle, and Bifidobacterium infantis, which increase expression of proteins forming tight junctions, and as a result, strengthen the intestinal barrier [318]. Although bacteria have the leading role in the gut composition maintenance in the elderly, the methanogenic archaea also play important role in human ageing. The diversity of methanogens is increased in the elderly as compared to young adults [319], however there is no significant increase in prevalence. However, methane formed by these archea is more abundant in the elderly, which affects smooth muscle cells and results in slowed bowel transit [320]. This also impacts the intestinal pH. In the elderly, the pH is increased compared to young controls, and has deleterious impact on the beneficial bacteria.
7.2. Other than Gut Microbiota
Ageing has a tremendous influence on the microbial composition within all human body. For instance, human skin harbors fairly different colonies between dry volar forearm, quite moist axillary vault and sebaceous face [321], where the bacterial abundancy is extremely age-dependent [322]. In the elderly, higher species richness is noted in palms, forearms and forehead than in young controls, while young display greater diversity on nares, glabella or back [323]. With age, the total number of aerobes (especially Propionibacterium) on cheeks and forehead of healthy elderly decreases as compared to young adults [323,324], which tightly correlates with lower secretion of sebum [324]. However, in some skin conditions the sebum can be overproduced and lead to seborrheic dermatitis (SD), which is also a non-motor symptom of Parkinson’s Disease [325]. The skin of PD patients with SD have increased densities of fungi Malassezia spp. and bacteria Streptomyces spp., that produce complex volatile organic compounds, which in turn may serve as a noninvasive diagnostic tool of early Parkinson’s disease [326]. Similarly, breath analysis of microbial volatile organic compounds may also be used for diagnosis of Chronic Obstructive Pulmonary Disease (COPD) in the elderly [327]. Patients with COPD display significant increase in pathogenic Gram-negative bacteria [328] with commensurate decrease in diversity of resident bacterial communities [329,330] as compared to healthy subjects and young adults [331]. Elderly also exhibit profound changes in intraoral microflora [332], notably on the dental and subgingival plaque. These often result in inflammatory conditions like periodontitis, in which the rise in Gram-negative bacteria is observed [333,334]. The main pathogen of chronic periodontitis, Porphyromonas gingivalis, is a serious risk factor for developing Alzheimer’s disease [335].
8. Impact of the Diet on Microbiota
As mentioned earlier, the role of the microbiome is strongly associated with the pace of ageing and the quality of life of the elderly. Some data show that gut microbiota is implicated in the pathogenesis of metabolic disorders, such as diabetes and obesity, and promotes atherosclerosis, and vascular endothelial dysfunction [336,337,338,339]. On the other hand, the microbiome is also involved in atherosclerosis retardation by generating metabolites of dietary flavonoids, which stimulate reverse cholesterol transport in macrophages [340]. However, the most recognized impact of the microbiome is related to the symptoms of frailty [22,295].
The interaction between nutrients (micro and macro) and microbiota is very tight. Such a relation concerns, e.g., polyphenols [341,342,343,344]. The bioavailability of this group of compounds is very low. Gut microbiota metabolize them into active and bioavailable metabolites. On the other hand, polyphenols modulate microbiota because they can exert prebiotic-like effects and promote growth of some bacterial species (particularly hydrolysable and condensed tannins derived from polyphenol-rich foods, including cocoa, tea, grapes, wine, berries, pomegranates, and nuts). The impact is thus reciprocal [345,346]. Curcumin is also able to modulate the diversity of microbiota [341,342,343,347]. The composition of gut microbiota is very often the cause of some confusing results obtained from clinical trials, because of individual variability. It is suggested that the composition of bacteria is a key element in the response to polyphenol treatment and that gut microbiota are probably responsible for polyphenols’ health effects by regulating their bioavailability [345,348]. The bioavailability of most micronutrients is poor due to low absorption and fast metabolism leading to rapid elimination from the organism. One of the modifications related to the detoxication mechanism is glucuronidation, which has been well documented e.g., for curcumin, quercetin, genistein and ginger phenolics [349,350,351,352,353]. Mono- and diglucuronides are less active then free compounds [354]. One of the products of the gut bacteria is β-glucuronidase [355]. This enzyme is involved in hydrolysis of the glucuronide moiety [351,356], and has an impact on the level of free compounds.
Recent data have shown that SIRT1 can mediate the interaction between the host and the microbiome [357]. Mice with a deletion of SIRT1 in the intestinal epithelium exhibited increased activation of NFκB and inflammation and, moreover, had altered fecal microbiota due to altered bile acid metabolism. Additionally, the lower expression of SIRT1 was observed in intestinal tissues from patients suffering from colitis. It is suggested that SIRT1 prevents intestinal inflammation by the regulatory impact on gut microbiota. Similar role is attributed to SIRT3, the level of which decreases in colorectal cancer [358]. It interacts with gut microbiota, which plays a central role in the resistance to colon cancer, and in this manner acts as an anti-inflammatory and tumor-suppressing protein. On the other hand, SIRT1 can also be regulated by the microbiome [359]. The expression of SIRT1 is governed by microRNAs (e.g., miR-204) while expression of the latter is remotely controlled by the microbiome. The function of the endothelium is thus impaired since miR-204 downregulates the level of SIRT1 (antibiotics decrease miR-204 and increases Sirt1 expression). Additionally, sirtuins and AMPK can be regulated by SCFA produced by microbiota, reviewed in [292]. This can have an impact on chromatin structure and gene expression. SIRT1 is one of the proteins directly regulated by SCFA [360]. N-butyrate, activated several regulatory pathways including that involving AMPK signaling [361], and inhibited histone deacetylase (HDAC4), as was evidenced by the increased acetylation of H3K9 in the liver and brain [362]. A healthy gut microbiota produces significant amounts of folate and vitamin B12 [363], which are donors of the methyl group essential for DNA methylation. Moreover, gut bacteria synthesize amino acids, such as tryptophan, which stimulates the IGF-1/p70s6k/mTor pathway and in this manner activates gene expression [364]. An association has been documented between the intestinal microbiota composition and weight loss caused by CR. Moreover, it has been shown in animal models that CR-induced lifespan elongation was accompanied by a structural modulation of gut microbiota [365].
Maintaining a proper diet is important for individuals of all age. Moreover, the nutritional needs of elderly do not substantially differ from those of younger adults. However, it is particularly important to promote some nutritional strategies among those of older age because of the risk of malnutrition due to impaired absorption, loss of appetite and chewing difficulties, which may affect the proper nutritional status [366]. These strategies are elaborated to restore the diversity of microbiota in old people in order to reduce some adverse consequences of ageing, including frailty.
9. Conclusions
Well-recognized anti-ageing strategies have been for a long time linked to a proper diet. Recently, it has been well documented that diet can modify chromatin structure. Moreover, it was proven that certain epigenetic modifications are long-lasting and can be inherited while others can be quite easily reversed. Therefore, nutrients can determine our health and disease even if they were consumed a long time ago, e.g., in the prenatal period. They can play a pivotal role in the ageing process and longevity. To the puzzle composed of two elements, i.e., nutrients and epigenetics, the microbiome should also be added. The interaction of these three seemingly independent elements creates a coherent picture that may be useful for developing potential anti-ageing strategies. The awareness of this relationship allows the adoption of a holistic approach to ageing with only one aim in mind: to synchronize all strategies in order to optimize the conditions necessary to healthspan elongation. The age-related epigenetic changes can be reversed by some macromolecules from the diet, the bioavailability of which depends on the microbiome, which per se affects the age-related dysfunctions. The urgent issue to address is how the current knowledge can be transferred into practice. More broadly, it is a question of how to target the individual needs of people, based on their epigenetic history and the composition of the microbiome, taking simultaneously into account age and gender, and not neglecting the impact of environment, which modulates the epigenetic landscape. The current view suggests that the epigenetic age, defined as the presence of characteristic markers, could be a more powerful predictor of future health decline and ARD than chronological age. The most important scope for future studies is to develop the strategies and promote such dietary habits, which will be effective in modulating the epigenome (directly or by the impact on microbiome) to eliminate or delay the expression of genes associated with the initiation and progression of ARD and ageing disabilities (e.g., frailty). Such nutritional habits/strategies must be adapted to age. It is essential to identify and characterize the most potent dietary factors with a strong and defined impact on the epigenome and microbiome, and to recognize the mechanism of their action.
Author Contributions
All authors contributed equally to the review.
Funding
This study was supported by National Science Centre grants: UMO-2016/21/B/NZ3/00370.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Naylor, R.M.; Baker, D.J.; Van Deursen, J.M. Senescent cells: A novel therapeutic target for aging and age-related diseases. Clin. Pharmacol. Ther. 2013, 93, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.G.; Gibbs, M.E. Mechanisms and the clinical relevance of complex drug-drug interactions. Clin. Pharmacol. Adv. Appl. 2018, 10, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Bielak-Zmijewska, A.; Grabowska, W.; Ciolko, A.; Bojko, A.; Mosieniak, G.; Bijoch, Ł.; Sikora, E. The Role of Curcumin in the Modulation of Ageing. Int. J. Mol. Sci. 2019, 20, 1239. [Google Scholar] [CrossRef] [PubMed]
- Most, J.; Tosti, V.; Redman, L.M.; Fontana, L. Calorie restriction in humans: An update. Ageing Res. Rev. 2017, 39, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Fahy, G.M.; West, M.D.; Coles, L.S.; Harris, S.B. The Future of Aging: Pathways to Human Life Extension; Springer: New York, NY, USA, 2010. [Google Scholar]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447–476. [Google Scholar] [CrossRef] [Green Version]
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 2004, 6, 168–170. [Google Scholar] [CrossRef]
- Wang, C.; Jurk, D.; Maddick, M.; Nelson, G.; Martin-ruiz, C.; Von Zglinicki, T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 2009, 8, 311–323. [Google Scholar] [CrossRef]
- Jeyapalan, J.C.; Sedivy, J.M. Cellular senescence and organismal aging. Mech. Ageing Dev. 2008, 129, 467–474. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16 Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16 Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Yu, R.; Dang, W. Chromatin architectural changes during cellular senescence and aging. Genes 2018, 9, 211. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.L.; Vastolo, V.; Ciccarelli, M.; Albano, L.; Macchia, P.E.; Ungaro, P. Dietary polyphenols and chromatin remodeling. Crit. Rev. Food Sci. Nutr. 2015, 57, 2589–2599. [Google Scholar] [CrossRef] [PubMed]
- Salazar, N.; Valdés-Varela, L.; González, S.; Gueimonde, M.; de los Reyes-Gavilán, C.G. Nutrition and the gut microbiome in the elderly. Gut. Microbes 2017, 8, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Benayoun, B.A.; Pollina, E.A.; Brunet, A. Epigenetic regulation of ageing: Linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 2015, 16, 593–610. [Google Scholar] [CrossRef] [PubMed]
- Lillycrop, K.A.; Hoile, S.P.; Grenfell, L.; Burdge, G.C. DNA methylation, ageing and the influence of early life nutrition. Proce. Nutr. Soc. 2014, 73, 413–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, C.K.B. Functional foods, herbs and nutraceuticals: Towards biochemical mechanisms of healthy aging. Biogerontology 2004, 5, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S. Microbiota and aging. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 26–30. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, P.W.; Jeffery, I.B. Gut microbiota and aging. Science 2015, 350, 1214–1215. [Google Scholar] [CrossRef] [PubMed]
- Gorenne, I.; Kavurma, M.; Scott, S.; Bennett, M. Vascular smooth muscle cell senescence in atherosc. Cardiovasc. Res. 2006, 72, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Morbeck, D.E.; Von Zglinicki, T.; Van Deursen, J.; Lustgarten, J.; Scrable, H.; Khosla, S.; Jensen, M.D.; Kirkland, J.L. Fat tissue, aging, and cellular senescence. Aging Cell 2010, 9, 667–684. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, S.; Araya, J.; Numata, T.; Nojiri, S.; Hara, H.; Yumino, Y.; Kawaishi, M.; Odaka, M.; Morikawa, T.; Nishimura, S.L.; et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-β-induced senescence of human bronchial epithelial cells. Am. J. Physiol. Cell Mol. Physiol. 2011, 300, L391–L401. [Google Scholar] [CrossRef] [PubMed]
- McShea, A.; Harris, P.L.R.; Webster, K.R.; Wahl, A.F.; Smith, M.A. Abnormal Expression Of the Cell Cycle Regulators P16 and Cdk4 In Alzheimers-Disease. Am. J. Pathol. 1997, 150, 1933–1939. [Google Scholar] [PubMed]
- Cohen, G. The pathobiology of Parkinson’s disease: Biochemical aspects of dopamine neuron senescence. J. Neural Transm. Suppl. 1983, 19, 89–103. [Google Scholar] [PubMed]
- Price, J.S.; Waters, J.G.; Darrah, C.; Pennington, C.; Edwards, D.R.; Donell, S.T.; Clark, I.M. The role of chondrocyte senescence in osteoarthritis. Aging Cell 2002, 1, 57–65. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Evans, G.; Tonne, J.M.; Verzosa, G.C.; Stout, M.B.; Mazula, D.L.; Palmer, A.K.; Baker, D.J.; Jensen, M.D.; et al. Exercise prevents diet-induced cellular senescence in adipose tissue. Diabetes 2016, 65, 1606–1615. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, O.; Dumont, P.; Dierick, J.F.; Pascal, T.; Frippiat, C.; Chainiaux, F.; Sluse, F.; Eliaers, F.; Remacle, J. Stress-Induced Premature Senescence: Essence of Life, Evolution, Stress, and Aging. Ann. N. Y. Acad. Sci. 2010, 908, 85–98. [Google Scholar] [CrossRef]
- Fridlyanskaya, I.; Alekseenko, L.; Nikolsky, N. Senescence as a general cellular response to stress: A mini-review. Exp. Gerontol. 2015, 72, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Bielak-Zmijewska, A.; Mosieniak, G.; Sikora, E. Is DNA damage indispensable for stress-induced senescence? Mech. Ageing Dev. 2018, 170, 13–21. [Google Scholar] [CrossRef]
- Rebelo-Marques, A.; Lages, A.D.S.; Andrade, R.; Ribeiro, C.F.; Mota-Pinto, A.; Carrilho, F.; Espregueira-Mendes, J. Aging hallmarks: The benefits of physical exercise. Front. Endocrinol. 2018, 9, 258. [Google Scholar] [CrossRef]
- Sikora, E.; Bielak Zmijewska, A.; Mosieniak, G. What is and what is not cell senescence. Postepy Biochem. 2018, 64, 110–118. [Google Scholar] [CrossRef]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef] [Green Version]
- D’Adda Di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.J.; Chiang, Y.J.; Hathcock, K.S.; Horikawa, I.; Sedelnikova, O.A.; Hodes, R.J.; Bonner, W.M. Both telomeric and non-telomeric DNA damage are determinants of mammalian cellular senescence. Epigenet. Chromatin 2008, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, W.; Kucharewicz, K.; Wnuk, M.; Lewinska, A.; Suszek, M.; Przybylska, D.; Mosieniak, G.; Sikora, E.; Bielak-Zmijewska, A. Curcumin induces senescence of primary human cells building the vasculature in a DNA damage and ATM-independent manner. Age 2015, 37, 9744. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, P.; Howell, P.R.; Anderson, R.M. Aging and Caloric Restriction Research: A Biological Perspective With Translational Potential. EBioMedicine 2017, 21, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingram, D.K.; de Cabo, R. Calorie restriction in rodents: Caveats to consider. Ageing Res. Rev. 2017, 39, 15–28. [Google Scholar] [CrossRef]
- Weindruch, R. The retardation of aging by caloric restriction: Studies in rodents and primates. Toxicol. Pathol. 1996, 24, 742–745. [Google Scholar] [CrossRef]
- Lane, M.A.; Baer, D.J.; Rumpler, W.V.; Weindruch, R.; Ingram, D.K.; Tilmont, E.M.; Cutler, R.G.; Roth, G.S. Calorie restriction lowers body temperature in rhesus monkeys, consistent with a postulated anti-aging mechanism in rodents. Proc. Natl. Acad. Sci. USA 1996, 93, 4159–4164. [Google Scholar] [CrossRef]
- Masoro, E.J. Overview of caloric restriction and ageing. Mech. Ageing Dev. 2005, 126, 913–922. [Google Scholar] [CrossRef]
- Hanjani, N.; Vafa, M. Protein restriction, epigenetic diet, intermittent fasting as new approaches for preventing age-associated diseases. Int. J. Prev. Med. 2018, 9, 58. [Google Scholar]
- Mattson, M.P.; Longo, V.D.; Harvie, M. Impact of intermittent fasting on health and disease processes. Ageing Res. Rev. 2017, 39, 46–58. [Google Scholar] [CrossRef]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.J.; Beasley, T.M.; Kemnitz, J.W.; Johnson, S.C.; Weindruch, R.; Anderson, R.M. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat. Commun. 2014, 5, 3557. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Antebi, A.; Bartke, A.; Barzilai, N.; Brown-Borg, H.M.; Caruso, C.; Curiel, T.J.; de Cabo, R.; Franceschi, C.; Gems, D.; et al. Interventions to slow aging in humans: Are we ready? Aging Cell 2015, 14, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Cava, E.; Fontana, L. Will calorie restriction work in humans? Aging 2013, 5, 507–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wątroba, M.; Szukiewicz, D. The role of sirtuins in aging and age-related diseases. Adv. Med. Sci. 2016, 61, 52–62. [Google Scholar] [CrossRef]
- Bielak-Zmijewska, A.; Wnuk, M.; Przybylska, D.; Grabowska, W.; Lewinska, A.; Alster, O.; Korwek, Z.; Cmoch, A.; Myszka, A.; Pikula, S.; et al. A comparison of replicative senescence and doxorubicin-induced premature senescence of vascular smooth muscle cells isolated from human aorta. Biogerontology 2014, 15, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Mohan, N.; Upadhyay, A.D.; Singh, A.P.; Sahu, V.; Dwivedi, S.; Dey, A.B.; Dey, S. Identification of serum sirtuins as novel noninvasive protein markers for frailty. Aging Cell 2014, 13, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.; Bergholz, J.; Zhang, H.; He, H.; Wang, Y.; Zhang, Y.; Li, Q.; Kirkland, J.L.; Xiao, Z.X. Insulin-like growth factor-1 regulates the SIRT1-p53 pathway in cellular senescence. Aging Cell 2014, 13, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Webster, M.; Witkin, K.L.; Cohen-Fix, O. Sizing up the nucleus: Nuclear shape, size and nuclear-envelope assembly. J. Cell Sci. 2009, 122, 1477–1486. [Google Scholar] [CrossRef]
- Wilson, K.L.; Dawson, S.C. Functional evolution of nuclear structure. J. Cell Biol. 2011, 195, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Mckay, J.A.; Mathers, J.C. Diet induced epigenetic changes and their implications for health. Acta Physiol. 2011, 202, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Csoka, A.B.; Szyf, M. Epigenetic side-effects of common pharmaceuticals: A potential new field in medicine and pharmacology. Med. Hypotheses 2009, 73, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Grazioli, E.; Dimauro, I.; Mercatelli, N.; Wang, G.; Pitsiladis, Y.; Di Luigi, L. Physical activity in the prevention of human diseases: Role of epigenetic modifications. BMC Genom. 2017, 18, 802. [Google Scholar] [CrossRef] [PubMed]
- Szarc vel Szic, K.; Ndlovu, M.N.; Haegeman, G.; Vanden Berghe, W. Nature or nurture: Let food be your epigenetic medicine in chronic inflammatory disorders. Biochem. Pharm. 2010, 15, 1816–1832. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Wade, P.A. Crosstalk between the microbiome and epigenome: Messages from bugs. J. Biochem. 2018, 163, 105–112. [Google Scholar] [CrossRef]
- Kim, M.; Costello, J. DNA methylation: An epigenetic mark of cellular memory. Exp. Mol. Med. 2017, 49, e322. [Google Scholar] [CrossRef]
- Bird, A. The essentials of DNA methylation. Cell 1992, 70, 5–8. [Google Scholar] [CrossRef]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Paabo, S.; Rebhan, M.; Schubeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Issa, J.P.J.; Ottaviano, Y.L.; Celano, P.; Hamilton, S.R.; Davidson, N.E.; Baylin, S.B. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat. Genet. 1994, 7, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Khalil, H.; Tazi, M.; Caution, K.; Ahmed, A.; Kanneganti, A.; Assani, K.; Kopp, B.; Marsh, C.; Dakhlallah, D.; Amer, A.O. Aging is assoiated with hypermethylation of autophagy genes in macrophages. Epigenetics 2016, 11, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Wilson, V.L.; Jones, P.A. DNA methylation decreases in aging but not in immortal cells. Science 1983, 220, 1055–1057. [Google Scholar] [CrossRef] [PubMed]
- Heyn, H.; Li, N.; Ferreira, H.J.; Moran, S.; Pisano, D.G.; Gomez, A.; Diez, J.; Sanchez-Mut, J.V.; Setien, F.; Carmona, F.J.; et al. Distinct DNA methylomes of newborns and centenarians. Proc. Natl. Acad. Sci. USA 2012, 109, 10522–10527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Brueckner, B.; Lyko, F. DNA methyltransferase inhibitors: Old and new drugs for an epigenetic cancer therapy. Trends Pharm. Sci. 2004, 25, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A.; Jurkowska, R.Z. New concepts in DNA methylation. Trends Biochem. Sci. 2014, 39, 310–318. [Google Scholar] [CrossRef]
- Ooi, S.K.T.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.P.; Allis, C.D.; et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef] [Green Version]
- Hermann, A.; Schmitt, S.; Jeltsch, A. The human Dnmt2 has residual DNA-(Cytosine-C5) methyltransferase activity. J. Biol. Chem. 2003, 278, 31717–31721. [Google Scholar] [CrossRef]
- Jeltsch, A.; Ehrenhofer-Murray, A.; Jurkowski, T.P.; Lyko, F.; Reuter, G.; Ankri, S.; Nellen, W.; Schaefer, M.; Helm, M. Mechanism and biological role of Dnmt2 in Nucleic Acid Methylation. RNA Biol. 2016, 14, 1108–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewinska, A.; Adamczyk-Grochala, J.; Kwasniewicz, E.; Deregowska, A.; Semik, E.; Zabek, T.; Wnuk, M. Reduced levels of methyltransferase DNMT2 sensitize human fibroblasts to oxidative stress and DNA damage that is accompanied by changes in proliferation-related miRNA expression. Redox Biol. 2018, 14, 20–34. [Google Scholar] [CrossRef]
- Lewinska, A.; Adamczyk-Grochala, J.; Kwasniewicz, E.; Wnuk, M. Downregulation of methyltransferase Dnmt2 results in condition-dependent telomere shortening and senescence or apoptosis in mouse fibroblasts. J. Cell Physiol. 2017, 232, 3714–3726. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Globisch, D.; Münzel, M.; Müller, M.; Michalakis, S.; Wagner, M.; Koch, S.; Brückl, T.; Biel, M.; Carell, T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE 2010, 5, e15367. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef]
- Jin, S.G.; Wu, X.; Li, A.X.; Pfeifer, G.P. Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res. 2011, 39, 5015–5024. [Google Scholar] [CrossRef]
- Chen, H.; Dzitoyeva, S.; Manev, H. Effect of aging on 5-hydroxymethylcytosine in the mouse hippocampus. Restor. Neurol. Neurosci. 2012, 30, 237–245. [Google Scholar] [Green Version]
- Fasolino, M.; Liu, S.; Wang, Y.; Zhou, Z. Distinct cellular and molecular environments support aging-related DNA methylation changes in the substantia nigra. Epigenomics 2017, 9, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Lehnertz, B.; Ueda, Y.; Derijck, A.A.H.A.; Braunschweig, U.; Perez-Burgos, L.; Kubicek, S.; Chen, T.; JenuweiN, T.; Peters, A. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 2003, 15, 1192–1200. [Google Scholar] [CrossRef]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Smith, M.M. Histone variants and epigenetics. Cold Spring Harb. Perspect. Biol. 2015, 7, a019364. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Garcia, B.A. Comprehensive catalog of currently documented histone modifications. Cold Spring Harb. Perspect. Biol. 2015, 7, a025064. [Google Scholar] [CrossRef] [PubMed]
- Sterner, D.E.; Berger, S.L. Acetylation of Histones and Transcription-Related Factors. Microbiol. Mol. Biol. Rev. 2003, 64, 435–459. [Google Scholar] [CrossRef]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone Lysine Methylation Dynamics: Establishment, Regulation, and Biological Impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raurell-Vila, H.; Bosch-Presegue, L.; Gonzalez, J.; Kane-Goldsmith, N.; Casal, C.; Brown, J.; Marazuela-Duque, A.; Singh, P.B.; Serrano, L.; Vaquero, A. An HP1 isoform-specific feedback mechanism regulates Suv39h1 activity under stress conditions. Epigenetics 2017, 12, 166–175. [Google Scholar] [CrossRef]
- Gillette, T.G.; Hill, J.A. Readers, writers, and erasers: Chromatin as the whiteboard of heart disease. Circ. Res. 2015, 116, 1245–1253. [Google Scholar] [CrossRef]
- Yun, M.; Wu, J.; Workman, J.L.; Li, B. Readers of histone modifications. Cell Res. 2011, 21, 564–578. [Google Scholar] [CrossRef] [Green Version]
- Zane, L.; Sharma, V.; Misteli, T. Common features of chromatin in aging and cancer: Cause or coincidence? Trends Cell Biol. 2014, 24, 686–694. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.H.; Sengupta, K.; Li, C.; Kim, H.S.; Cao, L.; Xiao, C.; Kim, S.; Xu, X.; Zheng, Y.; Chilton, B.; et al. Impaired DNA Damage Response, Genome Instability, and Tumorigenesis in SIRT1 Mutant Mice. Cancer Cell 2008, 14, 312–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Poulose, N.; Raju, R. Sirtuin regulation in aging and injury. Biochim. Biophys. Acta 2015, 1852, 2442–2455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaquero, A.; Scher, M.; Erdjument-Bromage, H.; Tempst, P.; Serrano, L.; Reinberg, D. SIRT1 regulates the histone methyl-transferase SUV39H1 during heterochromatin formation. Nature 2007, 450, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Presegué, L.; Vaquero, A. The dual role of sirtuins in cancer. Genes Cancer 2011, 2, 648–662. [Google Scholar] [CrossRef] [PubMed]
- Bouras, T.; Fu, M.; Sauve, A.A.; Wang, F.; Quong, A.A.; Perkins, N.D.; Hay, R.T.; Gu, W.; Pestell, R.G. SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J. Biol. Chem. 2005, 280, 10264–10276. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The Histone Deacetylase Sirt6 Regulates Glucose Homeostasis via Hif1α. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef]
- Cardus, A.; Uryga, A.K.; Walters, G.; Erusalimsky, J.D. SIRT6 protects human endothelial cells from DNA damage, telomere dysfunction, and senescence. Cardiovasc. Res. 2013, 97, 571–579. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Krek, A.; Grün, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet. 2005, 37, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Boehm, M.; Slack, F. A developmental timing microRNA and its target regulate life span in C. elegans. Science 2005, 310, 1954–1957. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- van Steensel, B.; Belmont, A.S. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Drinkwater, R.D.; Blake, T.J.; Morley, A.A.; Turner, D.R. Human lymphocytes aged in vivo have reduced levels of methylation in transcriptionally active and inactive DNA. Mutat. Res. 1989, 219, 29–37. [Google Scholar] [CrossRef]
- Singhal, R.P.; Mays-Hoopes, L.L.; Eichhorn, G.L. DNA methylation in aging of mice. Mech. Ageing Dev. 1987, 41, 199–210. [Google Scholar] [CrossRef]
- Bollati, V.; Schwartz, J.; Wright, R.; Litonjua, A.; Tarantini, L.; Suh, H.; Sparrow, D.; Vokonas, P.; Baccarelli, A. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech. Ageing Dev. 2009, 130, 234–239. [Google Scholar] [CrossRef] [Green Version]
- Casillas, M.A.; Lopatina, N.; Andrews, L.G.; Tollefsbol, T.O. Transcriptional control of the DNA methyltransferases is altered in aging and neoplastically-transformed human fibroblasts. Mol. Cell Biochem. 2003, 252, 33–43. [Google Scholar] [CrossRef]
- Ahuja, N.; Issa, J.P.J. Aging, methylation and cancer. Histol. Histopathol. 2000, 15, 835–842. [Google Scholar]
- Hu, Z.; Chen, K.; Xia, Z.; Chavez, M.; Pal, S.; Seol, J.H.; . Chen, C.C.; Li, W.; Tyler, J.K. Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev. 2014, 28, 396–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, R.J.; Karlseder, J. The great unravelling: Chromatin as a modulator of the aging process. Trends Biochem. Sci. 2012, 37, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Xu, C.; Donahue, G.; Shimi, T.; Pan, J.A.; Zhu, J.; Ivanov, A.; Capell, B.C.; Drake, A.M.; Shah, P.P.; et al. Autophagy mediates degradation of nuclear lamina. Nature 2015, 527, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; . Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.P.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 2013, 202, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Sekeri-Pataryas, K.E. Histone variants of H2A and H3 families are regulated during in vitro aging in the same manner as during differentiation. Exp. Gerontol. 1999, 34, 741–754. [Google Scholar] [CrossRef]
- Contrepois, K.; Coudereau, C.; Benayoun, B.A.; Schuler, N.; Roux, P.F.; Bischof, O.; Courbeyrette, R.; Carvalho, C.; Thuret, J.Y.; Ma, Z.; et al. Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression. Nat. Commun. 2017, 8, 4995. [Google Scholar] [CrossRef]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. γH2AX as a molecular marker of aging and disease. Epigenetics 2010, 5, 129–136. [Google Scholar] [CrossRef]
- Takahashi, A.; Imai, Y.; Yamakoshi, K.; Kuninaka, S.; Ohtani, N.; Yoshimoto, S.; Hori, S.; Tachibana, M.; Anderton, E.; Takeuchi, T.; et al. DNA damage signaling triggers degradation of histone methyltransferases through APC/C Cdh1 in senescent cells. Mol. Cell 2012, 45, 123–131. [Google Scholar] [CrossRef]
- Hayakawa, T.; Iwai, M.; Aoki, S.; Takimoto, K.; Maruyama, M.; Maruyama, W.; Motoyama, N. SIRT1 suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PLoS ONE 2015, 10, e0116480. [Google Scholar] [CrossRef]
- Kida, Y.; Goligorsky, M.S. Sirtuins, Cell Senescence, and Vascular Aging. Can. J. Cardiol. 2016, 32, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Dang, W.; Steffen, K.K.; Perry, R.; Dorsey, J.A.; Johnson, F.B.; Shilatifard, A.; Kaeberlein, M.; Kennedy, B.K.; Berger, S.L. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 2009, 459, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Elliott, G.O.; Murphy, K.J.; Hayes, J.J.; Thiriet, C. Replication-independent nucleosome exchange is enhanced by local and specific acetylation of histone H4. Nucleic Acids Res. 2013, 41, 2228–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, R.J.; Kubicek, S.; Schreiber, S.L.; Karlseder, J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 2010, 17, 1218–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michishita, E.; McCord, R.A.; Berber, E.; Kioi, M.; Padilla-Nash, H.; Damian, M.; Cheung, P.; Kusumoto, R.; Kawahara, T.L.A.; Barrett, J.C.; et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 2008, 452, 492–496. [Google Scholar] [CrossRef]
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age related changes in NAD+ metabolism oxidative stress and sirt1 activity in wistar rats. PLoS ONE 2011, 6, e19194. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Vanhoutte, P.M.; Wang, Y. Loss-of-SIRT1 function during vascular ageing: Hyperphosphorylation mediated by cyclin-dependent kinase 5. Trends Cardiovasc. Med. 2014, 24, 81–84. [Google Scholar] [CrossRef]
- Zhang, W.; Li, J.; Suzuki, K.; Qu, J.; Wang, P.; Zhou, J.; Liu, X.; Ren, R.; Xu, X.; Ocampo, A.; et al. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 2015, 348, 1160–1163. [Google Scholar] [CrossRef] [Green Version]
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef]
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; dal Zuffo, R.; Matti, V.; d’Ario, G.; Montani, E.; et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011, 13, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Lenain, C.; De Graaf, C.A.; Pagie, L.; Visser, N.L.; De Haas, M.; De Vries, S.S.; Peric-Hupkes, D.; van Steensel, B.; Peeper, D.S. Massive reshaping of genome-nuclear lamina interactions during oncogene-induced senescence. Genome Res. 2017, 27, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Poustovoitov, M.V.; Ye, X.; Santos, H.A.; Chen, W.; Daganzo, S.M.; Erzberger, J.P.; Serebriiskii, I.G.; Canutescu, A.A.; Dunbrack, R.L.; et al. Formation of macroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell 2005, 8, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Chandra, T.; Kirschner, K.; Thuret, J.Y.; Pope, B.D.; Ryba, T.; Newman, S.; Ahmed, K.; Samarajiwa, S.A.; Salama, R.; Carroll, T.; et al. Independence of Repressive Histone Marks and Chromatin Compaction during Senescent Heterochromatic Layer Formation. Mol. Cell 2012, 47, 203–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, T.; Kirschner, K. Chromosome organisation during ageing and senescence. Curr. Opin. Cell Biol. 2016, 40, 161–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boumendil, C.; Hari, P.; Olsen, K.C.F.; Acosta, J.C.; Bickmore, W.A. Nuclear pore density controls heterochromatin reorganization during senescence. Genes Dev. 2019, 33, 144–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schermelleh, L.; Carlton, P.M.; Haase, S.; Shao, L.; Winoto, L.; Kner, P.; Burke, B.; Cardoso, M.C.; Agard, D.A.; Gustafsson, M.G.L.; et al. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 2008, 320, 1332–1336. [Google Scholar] [CrossRef]
- Burgess, R.C.; Burman, B.; Kruhlak, M.J.; Misteli, T. Activation of DNA Damage Response Signaling by Condensed Chromatin. Cell Rep. 2014, 9, 1703–1717. [Google Scholar] [CrossRef] [Green Version]
- Prieur, A.; Besnard, E.; Babled, A.; Lemaitre, J.M. P53 and p16INK4A independent induction of senescence by chromatin-dependent alteration of S-phase progression. Nat. Commun. 2011, 2, 473. [Google Scholar] [CrossRef]
- Olivieri, F.; Capri, M.; Bonafè, M.; Morsiani, C.; Jung, H.J.; Spazzafumo, L.; Viña, J.; Suh, Y. Circulating miRNAs and miRNA shuttles as biomarkers: Perspective trajectories of healthy and unhealthy aging. Mech. Ageing Dev. 2017, 165, 162–170. [Google Scholar] [CrossRef]
- Huang, X.; Luo, Y.; Mao, Y.S.; Ji, J.L. The link between long noncoding RNAs and depression. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2017, 73, 73–78. [Google Scholar] [CrossRef]
- Szafranski, K.; Abraham, K.J.; Mekhail, K. Non-coding RNA in neural function, disease, and aging. Front. Genet. 2015, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Karnati, H.K.; Panigrahi, M.K.; Gutti, R.K.; Greig, N.H.; Tamargo, I.A. MiRNAs: Key Players in Neurodegenerative Disorders and Epilepsy. J. Alzheimer’s Dis. 2015, 48, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Salta, E.; De Strooper, B. Non-coding RNAs with essential roles in neurodegenerative disorders. Lancet Neurol. 2012, 11, 189–200. [Google Scholar] [CrossRef]
- Caravia, X.M.; López-Otín, C. Regulatory roles of miRNAs in aging. Adv. Expmed. Biol. 2015, 887, 213–230. [Google Scholar]
- Kato, M.; Slack, F.J. Ageing and the small, non-coding RNA world. Ageing Res. Rev. 2013, 12, 429–435. [Google Scholar] [CrossRef] [PubMed]
- De Lencastre, A.; Pincus, Z.; Zhou, K.; Kato, M.; Lee, S.S.; Slack, F.J. MicroRNAs both promote and antagonize longevity in C. elegans. Curr. Biol. 2010, 20, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liu, J.; Xie, F.; Gao, X.; Ye, J.H.; Sun, L.Y.; Wei, R.; Ai, J. miR-124/ATF-6, a novel lifespan extension pathway of Astragalus polysaccharide in Caenorhabditis elegans. J. Cell Biochem. 2015, 116, 242–251. [Google Scholar] [CrossRef]
- Du, W.W.; Yang, W.; Fang, L.; Xuan, J.; Li, H.; Khorshidi, A.; Gupta, S.; Li, X.; Yang, B.B. MiR-17 extends mouse lifespan by inhibiting senescence signaling mediated by MKP7. Cell Death Dis. 2014, 5, e1355. [Google Scholar] [CrossRef]
- Boehm, M.; Slack, F.J. MicroRNA control of lifespan and metabolism. Cell Cycle 2006, 5, 837–840. [Google Scholar] [CrossRef]
- Poy, M.N.; Eliasson, L.; Krutzfeldt, J.; Kuwajima, S.; Ma, X.; Macdonald, P.E.; Pfeffer, S.; Tuschl, T.; Rajewsky, N.; Rorsman, P.; et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature 2004, 432, 226–230. [Google Scholar] [CrossRef]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Cobb, A.M.; Larrieu, D.; Warren, D.T.; Liu, Y.; Srivastava, S.; Smith, A.J.O.; Bowater, R.P.; Jackson, S.P.; Shanahan, C.M. Prelamin A impairs 53BP1 nuclear entry by mislocalizing NUP153 and disrupting the Ran gradient. Aging Cell 2016, 15, 1039–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shumaker, D.K.; Dechat, T.; Kohlmaier, A.; Adam, S.A.; Bozovsky, M.R.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Khuon, S.; Collins, F.S.; et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. USA 2006, 103, 8703–8708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaina, S.; Heyn, H.; Carmona, F.J.; Varol, N.; Sayols, S.; Condom, E.; Ramirez-Ruz, J.; Gomez, A.; Goncalves, I.; Moran, S.; et al. DNA methylation map of human atherosclerosis. Circ. Cardiovasc. Genet. 2014, 7, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.G.; Teschendorff, A.E.; Rakyan, V.K.; Maxwell, A.P.; Beck, S.; Savage, D.A. Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med. Genom. 2010, 3, 33. [Google Scholar] [CrossRef]
- Hansen, K.D.; Timp, W.; Bravo, H.C.; Sabunciyan, S.; Langmead, B.; McDonald, O.G.; Wen, B.; Wu, H.; Liu, Y.; Diep, D.; et al. Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 2011, 43, 768–775. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Long, T.I.; Arakawa, K.; Wang, R.; Yu, M.C.; Laird, P.W. DNA methylation as a biomarker for cardiovascular disease risk. PLoS ONE 2010, 5, e9692. [Google Scholar] [CrossRef]
- Heerboth, S.; Lapinska, K.; Snyder, N.; Leary, M.; Rollinson, S.; Sarkar, S. Use of epigenetic drugs in disease: An overview. Genet. Epigenet. 2014, 6, 9–19. [Google Scholar] [CrossRef]
- Godfrey, K.M.; Sheppard, A.; Gluckman, P.D.; Lillycrop, K.A.; Burdge, G.C.; McLean, C.; Rodford, J.; Slater-Jefferies, J.L.; Garratt, E.; Crozier, S.R.; et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes 2011, 60, 1528–1534. [Google Scholar] [CrossRef]
- Clarke-Harris, R.; Wilkin, T.J.; Hosking, J.; Pinkney, J.; Jeffery, A.N.; Metcalf, B.S.; Godfrey, K.M.; Voss, L.D.; Lillycrop, K.A.; Burdge, G.C. PGC1α promoter methylation in blood at 5-7 years predicts adiposity from 9 to 14 years (EarlyBird 50). Diabetes 2014, 63, 2528–2537. [Google Scholar] [CrossRef] [PubMed]
- Shiels, P.G.; McGuinness, D.; Eriksson, M.; Kooman, J.P.; Stenvinkel, P. The role of epigenetics in renal ageing. Nat. Rev. Nephrol 2017, 13, 471–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jasiulionis, M.G. Abnormal epigenetic regulation of immune system during aging. Front. Immunol. 2018, 9, 197. [Google Scholar] [CrossRef] [PubMed]
- Saccani, S.; Natoli, G. Dynamic changes in histone H3 Lys 9 methylation occurring at tightly regulated inducible inflammatory genes. Genes Dev. 2002, 16, 2219–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidler, C.; Wóycicki, R.; Ilnytskyy, Y.; Metz, G.; Kovalchuk, I.; Kovalchuk, O. Immunosenescence is associated with altered gene expression and epigenetic regulation in primary and secondary immune organs. Front. Genet. 2013, 4, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, K.; Ryan, D.P.; Pearson, B.L.; Henzel, K.S.; Neff, F.; Vidal, R.O.; Hennion, M.; Lehmann, I.; Schleif, M.; Schröder, S.; et al. Epigenetic alterations in longevity regulators, reduced life span, and exacerbated aging-related pathology in old father offspring mice. Proc. Natl. Acad. Sci. USA 2018, 115, E2348–E2357. [Google Scholar] [CrossRef] [Green Version]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [Green Version]
- Tobi, E.W.; Lumey, L.H.; Talens, R.P.; Kremer, D.; Putter, H.; Stein, A.D.; Slagboom, P.E.; Heijmans, B.T. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum. Mol. Genet. 2009, 18, 4046–4053. [Google Scholar] [CrossRef] [PubMed]
- Steegers-Theunissen, R.P.; Obermann-Borst, S.A.; Kremer, D.; Lindemans, J.; Siebel, C.; Steegers, E.A.; Slagboom, P.E.; Heijmans, B.T. Periconceptional maternal folic acid use of 400 μg per day is related to increased methylation of the IGF2 gene in the very young child. PLoS ONE 2009, 4, e7845. [Google Scholar] [CrossRef] [PubMed]
- Hahn, O.; Grönke, S.; Stubbs, T.M.; Ficz, G.; Hendrich, O.; Krueger, F.; Andrews, S.; Zhang, Q.; Wakelam, J.M.; Beyer, A.; et al. Dietary restriction protects from age-associated DNA methylation and induces epigenetic reprogramming of lipid metabolism. Genome Biol. 2017, 18, 56. [Google Scholar] [CrossRef]
- Hass, B.S.; Hart, R.W.; Lu, M.H.; Lyn-Cook, B.D. Effects of caloric restriction in animals on cellular function, oncogene expression, and DNA methylation in vitro. Mutat. Res. 1993, 295, 281–289. [Google Scholar] [CrossRef]
- Chouliaras, L.; van den Hove, D.L.A.; Kenis, G.; Dela Cruz, J.; Lemmens, M.A.; van Os, J.; Steinbusch, H.W.; Schmitz, C.; Rutten, B.P. Caloric restriction attenuates age-related changes of DNA methyltransferase 3a in mouse hippocampus. Brain Behav. Immun. 2011, 25, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, L.; Tollefsbol, T.O. Glucose restriction can extend normal cell lifespan and impair precancerous cell growth through epigenetic control of hTERT and p16 expression. FASEB J. 2010, 24, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Torrellas, C. Epigenetics of aging and Alzheimer’s disease: Implications for pharmacogenomics and drug response. Int. J. Mol. Sci. 2015, 16, 30483–30543. [Google Scholar] [CrossRef] [PubMed]
- Miyamura, Y.; Tawa, R.; Koizumi, A.; Uehara, Y.; Kurishita, A.; Sakurai, H.; Kamiyama, S.; Ono, T. Effects of energy restriction on age-associated changes of DNA methylation in mouse liver. Mutat. Res. 1993, 295, 63–69. [Google Scholar] [CrossRef]
- Van Straten, E.M.; Bloks, V.W.; Huijkman, N.C.; Baller, J.F.; Meer, H.; Lutjohann, D.; Kuipers, F.; Plosch, T. The liver X-receptor gene promoter is hypermethylated in a mouse model of prenatal protein restriction. Am. J. Physiol. Integr. Comp. Physiol. 2010, 298, R275–R282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, C.D.; Uthus, E.O. Dietary Folate and Selenium Affect Dimethylhydrazine-Induced Aberrant Crypt Formation, Global DNA Methylation and One-Carbon Metabolism in Rats. J. Nutr. 2003, 133, 2907–2914. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.D.; Uthus, E.O.; Finley, J.W. Dietary Selenium and Arsenic Affect DNA Methylation In Vitro in Caco-2 Cells and In Vivo in Rat Liver and Colon. J. Nutr. 2000, 130, 2903–2909. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, V.; Vaid, M.; Katiyar, S.K. (−)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis 2011, 32, 537–544. [Google Scholar] [CrossRef]
- Chung, S.; Yao, H.; Caito, S.; Hwang, J.W.; Arunachalam, G.; Rahman, I. Regulation of SIRT1 in cellular functions: Role of polyphenols. Arch Biochem. Biophys. 2010, 501, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Xie, Z.; Jones, W.; Pavlovicz, R.E.; Liu, S.; Yu, J.; Li, P.K.; Lin, J.; Fuchs, J.R.; Marcucci, G.; et al. Curcumin is a potent DNA hypomethylation agent. Bioorganic Med. Chem. Lett. 2009, 19, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Ayissi, V.B.O.; Ebrahimi, A.; Schluesenner, H. Epigenetic effects of natural polyphenols: A focus on SIRT1-mediated mechanisms. Mol. Nutr. Food Res. 2014, 58, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.Z.; Wang, Y.; Ai, N.; Hou, Z.; Sun, Y.; Lu, H.; Welsh, W.; Yang, C.S. Tea Polyphenol (−)-Epigallocatechin-3-Gallate Inhibits DNA Methyltransferase and Reactivates Methylation-Silenced Genes in Cancer Cell Lines. Cancer Res. 2003, 63, 7563–7570. [Google Scholar] [PubMed]
- Handel, M.L.; Watts, C.K.; DeFazio, A.; Day, R.O.; Sutherland, R.L. Inhibition of AP-1 binding and transcription by gold and selenium involving conserved cysteine residues in Jun and Fos. Proc. Natl. Acad. Sci. USA 1995, 92, 4497–4501. [Google Scholar] [CrossRef] [PubMed]
- Spyrou, G.; Björnstedt, M.; Kumar, S.; Holmgren, A. AP-1 DNA-binding activity is inhibited by selenite and selenodiglutathione. FEBS Lett. 1995, 368, 59–63. [Google Scholar] [CrossRef] [Green Version]
- Xiang, N.; Zhao, R.; Song, G.; Zhong, W. Selenite reactivates silenced genes by modifying DNA methylation and histones in prostate cancer cells. Carcinogenesis 2008, 29, 2175–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.J. Mechanisms for the Inhibition of DNA Methyltransferases by Tea Catechins and Bioflavonoids. Mol. Pharm. 2005, 68, 1018–1030. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Wang, C.; Lu, C.; Zhao, B.; Cui, Y.; Shi, X. and Ma, X. Quercetin is able to demethylate the p16INK4a gene promoter. Chemotherapy 2009, 55, 6–10. [Google Scholar] [CrossRef]
- Wolff, G.L.; Kodell, R.L.; Moore, S.R.; Cooney, C.A. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J. 1998, 12, 949–957. [Google Scholar] [CrossRef]
- Dolinoy, D.C.; Weidman, J.R.; Waterland, R.A.; Jirtle, R.L. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ. Health Perspect. 2006, 114, 567–572. [Google Scholar] [CrossRef]
- Waterland, R.A.; Travisano, M.; Tahiliani, K.G.; Rached, M.T.; Mirza, S. Methyl donor supplementation prevents transgenerational amplification of obesity. Int. J. Obes. 2008, 32, 1373–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harkness, R.W.; Mittermaier, A.K. G-quadruplex dynamics. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- François, M.; Leifert, W.; Tellam, R.; Fenech, M. G-quadruplexes: A possible epigenetic target for nutrition. Mutat. Res. 2015, 764, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Zhu, W.; Shi, H.; Hewett, J.E.; Ruhlen, R.L.; MacDonald, R.S.; Rottinghaus, G.E.; Chen, Y.C.; Sauter, E.R. Soy isoflavones have an antiestrogenic effect and alter mammary promoter hypermethylation in healthy premenopausal women. Nutr. Cancer 2009, 61, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Majid, S.; Dar, A.A.; Shahryari, V.; Hirata, H.; Ahmad, A.; Saini, S.; Tanaka, Y.; Dahiya, A.V.; Dahiya, R. Genistein reverses hypermethylation and induces active histone modifications in tumor suppressor gene B-cell translocation gene 3 in prostate cancer. Cancer 2010, 116, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, M.K.; Ansari, M.Z.; Al Mutery, A.; Ashraf, M.; Nasab, R.; Rai, S.; Rais, N.; Hussain, A. Genistein induces alterations of epigenetic modulatory signatures in human cervical cancer cells. Anticancer Agents Med. Chem. 2018, 18, 412–421. [Google Scholar] [CrossRef]
- Priyadarsini, R.V.; Vinothini, G.; Murugan, R.S.; Manikandan, P.; Nagini, S. The flavonoid quercetin modulates the hallmark capabilities of hamster buccal pouch tumors. Nutr. Cancer 2011, 63, 218–226. [Google Scholar] [CrossRef]
- Hong, K.S.; Park, J.I.; Kim, M.J.; Kim, H.B.; Lee, J.W.; Dao, T.T.; Oh, W.K.; Kang, C.D.; Kim, S.H. Involvement of SIRT1 in hypoxic down-regulation of c-Myc and β-catenin and hypoxic preconditioning effect of polyphenols. Toxicol. Appl. Pharm. 2012, 259, 210–218. [Google Scholar] [CrossRef]
- Attoub, S.; Hassan, A.H.; Vanhoecke, B.; Iratni, R.; Takahashi, T.; Gaben, A.M.; Bracke, M.; Awad, S.; John, A.; Kamalboor, H.A.; et al. Inhibition of cell survival, invasion, tumor growth and histone deacetylase activity by the dietary flavonoid luteolin in human epithelioid cancer cells. Eur. J. Pharm. 2011, 651, 18–25. [Google Scholar] [CrossRef]
- Kang, S.K.; Cha, S.H.; Jeon, H.G. Curcumin-induced Histone Hypoacetylation Enhances Caspase-3-dependent Glioma Cell Death and Neurogenesis of Neural Progenitor Cells. Stem Cells Dev. 2006, 15, 165–174. [Google Scholar] [CrossRef]
- Teiten, M.H.; Dicato, M.; Diederich, M. Curcumin as a regulator of epigenetic events. Mol. Nutr. Food Res. 2013, 57, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, W.; Suszek, M.; Wnuk, M.; Lewinska, A.; Wasiak, E.; Sikora, E.; Bielak-Zmijewska, A. Curcumin elevates sirtuin level but does not postpone in vitro senescence of human cells building the vasculature. Oncotarget 2016, 7, 19201–19213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, K.C.; Myung, G.J.; Lee, Y.H.; Yoon, J.C.; Kwon, S.H.; Kang, H.B.; Kim, M.J.; Cha, J.H.; Kim, Y.J.; Jun, W.J.; et al. Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Res. 2009, 69, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.; Shukla, S.; Gupta, S. Promoter demethylation and chromatin remodeling by green tea polyphenols leads to re-expression of GSTP1 in human prostate cancer cells. Int. J. Cancer 2010, 126, 2520–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.; Shi, D.; Liu, L.; Wang, J.; Xie, X.; Kang, T.; Deng, W. Quercetin suppresses cyclooxygenase-2 expression and angiogenesis through inactivation of P300 signaling. PLoS ONE 2011, 6, e22934. [Google Scholar] [CrossRef]
- Lee, W.J.; Chen, Y.R.; Tseng, T.H. Quercetin induces FasL-related apoptosis, in part, through promotion of histone H3 acetylation in human leukemia HL-60 cells. Oncol. Rep. 2011, 25, 583–591. [Google Scholar] [PubMed]
- Lin, C.; Kang, J.; Zheng, R. Oxidative stress is involved in inhibition of copper on histone acetylation in cells. Chem. Biol. Interact 2005, 151, 167–176. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Adhikary, G.; Eckert, R.L. The Bmi-1 polycomb protein antagonizes the (-)-epigallocatechin-3-gallate-dependent suppression of skin cancer cell survival. Carcinogenesis 2010, 31, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.P.; Tryndyak, V.P.; Muskhelishvili, L.; Rusyn, I.; Ross, S.A. Methyl deficiency, alterations in global histone modifications, and carcinogenesis. J. Nutr. 2007, 137, 216S–222S. [Google Scholar] [CrossRef]
- Giblin, W.; Skinner, M.E.; Lombard, D.B. Sirtuins: Guardians of mammalian healthspan. Trends Genet. 2014, 30, 271–286. [Google Scholar] [CrossRef]
- Jayasena, T.; Poljak, A.; Smythe, G.; Braidy, N.; Münch, G.; Sachdev, P. The role of polyphenols in the modulation of sirtuins and other pathways involved in Alzheimer’s disease. Ageing Res. Rev. 2013, 12, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, B.P.; Sinclair, D.A. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharm. Sci. 2014, 35, 146–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greathouse, K.L.; Samuels, M.; DiMarco, N.M.; Criswell, D.S. Effects of increased dietary fat and exercise on skeletal muscle lipid peroxidation and antioxidant capacity in male rats. Eur. J. Nutr. 2005, 44, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Chung, H.Y.; Koltai, E.; Taylor, A.W.; Goto, S. Exercise, oxidative stress and hormesis. Ageing Res. Rev. 2008, 44, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Chung, H.Y.; Goto, S. Systemic adaptation to oxidative challenge induced by regular exercise. Free Radic Biol. Med. 2008, 44, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Vaiserman, A.M.; Lushchak, O.V.; Koliada, A.K. Anti-aging pharmacology: Promises and pitfalls. Ageing Res. Rev. 2016, 31, 9–35. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.L.; Benzer, S.; Min, K.T. Life extension in Drosophila by feeding a drug. Proc. Natl. Acad. Sci. USA 2002, 99, 838–843. [Google Scholar] [CrossRef]
- Tao, D.; Lu, J.; Sun, H.; Zhao, Y.M.; Yuan, Z.G.; Li, X.X.; Huang, B.Q. Trichostatin A Extends the Lifespan of Drosophila melanogaster by Elevating hsp22 Expression. Acta Biochim. Biophys. Sin. 2004, 36, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Sun, H.; Lu, J.; Li, X.; Chen, X.; Tao, D.; Huang, W.; Huang, B. Lifespan extension and elevated hsp gene expression in Drosophila caused by histone deacetylase inhibitors. J. Exp. Biol. 2005, 208, 697–705. [Google Scholar] [CrossRef]
- Christensen, D.P.; Dahllöf, M.; Lundh, M.; Rasmussen, D.N.; Nielsen, M.D.; Billestrup, N.; Grunnet, L.G.; Mandrup-Poulsen, T. Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol. Med. Camb. Mass 2011, 17, 378–390. [Google Scholar] [CrossRef]
- Penney, J.; Tsai, L.H. Histone deacetylases in memory and cognition. Sci. Signal 2014, 7, re12. [Google Scholar] [CrossRef]
- Ganai, S.A.; Ramadoss, M.; Mahadevan, V. Histone Deacetylase (HDAC) Inhibitors—Emerging roles in neuronal memory, learning, synaptic plasticity and neural regeneration. Curr. Neuropharmacol. 2016, 14, 55–71. [Google Scholar] [CrossRef]
- Wang, L.; Yu, C.; Yu, G.; Lu, X.; Yu, D. Histone Acetylation Modifiers in the Pathogenesis of Alzheimer’s Disease. Front. Cell Neurosci. 2015, 9, 226. [Google Scholar]
- Sharma, S.; Taliyan, R. Targeting Histone Deacetylases: A Novel Approach in Parkinson’s Disease. Parkinsons Dis. 2015, 2015, 303294. [Google Scholar] [CrossRef]
- Chakrabarti, M.; Khandkar, M.; Banik, N.L.; Ray, S.K. Alterations in expression of specific microRNAs by combination of 4-HPR and EGCG inhibited growth of human malignant neuroblastoma cells. Brain Res. 2012, 1454, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Boesch-Saadatmandi, C.; Wagner, A.E.; Wolffram, S.; Rimbach, G. Effect of quercetin on inflammatory gene expression in mice liver in vivo—Role of redox factor 1, miRNA-122 and miRNA-125b. Pharm. Res. 2012, 65, 523–530. [Google Scholar] [CrossRef]
- Li, Y.; Vandenboom, T.G.; Kong, D.; Wang, Z.; Ali, S.; Philip, P.A.; Sarkar, F.H. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009, 69, 6. [Google Scholar] [CrossRef]
- Sun, M.; Estrov, Z.; Ji, Y.; Coombes, K.R.; Harris, D.H.; Kurzrock, R. Curcumin (diferuloylmethane) alters the expression profiles of microRNAs in human pancreatic cancer cells. Mol. Cancer 2008, 7, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.Z.; Zang, J.N.; Deng, L.L.; Wang, Z.Y.; Yu, C.H. Pentamethylquercetin reduces fat deposition via Sirt1-mediated pathways in male obese mice induced by a high fat diet. Food Chem. Toxicol. 2013, 62, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Barrès, R.; Yan, J.; Egan, B.; Treebak, J.T.; Rasmussen, M.; Fritz, T.; Caidahl, K.; Krook, A.; O’Gorman, D.J.; Zierath, J.R. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012, 15, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Barrès, R.; Kirchner, H.; Rasmussen, M.; Yan, J.; Kantor, F.R.; Krook, A.; Näslund, E.; Zierath, J.R. Weight Loss after Gastric Bypass Surgery in Human Obesity Remodels Promoter Methylation. Cell Rep. 2013, 3, 1020–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobsen, S.C.; Gillberg, L.; Bork-Jensen, J.; Ribel-Madsen, R.; Lara, E.; Calvanese, V.; Ling, C.; Fernandez, A.F.; Fraga, M.F.; Poulsen, P.; et al. Young men with low birthweight exhibit decreased plasticity of genome-wide muscle DNA methylation by high-fat overfeeding. Diabetologia 2014, 57, 1154–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rönn, T.; Volkov, P.; Davegárdh, C.; Dayeh, T.; Hall, E.; Olsson, A.H.; Nilsson, E.; Tornberg, A.; Dekker Nitert, M.; Eriksson, K.F.; et al. A Six Months Exercise Intervention Influences the Genome-wide DNA Methylation Pattern in Human Adipose Tissue. PLoS Genet. 2013, 9, e1003572. [Google Scholar] [CrossRef] [PubMed]
- Weems, J.C.; Griesel, B.A.; Olson, A.L. Class II histone deacetylases downregulate GLUT4 transcription in response to increased cAMP signaling in cultured adipocytes and fasting mice. Diabetes 2012, 61, 1404–1414. [Google Scholar] [CrossRef] [PubMed]
- McGee, S.L.; Fairlie, E.; Garnham, A.P.; Hargreaves, M. Exercise-induced histone modifications in human skeletal muscle. J. Physiol. 2009, 587, 5951–5958. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.J.; Fairall, L.; Schwabe, J.W.R. Nuclear hormone receptor co-repressors: Structure and function. Mol. Cell Endocrinol. 2012, 348, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Franks, A.L.; Slansky, J.E. Multiple associations between a broad spectrum of autoimmune diseases, chronic inflammatory diseases and cancer. Anticancer Res. 2012, 32, 41119–41136. [Google Scholar]
- Flowers, E.; Won, G.Y.; Fukuoka, Y. MicroRNAs associated with exercise and diet: A systematic review. Physiol. Genom. 2014, 47, 1–11. [Google Scholar] [CrossRef]
- Cummins, J.M.; He, Y.; Leary, R.J.; Pagliarini, R.; Diaz, L.A.; Sjoblom, T.; Barad, O.; Bentwich, Z.; Szafranska, A.E.; Labourier, E.; et al. The colorectal microRNAome. Proc. Natl. Acad. Sci. USA 2006, 103, 3687–3692. [Google Scholar] [CrossRef] [Green Version]
- Rowlands, D.S.; Page, R.A.; Sukala, W.R.; Giri, M.; Ghimbovschi, S.D.; Hayat, I.; Cheema, B.S.; Lys, I.; Leikis, M.; Sheard, P.W.; et al. Multi-omic integrated networks connect DNA methylation and miRNA with skeletal muscle plasticity to chronic exercise in Type 2 diabetic obesity. Physiol. Genom. 2014, 46, 747–765. [Google Scholar] [CrossRef]
- Denham, J.; O’Brien, B.J.; Marques, F.Z.; Charchar, F.J. Changes in the leukocyte methylome and its effect on cardiovascular-related genes after exercise. J. Appl. Physiol. 2015, 118, 475–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laker, R.C.; Wlodek, M.E.; Connelly, J.J.; Yan, Z. Epigenetic origins of metabolic disease: The impact of the maternal condition to the offspring epigenome and later health consequences. Food Sci. Hum. Wellness 2013, 2, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Chen, N. Regulatory role of microRNAs in muscle atrophy during exercise intervention. Int. J. Mol. Sci. 2018, 19, 405. [Google Scholar] [CrossRef] [PubMed]
- Mancini, A.; Vitucci, D.; Labruna, G.; Imperlini, E.; Randers, M.B.; Schmidt, J.F.; Hagman, M.; Andersen, T.R.; Russo, R.; Orrù, S.; et al. Effect of lifelong football training on the expression of muscle molecular markers involved in healthy longevity. Eur. J. Appl. Physiol. 2017, 117, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Lew, J.K.S.; Pearson, J.T.; Schwenke, D.O.; Katare, R. Exercise mediated protection of diabetic heart through modulation of microRNA mediated molecular pathways. Cardiovasc. Diabetol. 2017, 16, 10. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, L.R.; Kumsta, C.; Sandri, M.; Ballabio, A.; Hansen, M. Transcriptional and epigenetic regulation of autophagy in aging. Autophagy 2015, 6, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K. SIRT1: Regulation of longevity via autophagy. Cell Signal 2009, 21, 1356–1360. [Google Scholar] [CrossRef] [PubMed]
- Artal-Martinez de Narvajas, A.; Gomez, T.S.; Zhang, J.J.S.; Mann, A.O.; Taoda, Y.; Gorman, J.A.; Herreros-Villanueva, M.; Gress, T.M.; Ellenrieder, V.; Bujanda, L.; et al. Epigenetic Regulation of Autophagy by the Methyltransferase G9a. Mol. Cell Biol. 2013, 33, 3983–3993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Li, X.; Zhu, J.X.; Xie, W.; Le, W.; Fan, Z.; Jankovic, J.; Pan, T. Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. Neurosignals 2011, 19, 163–174. [Google Scholar] [CrossRef]
- Eisenberg, T.; Schroeder, S.; Andryushkova, A.; Pendl, T.; Küttner, V.; Bhukel, A.; Mariño, G.; Pietrocola, F.; Harger, A.; Zimmermann, S.; et al. Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme A stimulates autophagy and prolongs lifespan. Cell Metab. 2014, 19, 431–444. [Google Scholar] [CrossRef]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Büttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Khanna, A.; Muthusamy, S.; Liang, R.; Sarojini, H.; Wang, E. Gain of survival signaling by down-regulation of three key miRNAs in brain of calorie-restricted mice. Aging 2011, 3, 223–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Chen, D.; He, Y.; Melendez, A.; Feng, Z.; Hong, Q.; Bai, X.; Li, Q.; Cai, G.; Wang, J.; et al. MiR-34 modulates Caenorhabditis elegans lifespan via repressing the autophagy gene atg9. Age 2013, 35, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Muñoz, M.E.; Arrieta, M.C.; Ramer-Tait, A.E.; Walter, J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: Implications for research on the pioneer infant microbiome. Microbiome 2017, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.C.; Rautava, S.; Aakko, J.; Isolauri, E.; Salminen, S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 2016, 6, 23129. [Google Scholar] [CrossRef]
- Walker, R.W.; Clemente, J.C.; Peter, I.; Loos, R.J.F. The prenatal gut microbiome: Are we colonized with bacteria in utero? Pediatr. Obes. 2017, 12, 3–17. [Google Scholar] [CrossRef]
- Lim, E.S.; Rodriguez, C.; Holtz, L.R. Amniotic fluid from healthy term pregnancies does not harbor a detectable microbial community. Microbiome 2018, 6, 87. [Google Scholar] [CrossRef]
- Rehbinder, E.M.; Lødrup Carlsen, K.C.; Staff, A.C.; Angell, I.L.; Landrø, L.; Hilde, K.; Gaustad, P.; Rudi, K. Is amniotic fluid of women with uncomplicated term pregnancies free of bacteria? Am. J. Obs. Gynecol. 2018, 219, e1–e289. [Google Scholar] [CrossRef]
- Mackie, R.I.; Sghir, A.; Gaskins, H.R. Developmental microbial ecology of the neonatal gastrointestinal tract. Am. J. Clin. Nutr. 1999, 69, 1035S–1045S. [Google Scholar] [CrossRef]
- Li, H.; Wang, J.; Wu, L.; Luo, J.; Liang, X.; Xiao, B.; Zhu, Y. The impacts of delivery mode on infant’s oral microflora. Sci. Rep. 2018, 8, 11938. [Google Scholar] [CrossRef]
- Rodríguez, J.M.; Murphy, K.; Stanton, C.; Ross, R.P.; Kober, O.I.; Juge, N.; Avershina, E.; Rudi, K.; Narbad, A.; Jenmalm, M.C.; et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb. Ecol. Heal Dis. 2015, 26, 26050. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Zeng, D.N.; Chi, L.; Tan, Y.; Galzote, C.; Cardona, C.; Lax, S.; Gilbert, J.; Quan, Z.X. The influence of age and gender on skin-associated microbial communities in urban and rural human populations. PLoS ONE 2015, 10, e0141842. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Codella, R.; Luzi, L.; Terruzzi, I. Exercise has the guts: How physical activity may positively modulate gut microbiota in chronic and immune-based diseases. Dig. Liver Dis. 2018, 50, 331–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mach, N.; Fuster-Botella, D. Endurance exercise and gut microbiota: A review. J. Sport Heal Sci. 2017, 6, 179–197. [Google Scholar] [CrossRef] [PubMed]
- Palleja, A.; Mikkelsen, K.H.; Forslund, S.K.; Kashani, A.; Allin, K.H.; Nielsen, T.; Hansen, T.H.; Liang, S.; Feng, Q.; Zhang, C.; et al. Recovery of gut microbiota of healthy adults following antibiotic exposure. Nat. Microbiol. 2018, 3, 1255–1265. [Google Scholar] [CrossRef]
- Maier, L.; Pruteanu, M.; Kuhn, M.; Zeller, G.; Telzerow, A.; Anderson, E.E.; Brochado, A.R.; Fernandez, K.C.; Dose, H.; Mori, H.; et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 2018, 555, 623–628. [Google Scholar] [CrossRef]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Tasnim, N.; Abulizi, N.; Pither, J.; Hart, M.M.; Gibson, D.L. Linking the gut microbial ecosystem with the environment: Does gut health depend on where we live? Front. Microbiol. 2017, 8, 1935. [Google Scholar] [CrossRef] [PubMed]
- Salazar, N.; Arboleya, S.; Valdés, L.; Stanton, C.; Ross, P.; Ruiz, L.; Gueimonde, M.; de los Reyes-Gavilan, C.G. The human intestinal microbiome at extreme ages of life. Dietary intervention as a way to counteract alterations. Front. Genet. 2014, 5, 406. [Google Scholar] [CrossRef]
- Rolhion, N.; Chassaing, B. When pathogenic bacteria meet the intestinal microbiota. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascal, M.; Perez-Gordo, M.; Caballero, T.; Escribese, M.M.; Lopez Longo, M.N.; Luengo, O.; Manso, L.; Matheu, V.; Seoane, E.; Zamorano, M.; et al. Microbiome and allergic diseases. Front. Immunol. 2018, 9, 1584. [Google Scholar] [CrossRef] [PubMed]
- De la Cuesta-Zuluaga, J.; Corrales-Agudelo, V.; Velásquez-Mejía, E.P.; Carmona, J.A.; Abad, J.M.; Escobar, J.S. Gut microbiota is associated with obesity and cardiometabolic disease in a population in the midst of Westernization. Sci. Rep. 2018, 8, 11356. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Ling, Z.; Zhang, Y.; Mao, H.; Ma, Z.; Yin, Y.; Wang, W.; Tang, W.; Tan, Z.; Shi, J.; et al. Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav. Immun. 2015, 48, 186–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luna, R.A.; Foster, J.A. Gut brain axis: Diet microbiota interactions and implications for modulation of anxiety and depression. Curr. Opin. Biotechnol. 2015, 32, 35–41. [Google Scholar] [CrossRef]
- Kataoka, K. The intestinal microbiota and its role in human health and disease. J. Med. Investig. 2016, 63, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.; Mach, N. The crosstalk between the gut microbiota and mitochondria during exercise. Front. Physiol. 2017, 8, 319. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Gordon, J.I. The core gut microbiome, energy balance and obesity. J. Physiol. 2009, 587, 4153–4158. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ticinesi, A.; Lauretani, F.; Milani, C.; Nouvenne, A.; Tana, C.; Del Rio, D.; Maggio, M.; Ventura, M.; Meschi, T.; Lauretani, F.; et al. Aging gut microbiota at the cross-road between nutrition, physical frailty, and sarcopenia: Is there a gut–muscle axis? Nutrients 2017, 9, 1303. [Google Scholar] [CrossRef]
- Tran, L.; Greenwood-Van Meerveld, B. Age-associated remodeling of the intestinal epithelial barrier. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Holt, P.R. Intestinal malabsorption in the elderly. Dig. Dis. 2007, 25, 144–150. [Google Scholar] [CrossRef]
- Bottazzi, B.; Riboli, E.; Mantovani, A. Aging, inflammation and cancer. Semin Immunol. 2018, 40, 74–82. [Google Scholar] [CrossRef]
- Zinger, A.; Cho, W.C.; Ben-Yehuda, A. Cancer and Aging—The Inflammatory Connection. Aging Dis. 2018, 8, 611–627. [Google Scholar] [CrossRef]
- Mayer, E.A.; Tillisch, K.; Gupta, A. Gut/brain axis and the microbiota. J. Clin. Investig. 2015, 125, 926–938. [Google Scholar] [CrossRef]
- Dinan, T.G.; Cryan, J.F. The Microbiome-Gut-Brain Axis in Health and Disease. Gastroenterol. Clin. North Am. 2017, 46, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Claesson, M.J.; Cusack, S.; O’Sullivan, O.; Greene-Diniz, R.; de Weerd, H.; Flannery, E.; Marchesi, J.R.; Falush, D.; Dinan, T.; Fitzgerald, G.; et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc. Natl. Acad. Sci. USA 2011, 108, 4586–4591. [Google Scholar] [CrossRef] [PubMed]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Odamaki, T.; Kato, K.; Sugahara, H.; Hashikura, N.; Takahashi, S.; Xiao, J.Z.; Abe, F.; Osawa, R. Age-related changes in gut microbiota composition from newborn to centenarian: A cross-sectional study. BMC Microbiol. 2016, 16, 90. [Google Scholar] [CrossRef]
- Jackson, M.; Jeffery, I.B.; Beaumont, M.; Bell, J.T.; Clark, A.G.; Ley, R.E.; O’Toole, P.W.; Spector, T.D.; Steves, C.J. Signatures of early frailty in the gut microbiota. Genome Med. 2016, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Aidy, S.E.; van den Bogert, B.; Kleerebezem, M. The small intestine microbiota, nutritional modulation and relevance for health. Curr. Opin. Biotechnol. 2015, 32, 14–20. [Google Scholar] [CrossRef]
- Amer, S.; Manzar, H.S. Small intestinal bacterial overgrowth in older people. Rev. Clin. Gerontol. 2015, 25, 81–85. [Google Scholar] [CrossRef]
- Sicard, J.F.; Le Bihan, G.; Vogeleer, P.; Jacques, M.; Harel, J. Interactions of Intestinal Bacteria with Components of the Intestinal Mucus. Front. Cell Infect. Microbiol. 2017, 7, 387. [Google Scholar] [CrossRef]
- Quigley, E.M.M. Gut bacteria in health and disease. Gastroenterol. Hepatol. 2013, 9, 560–569. [Google Scholar]
- O’Toole, P.W.; Claesson, M.J. Gut microbiota: Changes throughout the lifespan from infancy to elderly. Int. Dairy J. 2010, 20, 281–291. [Google Scholar] [CrossRef]
- Hopkins, M.J.; Sharp, R.; Macfarlane, G.T. Variation in human intestinal microbiota with age. Dig. Liver Dis. 2002, 34, S12–S18. [Google Scholar] [CrossRef]
- König, J.; Wells, J.; Cani, P.D.; García-Ródenas, C.L.; MacDonald, T.; Mercenier, A.; Whyte, J.; Troost, F.; Brummer, R.J. Human intestinal barrier function in health and disease. Clin. Transl. Gastroenterol. 2016, 7, e196. [Google Scholar] [CrossRef] [PubMed]
- Herrou, J.; Choi, V.M.; Bubeck Wardenburg, J.; Crosson, S. Activation Mechanism of the Bacteroides fragilis Cysteine Peptidase, Fragipain. Biochemistry 2016, 55, 4077–4084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoni, L.; Nuding, S.; Wehkamp, J.; Stange, E.F. Intestinal barrier in inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 1165–1179. [Google Scholar] [CrossRef] [PubMed]
- Mulak, A.; Bonaz, B. Brain-gut-microbiota axis in Parkinson’s disease. World J. Gastroenterol. 2015, 21, 10609–10620. [Google Scholar] [CrossRef]
- Kowalski, K.; Mulak, A. Brain-gut-microbiota axis in Alzheimer’s disease. J. Neurogastroenterol. Motil. 2019, 25, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Raskov, H.; Burcharth, J.; Pommergaard, H.C. Linking gut microbiota to colorectal cancer. J. Cancer 2017, 8, 3378–3395. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal permeability—A new target for disease prevention and therapy. BMC Gastroenterol. 2014, 14, 189. [Google Scholar] [CrossRef] [PubMed]
- Mihajlovski, A.; Doré, J.; Levenez, F.; Alric, M.; Brugère, J.F. Molecular evaluation of the human gut methanogenic archaeal microbiota reveals an age-associated increase of the diversity. Environ. Microbiol. Rep. 2010, 2, 272–280. [Google Scholar] [CrossRef]
- Pimentel, M.; Lin, H.C.; Enayati, P.; van den Burg, B.; Lee, H.R.; Chen, J.H.; Park, S.; Kong, Y.; Conklin, J. Methane, a gas produced by enteric bacteria, slows intestinal transit and augments small intestinal contractile activity. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G1089–G1095. [Google Scholar] [CrossRef] [Green Version]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B. Topographical and temporal diversity of the human skin microbiome. Science 2009, 324, 1190–1192. [Google Scholar] [CrossRef]
- Jugé, R.; Rouaud-Tinguely, P.; Breugnot, J.; Servaes, K.; Grimaldi, C.; Roth, M.P.; Coppin, H.; Closs, B. Shift in skin microbiota of Western European women across aging. J. Appl. Microbiol. 2018, 125, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Shibagaki, N.; Suda, W.; Clavaud, C.; Bastien, P.; Takayasu, L.; Iioka, E.; Kurokawa, R.; Yamashita, N.; Hattori, Y.; Shindo, C.; et al. Aging-related changes in the diversity of women’s skin microbiomes associated with oral bacteria. Sci. Rep. 2017, 7, 10567. [Google Scholar] [CrossRef] [PubMed]
- Leyden, J.J.; McGinley, K.J.; Mills, O.H.; Kligman, A.M. Age related changes in the resident bacterial flora of the human face. J. Investig. Derm. 1975, 65, 379–381. [Google Scholar] [CrossRef]
- Ravn, A.H.; Thyssen, J.P.; Egeberg, A. Skin disorders in Parkinson’s disease: Potential biomarkers and risk factors. Clin. Cosmet. Investig. Derm. 2017, 10, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, D.K.; Sinclair, E.; Xu, Y.; Sarkar, D.; Walton-Doyle, C.; Liscio, C.; Banks, P.; Milne, J.; Silverdale, M.; Kunath, T.; et al. Discovery of volatile biomarkers of Parkinson ’ s disease from sebum. ACS Central Sci. 2019, 5, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Besa, V.; Teschler, H.; Kurth, I.; Khan, A.M.; Zarogoulidis, P.; Baumbach, J.I.; Sommerwerck, U.; Freitag, L.; Darwiche, K. Exhaled volatile organic compounds discriminate patients with chronic obstructive pulmonary disease from healthy subjects. Int. J. Chronic Obstr. Pulm. Dis. 2015, 10, 399–406. [Google Scholar] [Green Version]
- Bari, M.R.; Hiron, M.M.; Zaman, S.M.; Rahman, M.M.; Ganguly, K.C. Microbes responsible for acute exacerbation of COPD. Mymensingh Med. J. 2010, 19, 576–585. [Google Scholar]
- Shaw, J.G.; Vaughan, A.; Dent, A.G.; O’Hare, P.E.; Goh, F.; Bowman, R.V.; Fong, K.M.; Yang, I.A. Biomarkers of progression of chronic obstructive pulmonary disease (COPD). J. Thorac. Dis. 2014, 6, 1532–1547. [Google Scholar]
- Shukla, S.D.; Budden, K.F.; Neal, R.; Hansbro, P.M. Microbiome effects on immunity, health and disease in the lung. Clin. Transl. Immunol. 2017, 6, e133. [Google Scholar] [CrossRef]
- Coburn, B.; Wang, P.W.; Diaz Caballero, J.; Clark, S.T.; Brahma, V.; Donaldson, S.; Zhang, Y.; Surendra, A.; Gong, Y.; Elizabeth Tullis, D.; et al. Lung microbiota across age and disease stage in cystic fibrosis. Sci. Rep. 2015, 5, 10241. [Google Scholar] [CrossRef]
- Preza, D.; Olsen, I.; Willumsen, T.; Grinde, B.; Paster, B.J. Diversity and site-specificity of the oral microflora in the elderly. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 1033–1040. [Google Scholar] [CrossRef] [Green Version]
- Socransky, S.S.; Haffajee, A.D.; Cugini, M.A.; Smith, C.; Kent, R.L. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 1998, 25, 134–144. [Google Scholar] [CrossRef]
- Feres, M.; Teles, F.; Teles, R.; Figueiredo, L.C.; Faveri, M. The subgingival periodontal microbiota of the aging mouth. Periodontol. 2000 2016, 72, 30–53. [Google Scholar] [CrossRef] [PubMed]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Backhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.H.W.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal Microbial Metabolism of Phosphatidylcholine and Cardiovascular Risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Xia, M.; Yan, X.; Li, D.; Wang, L.; Xu, Y.; Jin, T.; Ling, W. Gut microbiota metabolism of anthocyanin promotes reverse cholesterol transport in mice via repressing miRNA-10b. Circ. Res. 2012, 111, 967–981. [Google Scholar] [CrossRef]
- Ohno, M.; Nishida, A.; Sugitani, Y.; Nishino, K.; Inatomi, O.; Sugimoto, M.; Kawahara, M.; Andoh, A. Nanoparticle curcumin ameliorates experimental colitis via modulation of gut microbiota and induction of regulatory T cells. PLoS ONE 2017, 12, e0185999. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, Y.; Xiang, L.; Wang, Z.; Xiao, G.G.; Hu, J. Effect of curcumin on the diversity of gut microbiota in ovariectomized rats. Nutrients 2017, 9, E1146. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Liu, L.; Ji, H.F. Regulative effects of curcumin spice administration on gut microbiota and its pharmacological implications. Food Nutr. Res. 2017, 61, 1361780. [Google Scholar] [CrossRef] [PubMed]
- Mayta-Apaza, A.C.; Pottgen, E.; De Bodt, J.; Papp, N.; Marasini, D.; Howard, L.; Abranko, L.; Van de Wiele, T.; Lee, S.O.; Carbonero, F. Impact of tart cherries polyphenols on the human gut microbiota and phenolic metabolites in vitro and in vivo. J. Nutr. Biochem. 2018, 59, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Tomás-Barberán, F.A.; Selma, M.V.; Espín, J.C. Interactions of gut microbiota with dietary polyphenols and consequences to human health. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Dueñas, M.; Muñoz-González, I.; Cueva, C.; Jiménez-Girón, A.; Sánchez-Patán, F.; Santos-Buelga, C.; Moreno-Arribas, M.V.; Bartolomé, B. A survey of modulation of gut microbiota by dietary polyphenols. BioMed Res. Int. 2015, 2015, 850902. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.T.; Vaughn, A.R.; Sharma, V.; Chopra, D.; Mills, P.J.; Peterson, S.N.; Sivamani, R.K. Effects of Turmeric and Curcumin Dietary Supplementation on Human Gut Microbiota: A Double-Blind, Randomized, Placebo-Controlled Pilot Study. J. Evid. Based Integr. Med. 2018, 23, 399–406. [Google Scholar] [CrossRef]
- Zam, W. Gut Microbiota as a Prospective Therapeutic Target for Curcumin: A Review of Mutual Influence. J. Nutr. Metab. 2018, 2018, 1367984. [Google Scholar] [CrossRef]
- Perez, A.; Gonzalez-Manzano, S.; Jimene, z.R.; Perez-Abud, R.; Haro, J.M.; Osuna, A.; Santos-Buelga, C.; Duarte, J.; Perez-Vizcaino, F. The flavonoid quercetin induces acute vasodilator effects in healthy volunteers: Correlation with beta-glucuronidase activity. Pharm. Res. 2014, 89, 11–18. [Google Scholar] [CrossRef]
- Menendez, C.; Dueñas, M.; Galindo, P.; González-Manzano, S.; Jimenez, R.; Moreno, L.; Zarzuelo, M.J.; Rodríguez-Gómez, I.; Duarte, J.; Santos-Buelga, C.; et al. Vascular deconjugation of quercetin glucuronide: The flavonoid paradox revealed? Mol. Nutr. Food Res. 2011, 55, 1780–1790. [Google Scholar] [CrossRef]
- Sasaki, H.; Sunagawa, Y.; Takahashi, K.; Imaizumi, A.; Fukuda, H.; Hashimoto, T.; Wada, H.; Katanasaka, Y.; Kakeya, H.; Fujita, M.; et al. Innovative preparation of curcumin for improved oral bioavailability. Biol. Pharm. Bull. 2011, 34, 660–665. [Google Scholar] [CrossRef]
- Mukkavilli, R.; Yang, C.; Tanwar, R.S.; Saxena, R.; Gundala, S.R.; Zhang, Y.; Ghareeb, A.; Floyd, S.D.; Vangala, S.; Kuo, W.W.; et al. Pharmacokinetic-pharmacodynamic correlations in the development of ginger extract as an anticancer agent. Sci. Rep. 2018, 8, 3056. [Google Scholar] [CrossRef]
- Kaneko, A.; Matsumoto, T.; Matsubara, Y.; Sekiguchi, K.; Koseki, J.; Yakabe, R.; Aoki, K.; Aiba, S.; Yamasaki, K. Glucuronides of phytoestrogen flavonoid enhance macrophage function via conversion to aglycones by β-glucuronidase in macrophages. Immun. Inflamm. Dis. 2017, 5, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Szymusiak, M.; Hu, X.; Leon Plata, P.A.; Ciupinski, P.; Wang, Z.J.; Liu, Y. Bioavailability of curcumin and curcumin glucuronide in the central nervous system of mice after oral delivery of nano-curcumin. Int. J. Pharm. 2016, 511, 415–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntosh, F.M.; Maison, N.; Holtrop, G.; Young, P.; Stevens, V.J.; Ince, J.; Johnstone, A.M.; Lobley, G.E.; Flint, H.J.; Louis, P. Phylogenetic distribution of genes encoding β-glucuronidase activity in human colonic bacteria and the impact of diet on faecal glycosidase activities. Environ. Microbiol. 2012, 14, 1876–1887. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Uechi, S.; Takara, K.; Asikin, Y.; Wada, K. Evaluation of an oral carrier system in rats: Bioavailability and antioxidant properties of liposome-encapsulated curcumin. J. Agric Food Chem. 2009, 57, 9141–9146. [Google Scholar] [CrossRef] [PubMed]
- Wellman, A.S.; Metukuri, M.R.; Kazgan, N.; Xu, X.; Xu, Q.; Ren, N.S.X.; Czopik, A.; Shanahan, M.T.; Kang, A.; Chen, W.; et al. Intestinal Epithelial Sirtuin 1 Regulates Intestinal Inflammation During Aging in Mice by Altering the Intestinal Microbiota. Gastroenterology 2017, 153, 772–786. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.L.; Zhou, M.; Kang, C.; Lang, H.D.; Chen, M.T.; Hui, S.C.; Wang, B.; Mi, M.T. Crosstalk between gut microbiota and Sirtuin-3 in colonic inflammation and tumorigenesis. Exp. Mol. Med. 2018, 50, 21. [Google Scholar] [CrossRef]
- Vikram, A.; Kim, Y.R.; Kumar, S.; Li, Q.; Kassan, M.; Jacobs, J.S.; Irani, K. Vascular microRNA-204 is remotely governed by the microbiome and impairs endothelium-dependent vasorelaxation by downregulating Sirtuin1. Nat. Commun. 2016, 7, 12565. [Google Scholar] [CrossRef]
- Lakhan, S.E.; Kirchgessner, A. Gut inflammation in chronic fatigue syndrome. Nutr. Metab. 2010, 7, 79. [Google Scholar] [CrossRef]
- Den Besten, G.; Gerding, A.; Van Dijk, T.H.; Ciapaite, J.; Bleeker, A.; van Eunen, K.; Havinga, R.; Groen, A.K.; Reijngoud, D.J.; Bakker, B.M. Protection against the metabolic syndrome by guar gum-derived short-chain fatty acids depends on peroxisome proliferator-activated receptorγ And Glucagon-like peptide-1. PLoS ONE 2015, 10, e0136364. [Google Scholar] [CrossRef]
- Walsh, M.E.; Bhattacharya, A.; Sataranatarajan, K.; Qaisar, R.; Sloane, L.; Rahman, M.M.; Kinter, M.; Van Remmen, H. The histone deacetylase inhibitor butyrate improves metabolism and reduces muscle atrophy during aging. Aging Cell 2015, 14, 957–970. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, J.G.; Milani, C.; de Giori, G.S.; Sesma, F.; van Sinderen, D.; Ventura, M. Bacteria as vitamin suppliers to their host: A gut microbiota perspective. Curr. Opin. Biotechnol. 2013, 24, 160–168. [Google Scholar] [CrossRef]
- Dukes, A.; Davis, C.; El Refaey, M.; Upadhyay, S.; Mork, S.; Arounleut, P.; Johnson, M.H.; Hill, W.D.; Isales, C.M.; Hamrick, M.W. The aromatic amino acid tryptophan stimulates skeletal muscle IGF1/p70s6k/mTor signaling in vivo and the expression of myogenic genes in vitro. Nutrition 2015, 31, 1018–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Liu, J.; Xu, H.; Zhang, Y.; Chu, L.; Liu, Q.; Song, N.; Yang, C. Novel tumor-targeting, self-assembling peptide nanofiber as a carrier for effective curcumin delivery. Int. J. Nanomed. 2013, 9, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Salazar, N.; López, P.; Valdés, L.; Margolles, A.; Suárez, A.; Patterson, A.M.; Cuervo, A.; de los Reyes-Gavilán, C.G.; Ruas-Madiedo, P.; Gonzalez, S.; et al. Microbial Targets for the Development of Functional Foods Accordingly with Nutritional and Immune Parameters Altered in the Elderly. J. Am. Coll. Nutr. 2013, 32, 399–406. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Global changes and markers of senescent cells. During ageing, the accumulation of senescent cells is often observed. Senescent cells are characterized by permanent cell cycle arrest, increased size, elevated activity of senescence associated β galactosidase (SA-β-gal) and higher levels of cell cycle inhibitors, p16 and/or p21. Moreover, senescent cells secrete various proteins such as cytokines, growth factors and proteases. This phenomenon is generally referred as senescence associated secretory phenotype (SASP). Nuclear changes are manifested by disrupted structure of nuclear lamina (for instance, due to downregulation of Lamin B1), local condensation of chromatin in the form of senescent associated heterochromatin foci (SAHF) and by DNA-SCARS (DNA segments with chromatin alterations reinforcing senescence), which form in response to DNA damage.
Figure 1.
Global changes and markers of senescent cells. During ageing, the accumulation of senescent cells is often observed. Senescent cells are characterized by permanent cell cycle arrest, increased size, elevated activity of senescence associated β galactosidase (SA-β-gal) and higher levels of cell cycle inhibitors, p16 and/or p21. Moreover, senescent cells secrete various proteins such as cytokines, growth factors and proteases. This phenomenon is generally referred as senescence associated secretory phenotype (SASP). Nuclear changes are manifested by disrupted structure of nuclear lamina (for instance, due to downregulation of Lamin B1), local condensation of chromatin in the form of senescent associated heterochromatin foci (SAHF) and by DNA-SCARS (DNA segments with chromatin alterations reinforcing senescence), which form in response to DNA damage.

Figure 2.
Epigenetic mechanisms shaping chromatin. Chromatin structure is precisely controlled by DNA methylation and post-translational modifications (PTMs; methylation, acetylation, etc.) deposited onto histone tails. Incorporation of methyl and acetyl group is guided by histone methyltransferases (HMTs) and histone acetyltransferases (HATs), while removal of these residues is achieved by histone demethylases (HDMs) and histone deacetylases (HDACs and Sirtuins). The action of these enzymes determines chromatin arrangement and affects gene transcription. Chromatin can adopt two distinct conformations, i.e., heterochromatin and euchromatin. Heterochromatin is a condensed and transcriptionally inactive structure, maintained mainly by methylation of lysine residues. Typical modifications include H3K27me3, H3K20me3 and H3K9me3. Euchromatin represents an open and active conformation. This structure is abundant in acetylated histones H3 and H4, and H3K4me3, H3K36me3 or H3K79me3. On the DNA level, gene expression can be regulated by methylation of cytosine residues by DNA methyltransferases (DNMTs) and oxidation of methylated cytosines by TET enzymes.
Figure 2.
Epigenetic mechanisms shaping chromatin. Chromatin structure is precisely controlled by DNA methylation and post-translational modifications (PTMs; methylation, acetylation, etc.) deposited onto histone tails. Incorporation of methyl and acetyl group is guided by histone methyltransferases (HMTs) and histone acetyltransferases (HATs), while removal of these residues is achieved by histone demethylases (HDMs) and histone deacetylases (HDACs and Sirtuins). The action of these enzymes determines chromatin arrangement and affects gene transcription. Chromatin can adopt two distinct conformations, i.e., heterochromatin and euchromatin. Heterochromatin is a condensed and transcriptionally inactive structure, maintained mainly by methylation of lysine residues. Typical modifications include H3K27me3, H3K20me3 and H3K9me3. Euchromatin represents an open and active conformation. This structure is abundant in acetylated histones H3 and H4, and H3K4me3, H3K36me3 or H3K79me3. On the DNA level, gene expression can be regulated by methylation of cytosine residues by DNA methyltransferases (DNMTs) and oxidation of methylated cytosines by TET enzymes.

Figure 3.
Nuclear and chromatin architecture changes in ageing and senescent cells. The nucleus of a proliferating cell contains a highly condensed chromatin enriched in methylated histones and hypermethylated DNA. The chromatin is compartmentalized into topologically associated domains (TADs) enclosed by CTCF protein and cohesin (not shown). Heterochromatic and gene-poor regions, characterized by the presence of H3K9me3 and H3K27me3, are in close contact with nuclear lamina (NL) forming lamina associated domains (LADs) contributing to the overall stabilization of the nuclear structure. During ageing/senescence DNA becomes globally hypomethylated with exception of promoters of particular genes, which may be hypermethylated. As a result, the gene expression pattern is significantly altered. Similarly, nucleosomes display a general loss of histones leading to loosely compacted euchromatin. Reduced compaction is also a result of accumulation of acetylated residues on histone tails and reduction of their methylation. OIS cells exhibit local condensation and redistribution of heterochromatin in the form of SAHFs. SAHFs emerge from detachment of heterochromatin from the nuclear lamina (due to downregulation of Lamin B1 and accumulation of dysfunctional prelamin A and progerin) and have a layered structure with a H3K9me3 rich core encircled with H3K27me3. On the outside, fragments of active chromatin are found (H3K36me3). Moreover, the local accumulation of nucleoporins prevents deposition of heterochromatin near the nuclear envelope. Disturbances in NL structure also result in cytoplasmic chromatin fragments that contain DNA and repressive histone marks.
Figure 3.
Nuclear and chromatin architecture changes in ageing and senescent cells. The nucleus of a proliferating cell contains a highly condensed chromatin enriched in methylated histones and hypermethylated DNA. The chromatin is compartmentalized into topologically associated domains (TADs) enclosed by CTCF protein and cohesin (not shown). Heterochromatic and gene-poor regions, characterized by the presence of H3K9me3 and H3K27me3, are in close contact with nuclear lamina (NL) forming lamina associated domains (LADs) contributing to the overall stabilization of the nuclear structure. During ageing/senescence DNA becomes globally hypomethylated with exception of promoters of particular genes, which may be hypermethylated. As a result, the gene expression pattern is significantly altered. Similarly, nucleosomes display a general loss of histones leading to loosely compacted euchromatin. Reduced compaction is also a result of accumulation of acetylated residues on histone tails and reduction of their methylation. OIS cells exhibit local condensation and redistribution of heterochromatin in the form of SAHFs. SAHFs emerge from detachment of heterochromatin from the nuclear lamina (due to downregulation of Lamin B1 and accumulation of dysfunctional prelamin A and progerin) and have a layered structure with a H3K9me3 rich core encircled with H3K27me3. On the outside, fragments of active chromatin are found (H3K36me3). Moreover, the local accumulation of nucleoporins prevents deposition of heterochromatin near the nuclear envelope. Disturbances in NL structure also result in cytoplasmic chromatin fragments that contain DNA and repressive histone marks.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gadecka, A.; Bielak-Zmijewska, A. Slowing Down Ageing: The Role of Nutrients and Microbiota in Modulation of the Epigenome. Nutrients 2019, 11, 1251. https://doi.org/10.3390/nu11061251
AMA Style
Gadecka A, Bielak-Zmijewska A. Slowing Down Ageing: The Role of Nutrients and Microbiota in Modulation of the Epigenome. Nutrients. 2019; 11(6):1251. https://doi.org/10.3390/nu11061251
Chicago/Turabian StyleGadecka, Agnieszka, and Anna Bielak-Zmijewska. 2019. "Slowing Down Ageing: The Role of Nutrients and Microbiota in Modulation of the Epigenome" Nutrients 11, no. 6: 1251. https://doi.org/10.3390/nu11061251
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.