
An In Silico Framework to Mine Bioactive Peptides from Annotated Proteomes: A Case Study on Pancreatic Alpha Amylase Inhibitory Peptides from Algae and Cyanobacteria



Abstract
:1. Introduction
2. Materials and Methods
2.1. Bioactive Peptide Analysis
2.1.1. Data Retrieval
2.1.2. Searching Bioactive Peptides within Algae and Cyanobacteria Proteome
2.2. Molecular Modelling
2.2.1. Peptides and Protein Model Design
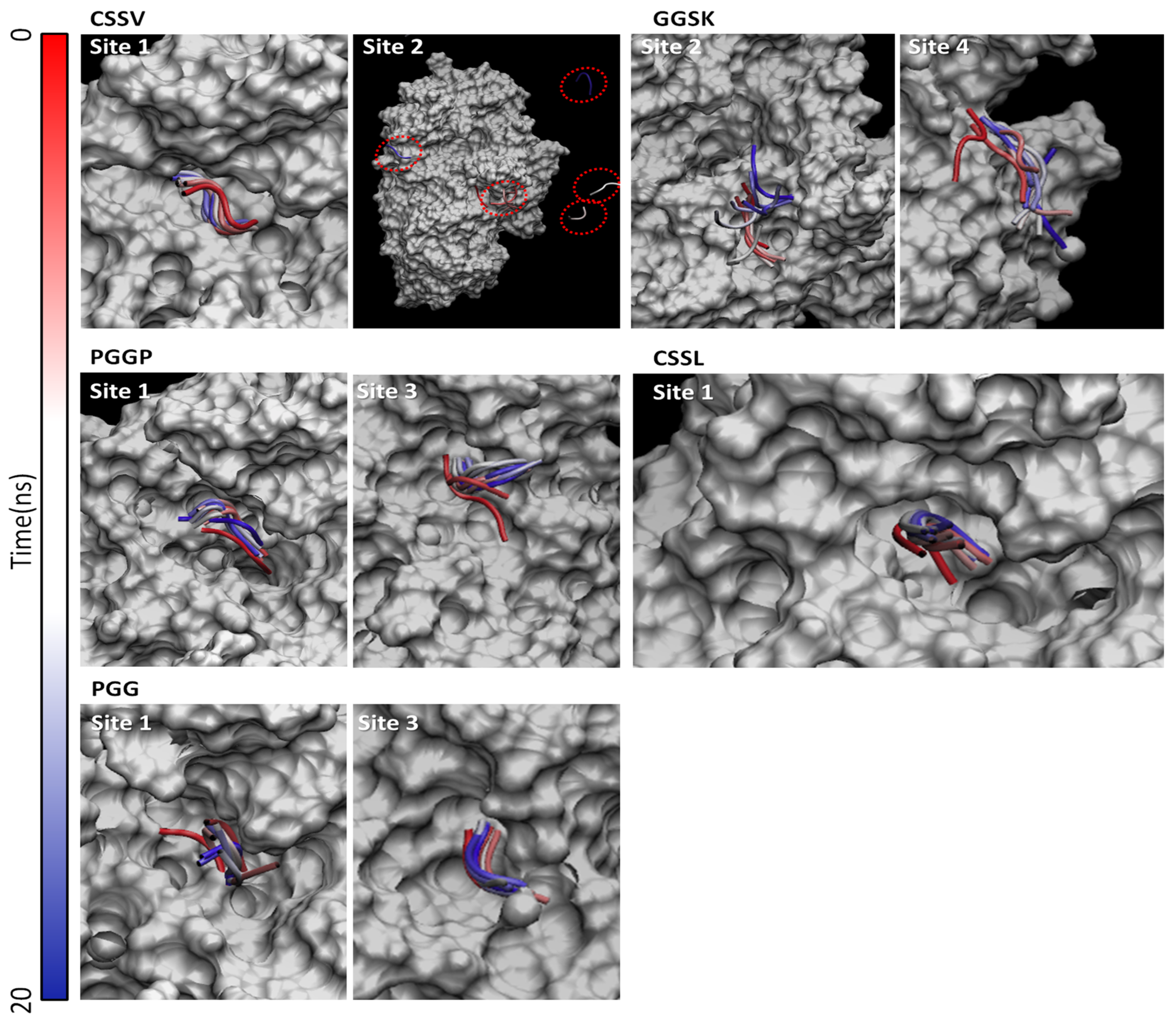
2.2.2. Pocket Scan and Docking Simulations
2.2.3. Molecular Dynamic Simulations
2.3. In Silico Digestion
3. Results and Discussion
3.1. Data Retrieval
3.2. Mining of Bioactive Sequences into Algal Proteomes

{kind=link}
{kind=link}
| Sequence | Activity | Source | References |
|---|---|---|---|
| FLS | 225 μg/mL a | Yellow field pea | [39] |
| YAL | |||
| TVF | |||
| IFS | |||
| FSL | |||
| ERA | |||
| EAR | |||
| NKN | |||
| KNN | |||
| NNK | |||
| PHY | |||
| WNP | |||
| GKGN | |||
| SLSD | |||
| VVSE | |||
| TFPG | |||
| ASFP | |||
| IARP | |||
| LQRF | |||
| RVLD | |||
| VDRI | |||
| INKQ | |||
| KQVQ | |||
| DLRV | |||
| VDRL | |||
| IVDR | |||
| KFFE | |||
| ACGP | 2.74 mM b | Bovine casein | [40] |
| CSSV | 34.88 mM c | Chinese giant salamander (Andrias davidianus) | [41] |
| YSFR | 18.93 mM c | ||
| SAAP | 12.95 mM c | ||
| PGGP | 12.96 mM c | ||
| ELS | 2.58 ± 0.08 mM c | Red seaweed (Porphyra spp.) | [42] |
| GGSK | 2.62 ± 0.05 mM c |
| Protein Name | UniProt AC | Corresponding Protein in Porphyra spp. Containing ELS |
|---|---|---|
| Translation initiation factor IF-1, chloroplastic a | P56290 | Homolog not detected |
| Photosistem II protein D1 a | P56318 | Photosistem II protein D1 (P51212) |
| ATP-dependent zinc metalloprotease FtsH homolog a | P56369 | Sequence not detected |
| ATP syntase subunit beta, chloroplastic a | P32978 | ATP synthase subunit beta, chloroplastic (P51259) |
| DNA direct RNA polymerase subunit alpha a | P56298 | DNA direct RNA polymerase subunit alpha (P51293) |
| Probable sulfate/thiosulfate import ATP-binding protein CysA a | P56344 | Homolog not detected |
| Photosystem I assembly protein Ycf4 a | P56312 | Photosystem I assembly protein Ycf3 (P51258) |
| Protein adenylyltransferase MntA b | A0A0B0QJN8 | Homolog not detected |
3.3. Molecular Modeling Results
3.4. In Silico Protein Digestion Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dellafiora, L.; Paolella, S.; Dall’Asta, C.; Dossena, A.; Cozzini, P.; Galaverna, G. Hybrid in Silico/in Vitro Approach for the Identification of Angiotensin I Converting Enzyme Inhibitory Peptides from Parma Dry-Cured Ham. J. Agric. Food Chem. 2015, 63, 6366–6375. [Google Scholar] [CrossRef] [PubMed]
- Moller, N.P.; Scholz-Ahrens, K.E.; Roos, N.; Schrezenmeir, J. Bioactive peptides and proteins from foods: Indication for health effects. Eur. J. Nutr. 2008, 47, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Valencia-Mejia, E.; Batista, K.A.; Fernandez, J.J.A.; Fernandes, K.F. Antihyperglycemic and hypoglycemic activity of naturally occurring peptides and protein hydrolysates from easy-to-cook and hard-to-cook beans (Phaseolus vulgaris L.). Food Res. Int. 2019, 121, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Antony, P.; Vijayan, R. Bioactive Peptides as Potential Nutraceuticals for Diabetes Therapy: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 9059. [Google Scholar] [CrossRef] [PubMed]
- Dandekar, P.D.; Kotmale, A.S.; Chavan, S.R.; Kadlag, P.P.; Sawant, S.V.; Dhavale, D.D.; RaviKumar, A. Insights into the Inhibition Mechanism of Human Pancreatic alpha-Amylase, a Type 2 Diabetes Target, by Dehydrodieugenol B Isolated from Ocimum tenuiflorum. Acs Omega 2021, 6, 1780–1786. [Google Scholar] [CrossRef] [PubMed]
- Grant, G.; Duncan, M.; Alonso, R.; Marzo, F. Peas and lentils. In Encyclopedia of Food Sciences and Nutrition, 2nd ed.; Caballero, B., Ed.; Academic Press: Cambridge, MA, USA, 2003; pp. 4433–4440. [Google Scholar]
- Aiello, G.; Lammi, C.; Boschin, G.; Zanoni, C.; Arnoldi, A. Exploration of Potentially Bioactive Peptides Generated from the Enzymatic Hydrolysis of Hempseed Proteins. J. Agric. Food Chem. 2017, 65, 10174–10184. [Google Scholar] [CrossRef]
- Schrader, M. Origins, Technological Development, and Applications of Peptidomics. In Peptidomics: Methods and Strategies; Methods in Molecular Biology; Schrader, M., Fricker, L., Eds.; Humana: New York, NY, USA, 2018; Volume 1719, pp. 3–39. [Google Scholar]
- Aramburo-Galvez, J.G.; Arvizu-Flores, A.A.; Cardenas-Torres, F.I.; Cabrera-Chavez, F.; Ramirez-Torres, G.I.; Flores-Mendoza, L.K.; Gastelum-Acosta, P.E.; Figueroa-Salcido, O.G.; Ontiveros, N. Prediction of ACE-I Inhibitory Peptides Derived from Chickpea (Cicer arietinum L.): In Silico Assessments Using Simulated Enzymatic Hydrolysis, Molecular Docking and ADMET Evaluation. Foods 2022, 11, 1576. [Google Scholar] [CrossRef]
- Jamalan, M.; Barzegari, E.; Gholami-Borujeni, F. Structure-Based Screening to Discover New Inhibitors for Papain-like Proteinase of SARS-CoV-2: An In Silico Study. J. Proteome Res. 2021, 20, 1015–1026. [Google Scholar] [CrossRef]
- Abuzinadah, M.F.; Ahmad, V.; Al-Thawdi, S.; Zakai, S.A.; Jamal, Q.M.S. Exploring the Binding Interaction of Active Compound of Pineapple against Foodborne Bacteria and Novel Coronavirus (SARS-CoV-2) Based on Molecular Docking and Simulation Studies. Nutrients 2022, 14, 3045. [Google Scholar] [CrossRef]
- Li, H.X.; Sun, X.Y.; Yu, F.; Xu, L.J.; Miu, J.H.; Xiao, P.G. In Silico Investigation of the Pharmacological Mechanisms of Beneficial Effects of Ginkgo biloba L. on Alzheimer’s Disease. Nutrients 2018, 10, 589. [Google Scholar] [CrossRef]
- Zhang, P.A.; Gao, J.H.; Che, H.L.; Xue, W.T.; Yang, D. Molecular Basis of IgE-Mediated Shrimp Allergy and Heat Desensitization. Nutrients 2021, 13, 3397. [Google Scholar] [CrossRef]
- Lin, K.; Zhang, L.W.; Han, X.; Xin, L.; Meng, Z.X.; Gong, P.M.; Cheng, D.Y. Yak milk casein as potential precursor of angiotensin I-converting enzyme inhibitory peptides based on in silico proteolysis. Food Chem. 2018, 254, 340–347. [Google Scholar] [CrossRef]
- Lammi, C.; Boschin, G.; Bollati, C.; Arnoldi, A.; Galaverna, G.; Dellafiora, L. A heuristic, computer-driven and top-down approach to identify novel bioactive peptides: A proof-of-principle on angiotensin I converting enzyme inhibitory peptides. Food Res. Int. 2021, 150, 110753. [Google Scholar] [CrossRef]
- Yu, Z.P.; Wang, Y.X.; Zhao, W.Z.; Li, J.R.; Shuian, D.; Liu, J.B. Identification of Oncorhynchus mykiss nebulin-derived peptides as bitter taste receptor TAS2R14 blockers by in silico screening and molecular docking. Food Chem. 2022, 368, 130839. [Google Scholar] [CrossRef]
- Nongonierma, A.B.; FitzGerald, R.J. An in silico model to predict the potential of dietary proteins as sources of dipeptidyl peptidase IV (DPP-IV) inhibitory peptides. Food Chem. 2014, 165, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela Zamudio, F.; Hidalgo-Figueroa, S.N.; Ortiz Andrade, R.R.; Hernandez Alvarez, A.J.; Segura Campos, M.R. Identification of antidiabetic peptides derived from in silico hydrolysis of three ancient grains: Amaranth, Quinoa and Chia. Food Chem 2022, 394, 133479. [Google Scholar] [CrossRef]
- Ji, D.W.; Xu, M.; Udenigwe, C.C.; Agyei, D. Physicochemical characterisation, molecular docking, and drug-likeness evaluation of hypotensive peptides encrypted in flaxseed proteome. Curr. Res. Food Sci. 2020, 3, 41–50. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, M.X.; Pan, F.; Li, J.Y.; Dou, R.; Wang, X.Y.; Wang, Y.Y.; He, Y.M.; Wang, S.X.; Cai, S.B. In silico analysis of novel dipeptidyl peptidase-IV inhibitory peptides released from Macadamia integrifolia antimicrobial protein 2 (MiAMP2) and the possible pathways involved in diabetes protection. Curr. Res. Food Sci. 2021, 4, 603–611. [Google Scholar] [CrossRef]
- Canelli, G.; Tarnutzer, C.; Carpine, R.; Neutsch, L.; Bolten, C.J.; Dionisi, F.; Mathys, A. Biochemical and Nutritional Evaluation of ChlorellaandAuxenochlorella Biomasses Relevant for Food Application. Front. Nutr. 2020, 7, 565996. [Google Scholar] [CrossRef]
- Hernandez, H.; Nunes, M.C.; Prista, C.; Raymundo, A. Innovative and Healthier Dairy Products through the Addition of Microalgae: A Review. Foods 2022, 11, 755. [Google Scholar] [CrossRef]
- Lauritano, C.; Ianora, A. Marine Organisms with Anti-Diabetes Properties. Mar. Drugs 2016, 14, 220. [Google Scholar] [CrossRef] [PubMed]
- Cunha, S.A.; Coscueta, E.R.; Nova, P.; Silva, J.L.; Pintado, M.M. Bioactive Hydrolysates from Chlorella vulgaris: Optimal Process and Bioactive Properties. Molecules 2022, 27, 2505. [Google Scholar] [CrossRef] [PubMed]
- Sanaei, M.; Ebrahimi, M.; Banazadeh, Z.; Shafiee, G.; Khatami, F.; Ahadi, Z.; Heshmat, R. Consequences of AphanizomenonFlos-aquae (AFA) extract (Stemtech(TM)) on metabolic profile of patients with type 2 diabetes. J. Diabetes Metab. Disord. 2015, 14, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batista, A.P.; Niccolai, A.; Bursic, I.; Sousa, I.; Raymundo, A.; Rodolfi, L.; Biondi, N.; Tredici, M.R. Microalgae as Functional Ingredients in Savory Food Products: Application to Wheat Crackers. Foods 2019, 8, 611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Qian, M.X.; Nahoum, V.; Bonicel, J.; Bischoff, H.; Henrissat, B.; Payan, F. Enzyme-catalyzed condensation reaction in a mammalian alpha-amylase. High-resolution structural analysis of an enzyme-inhibitor complex. Biochemistry 2001, 40, 7700–7709. [Google Scholar] [CrossRef]
- Gaudreault, F.; Morency, L.P.; Najmanovich, R.J. NRGsuite: A PyMOL plugin to perform docking simulations in real time using FlexAID. Bioinformatics 2015, 31, 3856–3858. [Google Scholar] [CrossRef] [Green Version]
- Maldonado-Rojas, W.; Olivero-Verbel, J. Potential interaction of natural dietary bioactive compounds with COX-2. J. Mol. Graph. Model. 2011, 30, 157–166. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Maillet, N. Rapid Peptides Generator: Fast and efficient in silico protein digestion. Nar Genom. Bioinform. 2020, 2, lqz004. [Google Scholar] [CrossRef] [Green Version]
- Ozorio, L.; Mellinger-Silva, C.; Cabral, L.M.C.; Jardin, J.; Boudry, G.; Dupont, D. The Influence of Peptidases in Intestinal Brush Border Membranes on the Absorption of Oligopeptides from Whey Protein Hydrolysate: An Ex Vivo Study Using an Ussing Chamber. Foods 2020, 9, 1415. [Google Scholar] [CrossRef]
- Van der Wielen, N.; Moughan, P.J.; Mensink, M. Amino Acid Absorption in the Large Intestine of Humans and Porcine Models. J. Nutr. 2017, 147, 1493–1498. [Google Scholar] [CrossRef] [Green Version]
- Patil, P.J.; Usman, M.; Zhang, C.N.; Mehmood, A.; Zhou, M.C.; Teng, C.; Li, X.T. An updated review on food-derived bioactive peptides: Focus on the regulatory requirements, safety, and bioavailability. Compr. Rev. Food Sci. Food Saf. 2022, 21, 1732–1776. [Google Scholar] [CrossRef]
- Tu, M.L.; Cheng, S.Z.; Lu, W.H.; Du, M. Advancement and prospects of bioinformatics analysis for studying bioactive peptides from food-derived protein: Sequence, structure, and functions. Trac-Trends Anal. Chem. 2018, 105, 7–17. [Google Scholar] [CrossRef]
- Nardo, A.E.; Anon, M.C.; Parisi, G. Large-scale mapping of bioactive peptides in structural and sequence space. PLoS ONE 2018, 13, e0191063. [Google Scholar] [CrossRef] [Green Version]
- Awosika, T.O.; Aluko, R.E. Inhibition of the in vitro activities of alpha-amylase, alpha-glucosidase and pancreatic lipase by yellow field pea (Pisum sativum L.) protein hydrolysates. Int. J. Food Sci. Technol. 2019, 54, 2021–2034. [Google Scholar] [CrossRef] [Green Version]
- Mudgil, P.; Kamal, H.; Kilari, B.P.; Salim, M.; Gan, C.Y.; Maqsood, S. Simulated gastrointestinal digestion of camel and bovine casein hydrolysates: Identification and characterization of novel anti-diabetic bioactive peptides. Food Chem. 2021, 353, 129374. [Google Scholar] [CrossRef]
- Ramadhan, A.H.; Nawas, T.; Zhang, X.W.; Pembe, W.M.; Xia, W.S.; Xu, Y.S. Purification and identification of a novel antidiabetic peptide from Chinese giant salamander (Andrias davidianus) protein hydrolysate against alpha-amylase and alpha-glucosidase. Int. J. Food Prop. 2018, 20, S3360–S3372. [Google Scholar] [CrossRef] [Green Version]
- Admassu, H.; Gasmalla, M.A.A.; Yang, R.J.; Zhao, W. Identification of Bioactive Peptides with alpha-Amylase Inhibitory Potential from Enzymatic Protein Hydrolysates of Red Seaweed (Porphyra spp.). J. Agric. Food Chem. 2018, 66, 4872–4882. [Google Scholar] [CrossRef]
- Lammi, C.; Aiello, G.; Dellafiora, L.; Bollati, C.; Boschin, G.; Ranaldi, G.; Ferruzza, S.; Sambuy, Y.; Galaverna, G.; Arnoldi, A. Assessment of the Multifunctional Behavior of Lupin Peptide P7 and Its Metabolite Using an Integrated Strategy. J. Agric. Food Chem. 2020, 68, 13179–13188. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Aiello, G.; Bollati, C.; Bartolomei, M.; Arnoldi, A.; Lammi, C. Phycobiliproteins from Arthrospira Platensis (Spirulina): A New Source of Peptides with Dipeptidyl Peptidase-IV Inhibitory Activity. Nutrients 2020, 12, 794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Castro, R.J.S.; Sato, H.H. Biologically active peptides: Processes for their generation, purification and identification and applications as natural additives in the food and pharmaceutical industries. Food Res. Int. 2015, 74, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, Z.; Cakir-Kiefer, C.; Roux, E.; Perrin, C.; Mido, L.; Dary-Mourot, A. Strategies of producing bioactive peptides from milk proteins to functionalize fermented milk products. Food Res. Int. 2014, 63, 71–80. [Google Scholar] [CrossRef]
- Toldra, F.; Reig, M.; Aristoy, M.C.; Mora, L. Generation of bioactive peptides during food processing. Food Chem. 2018, 267, 395–404. [Google Scholar] [CrossRef]
- Nongonierma, A.B.; Dellafiora, L.; Paolella, S.; Galaverna, G.; Cozzini, P.; FitzGerald, R.J. In Silico Approaches Applied to the Study of Peptide Analogs of Ile Pro-Ile in Relation to Their Dipeptidyl Peptidase IV Inhibitory Properties. Front. Endocrinol. 2018, 9, 329. [Google Scholar] [CrossRef] [Green Version]
- Lyon-Colbert, A.; Su, S.; Cude, C. A Systematic Literature Review for Evidence of Aphanizomenon flos-aquae Toxigenicity in Recreational Waters and Toxicity of Dietary Supplements: 2000–2017. Toxins 2018, 10, 254. [Google Scholar] [CrossRef] [Green Version]
- Ochiai, T.; Sugita, T.; Kato, R.; Okochi, M.; Honda, H. Screening of an alpha-Amylase Inhibitor Peptide by Photolinker-Peptide Array. Biosci. Biotechnol. Biochem 2012, 76, 819–824. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.P.; Yin, Y.G.; Zhao, W.Z.; Liu, J.B.; Chen, F. Anti-diabetic activity peptides from albumin against alpha-glucosidase and alpha-amylase. Food Chem. 2012, 135, 2078–2085. [Google Scholar] [CrossRef]
- Ngoh, Y.Y.; Lim, T.S.; Gan, C.Y. Screening and identification of five peptides from pinto bean with inhibitory activities against alpha-amylase using phage display technique. Enzyme Microb. Technol. 2016, 89, 76–84. [Google Scholar] [CrossRef]
- Ngoh, Y.Y.; Tye, G.J.; Gan, C.Y. The investigation of alpha-amylase inhibitory activity of selected Pinto bean peptides via preclinical study using AR42J cell. J. Funct. Foods 2017, 35, 641–647. [Google Scholar] [CrossRef]
- Mudgil, P.; Kamal, H.; Yuen, G.C.; Maqsood, S. Characterization and identification of novel antidiabetic and anti-obesity peptides from camel milk protein hydrolysates. Food Chem. 2018, 259, 46–54. [Google Scholar] [CrossRef]


| Peptide | Site 1 (Substrate Binding Site) | Site 2 | Site 3 | Site 4 |
|---|---|---|---|---|
| CSSV | 70 * | 70 * | 64 | 65 |
| GGSK | 67 | 72 * | 65 | 72 * |
| PGGP | 63 * | 54 | 64 * | 61 |
| CSSL | 70 * | np | np | np |
| PGG | 51 * | np | 53 * | np |
| Released Peptide | UniProt AC | Condition 1/Condition 2 |
|---|---|---|
| ELS | A0A0B0QJN8 | Endoproteinase Arg-C/Ficin |
| Endoproteinase Arg-C/Thermolysin | ||
| Clostripain/Ficin | ||
| Clostripain/Thermolysin | ||
| Ficin/Papain | ||
| Ficin/Trypsin | ||
| Papain/Thermolysin | ||
| Thermolysin/Trypsin | ||
| P32978 | Ficin/Formic Acid | |
| P56290 | Neutrophil-elastase/Ficin | |
| P56369 | Neutrophil-elastase/Ficin | |
| P56298 | Bromelain/Ficin | |
| Bromelain/Thermolysin | ||
| Neutrophil-elastase/Ficin | ||
| Neutrophil-elastase/Thermolysin | ||
| P56318 | BNPS-Skatole a/Ficin | |
| BNPS-Skatole a/Thermolysin | ||
| Chymotrypsin/Ficin | ||
| Chymotrypsin/Thermolysin | ||
| Iodosobenzoic acid/Ficin | ||
| Iodosobenzoic acid/Thermolysin | ||
| CSSL | P85869 | Endoproetinase Asp-N/Chymotrypsin |
| Endoproetinase Asp-N/Proteinase-K | ||
| Chymotrypsin/Formic acid | ||
| Chymotrypsin/Endoproteinase Glu-C | ||
| Chymotrypsin/NTCB b | ||
| Formic acid/Proteinase-K | ||
| Endoproteinase Glu-C/Proteinase-K | ||
| NTCB b/Proteinase-K |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedroni, L.; Perugino, F.; Galaverna, G.; Dall’Asta, C.; Dellafiora, L. An In Silico Framework to Mine Bioactive Peptides from Annotated Proteomes: A Case Study on Pancreatic Alpha Amylase Inhibitory Peptides from Algae and Cyanobacteria. Nutrients 2022, 14, 4680. https://doi.org/10.3390/nu14214680
Pedroni L, Perugino F, Galaverna G, Dall’Asta C, Dellafiora L. An In Silico Framework to Mine Bioactive Peptides from Annotated Proteomes: A Case Study on Pancreatic Alpha Amylase Inhibitory Peptides from Algae and Cyanobacteria. Nutrients. 2022; 14(21):4680. https://doi.org/10.3390/nu14214680
Chicago/Turabian StylePedroni, Lorenzo, Florinda Perugino, Gianni Galaverna, Chiara Dall’Asta, and Luca Dellafiora. 2022. "An In Silico Framework to Mine Bioactive Peptides from Annotated Proteomes: A Case Study on Pancreatic Alpha Amylase Inhibitory Peptides from Algae and Cyanobacteria" Nutrients 14, no. 21: 4680. https://doi.org/10.3390/nu14214680