Serum Amyloid A Production Is Triggered by Sleep Deprivation in Mice and Humans: Is That the Link between Sleep Loss and Associated Comorbidities?

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
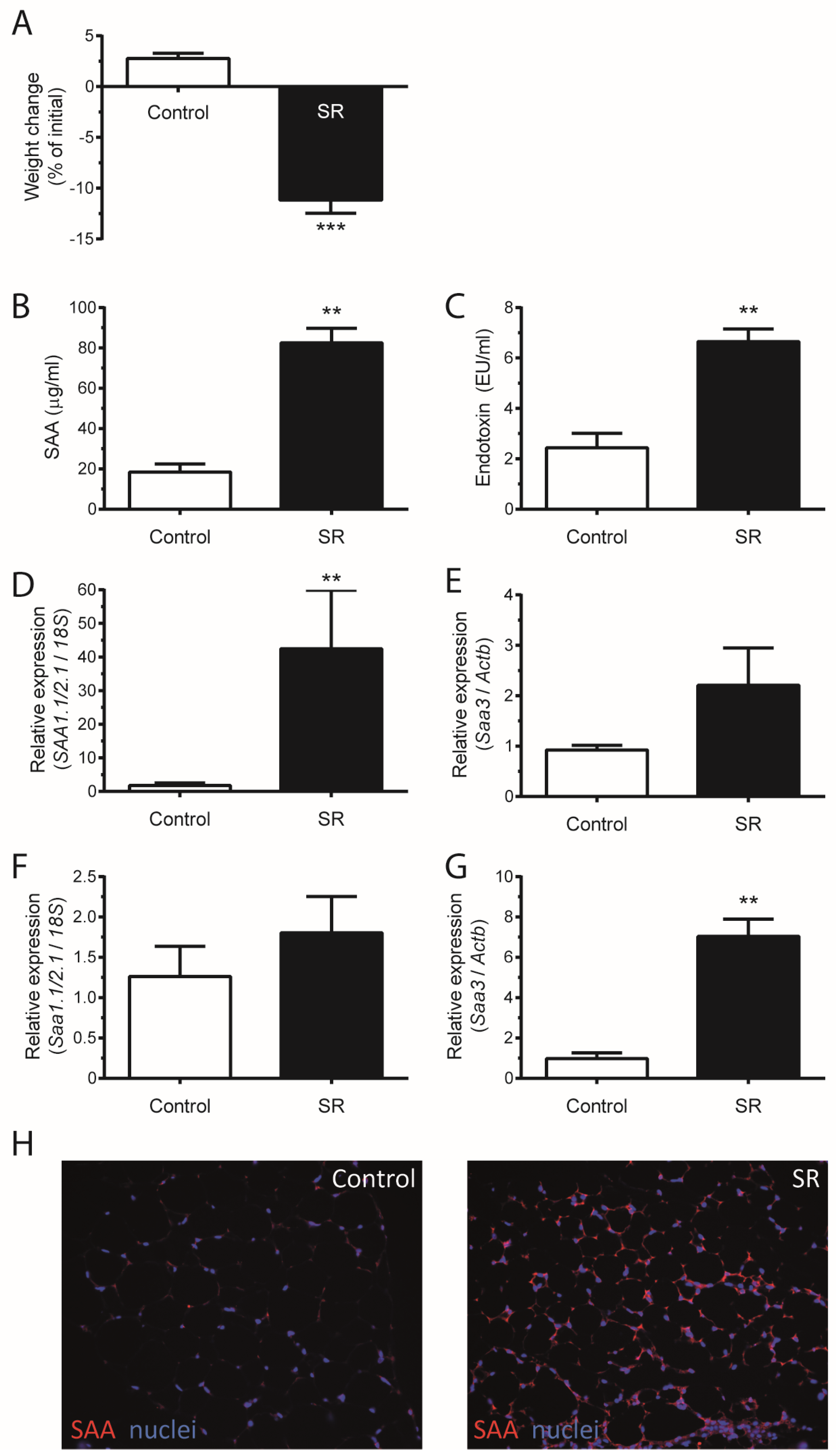
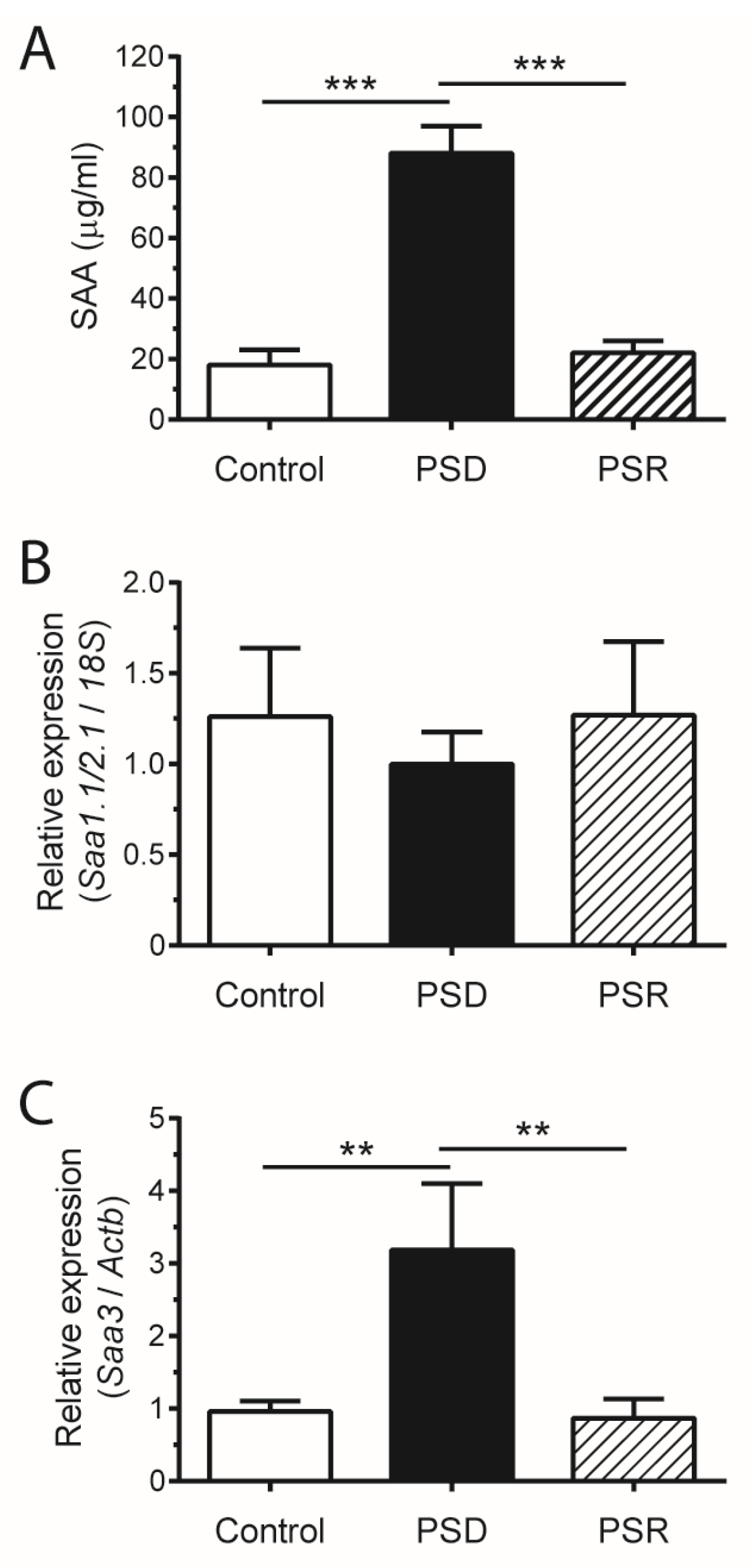
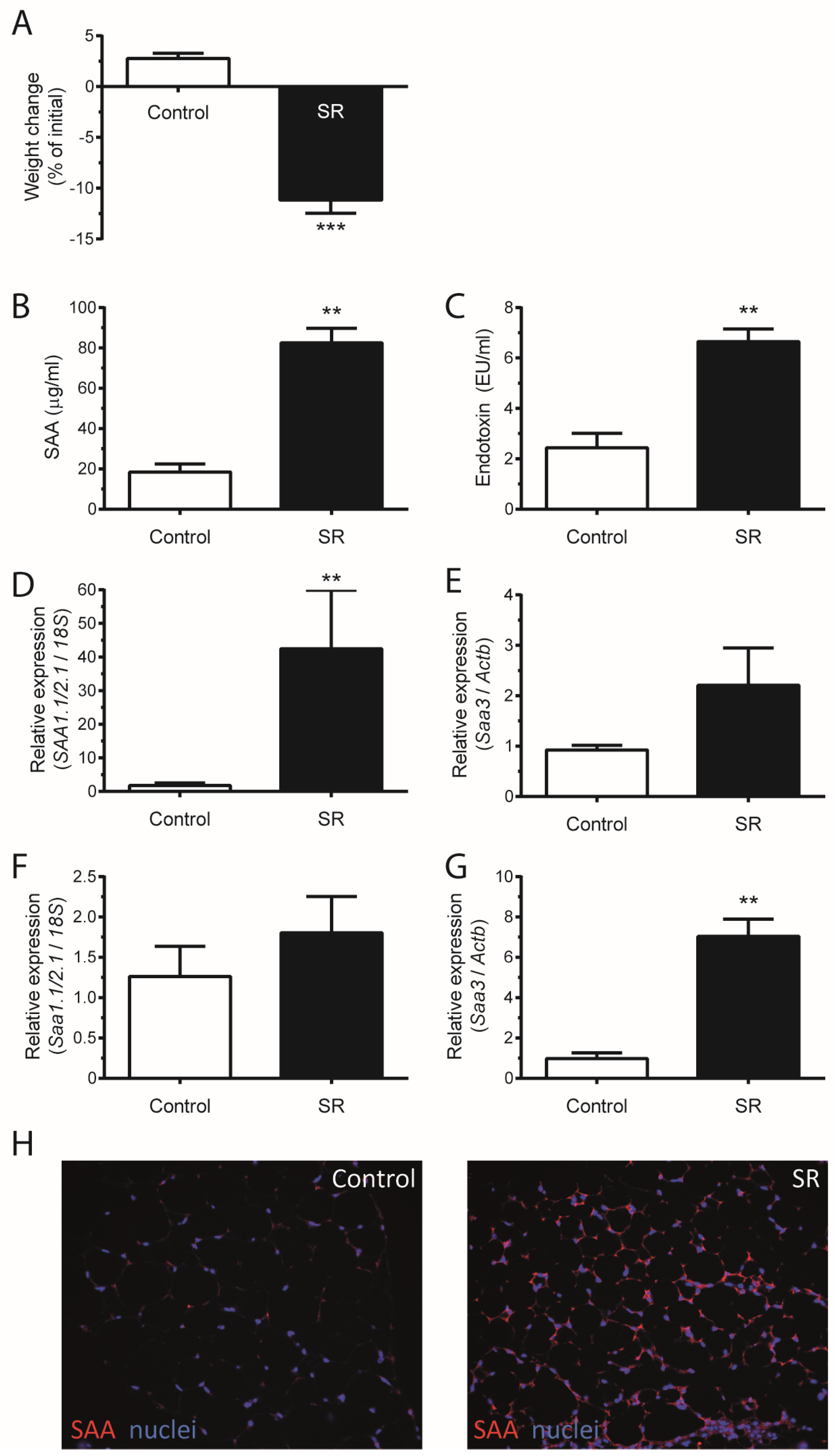
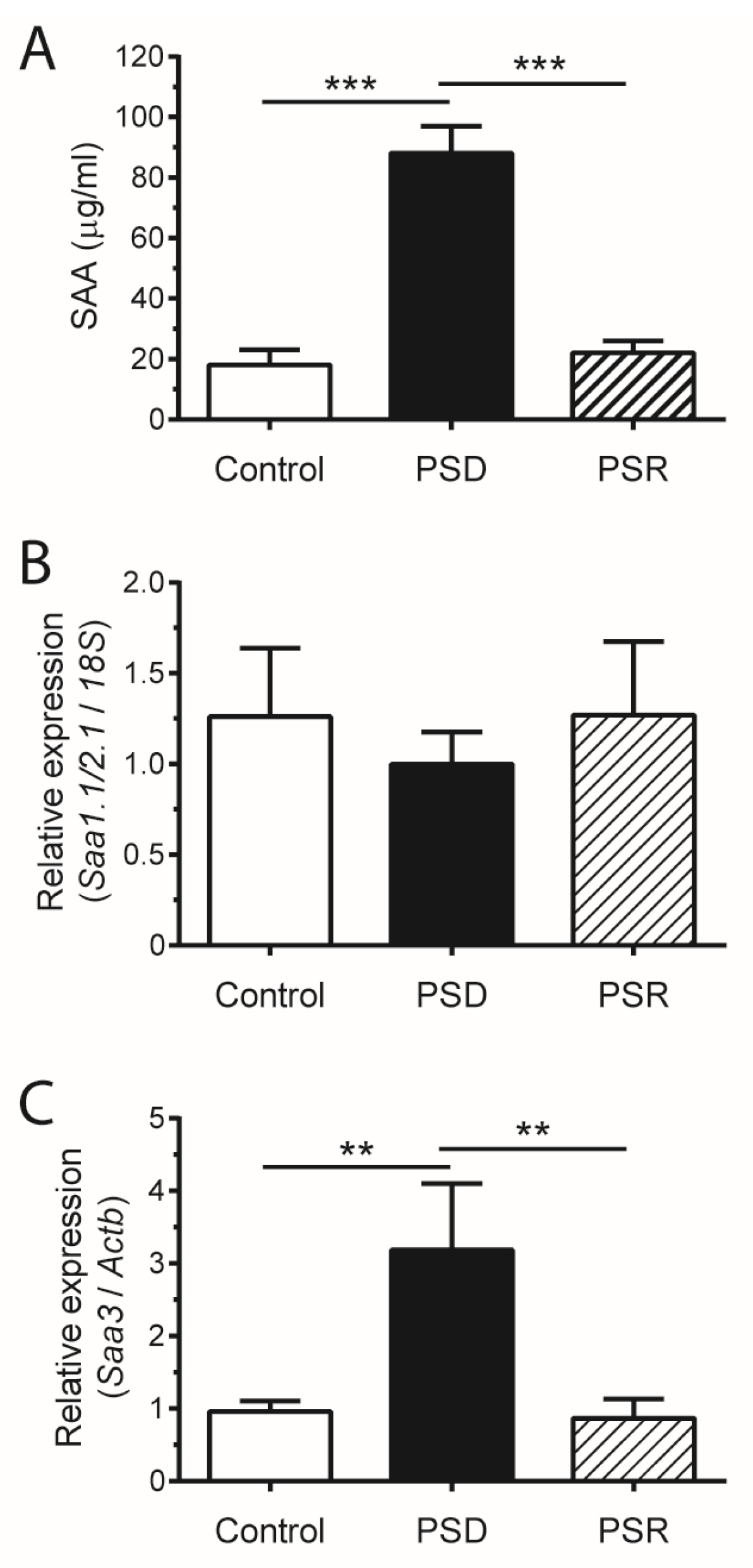
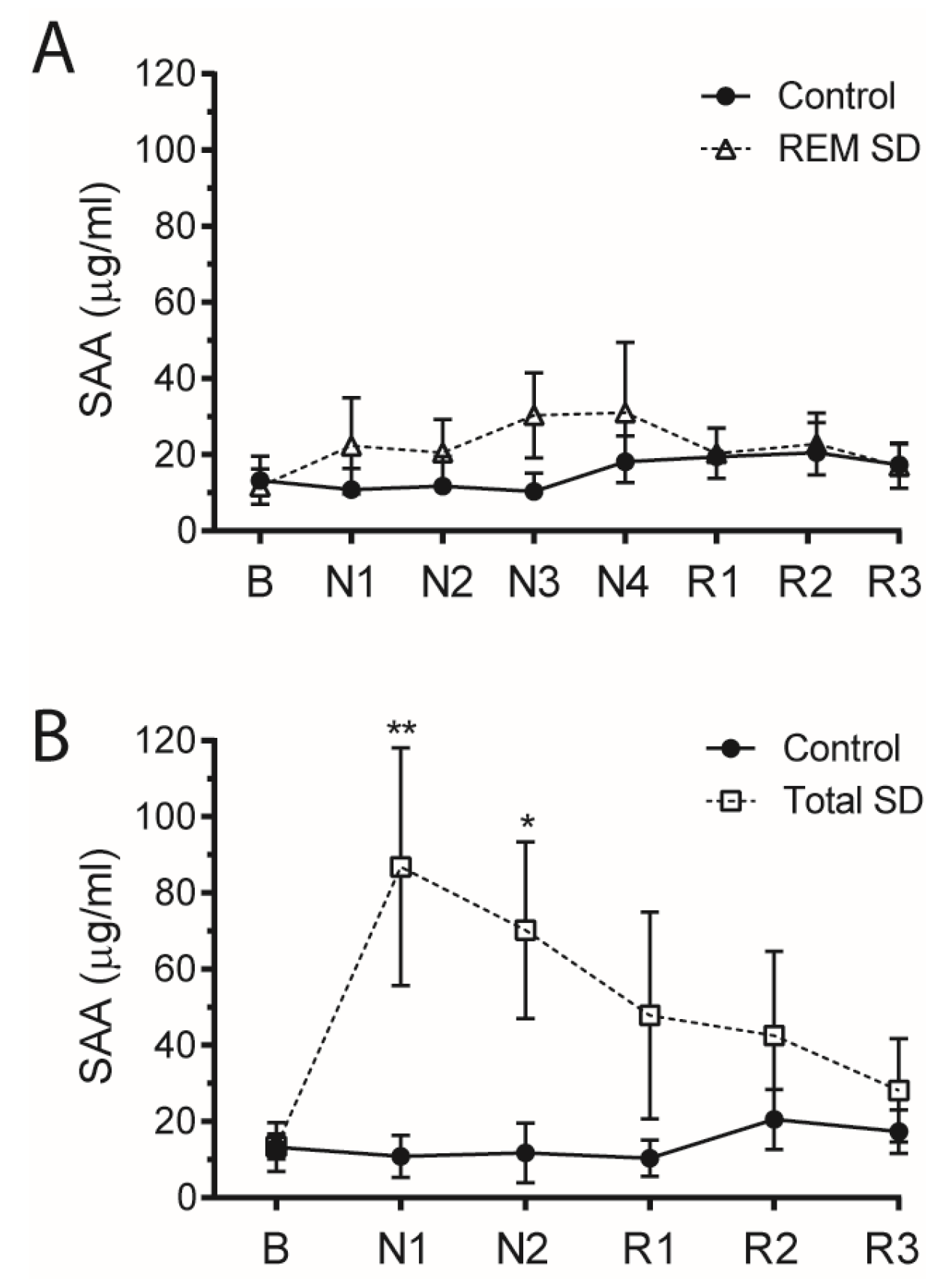
3. Results
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Knutson, K.L. Does inadequate sleep play a role in vulnerability to obesity? Am. J. Hum. Biol. 2012, 24, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Taheri, S.; Lin, L.; Austin, D.; Young, T.; Mignot, E. Short sleep duration is associated with reduced leptin, elevated ghrelin, and increased body mass index. PLoS Med. 2004, 1, 210–217. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, E.M.; Visniauskas, B.; Sandri, S.; Migliorini, S.; Andersen, M.L.; Tufik, S.; Fock, R.A.; Chagas, J.R.; Campa, A. Late effects of sleep restriction: Potentiating weight gain and insulin resistance arising from a high-fat diet in mice. Obesity 2015, 23, 391–398. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, E.M.; Ascar, T.P.; Silva, J.C.; Sandri, S.; Migliorini, S.; Fock, R.A.; Campa, A. Serum amyloid A links endotoxaemia to weight gain and insulin resistance in mice. Diabetologia 2016, 59, 1760–1768. [Google Scholar] [CrossRef] [PubMed]
- Eklund, K.K.; Niemi, K.; Kovanen, P.T. Immune functions of Serum Amyloid A. Crit. Rev. Immunol. 2012, 32, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Filippin-Monteiro, F.B.; de Oliveira, E.M.; Sandri, S.; Knebel, F.H.; Albuquerque, R.C.; Campa, A. Serum amyloid A is a growth factor for 3t3-l1 adipocytes, inhibits differentiation and promotes insulin resistance. Int. J. Obes. 2012, 36, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.P.; Freedman, S.B.; Witting, P.K. Serum amyloid A stimulates cultured endothelial cells to migrate and proliferate: Inhibition by the multikinase inhibitor BIBF1120. Clin. Exp. Pharm. Physiol. 2013, 40, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Sandri, S.; Rodriguez, D.; Gomes, E.; Monteiro, H.P.; Russo, M.; Campa, A. Is serum amyloid A an endogenous TLR4 agonist? J. Leukoc. Biol. 2008, 83, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; He, R.; Tian, J.; Ye, P.P.; Ye, R.D. Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid a. J. Immunol. 2008, 181, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.B.; Suchecki, D.; Tufik, S. Comparison of the sleep pattern throughout a protocol of chronic sleep restriction induced by two methods of paradoxical sleep deprivation. Brain Res. Bull. 2006, 70, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Kahan, V.; Ribeiro, D.A.; Egydio, F.; Barros, L.A.; Tomimori, J.; Tufik, S.; Andersen, M.L. Is lack of sleep capable of inducing dna damage in aged skin? Skin Pharm. Physiol. 2014, 27, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Maia, L.O.; Dias, W.; Carvalho, L.S.; Jesus, L.R.; Paiva, G.D.; Araujo, P.; Costa, M.F.O.; Andersen, M.L.; Tufik, S.; Mazaro-Costa, R. Association of methamidophos and sleep loss on reproductive toxicity of male mice. Environ. Toxicol. Pharm. 2011, 32, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.B.; Hipolide, D.C.; Benedito-Silva, A.A.; Tufik, S. Sleep deprivation induced by the modified multiple platform technique: Quantification of sleep loss and recovery. Brain Res. 2004, 1004, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Guariniello, L.D.; Vicari, P.; Lee, K.S.; De Oliveira, A.C.; Tufik, S. Bone marrow and peripheral white blood cells number is affected by sleep deprivation in a murine experimental model. J. Cell. Physiol. 2012, 227, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Zager, A.; Andersen, M.L.; Ruiz, F.S.; Antunes, I.B.; Tufik, S. Effects of acute and chronic sleep loss on immune modulation of rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R504–R509. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, F.S.; Andersen, M.L.; Martins, R.C.S.; Zager, A.; Lopes, J.D.; Tufik, S. Immune alterations after selective rapid eye movement or total sleep deprivation in healthy male volunteers. Innate Immun. 2012, 18, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.C.S.; Andersen, M.L.; Garbuio, S.A.; Bittencourt, L.R.; Guindalini, C.; Shih, M.C.; Hoexter, M.Q.; Bressan, R.A.; Castiglioni, M.L.V.; Tufik, S. Dopamine transporter regulation during four nights of rem sleep deprivation followed by recovery—An in vivo molecular imaging study in humans. Sleep 2010, 33, 243–251. [Google Scholar] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- De Beer, M.C.; Wroblewski, J.M.; Noffsinger, V.P.; Rateri, D.L.; Howatt, D.A.; Balakrishnan, A.; Ji, A.L.; Shridas, P.; Thompson, J.C.; van der Westhuyzen, D.R.; et al. Deficiency of endogenous acute phase serum amyloid a does not affect atherosclerotic lesions in apolipoprotein e-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Sommer, G.; Weise, S.; Kralisch, S.; Scherer, P.E.; Lossner, U.; Bluher, M.; Stumvoll, M.; Fasshauer, M. The adipokine SAA3 is induced by interleukin-1 beta in mouse adipocytes. J. Cell. Biochem. 2008, 104, 2241–2247. [Google Scholar] [CrossRef] [PubMed]
- Uhlar, C.M.; Whitehead, A.S. Serum amyloid A, the major vertebrate acute-phase reactant. Eur. J. Biochem. 1999, 265, 501–523. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, E.; Monteagudo, P.T.; Marrocos, M.S.M.; Campa, A. Interaction between serum amyloid A and leukocytes—A possible role in the progression of vascular complications in diabetes. Immunol. Lett. 2007, 108, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Han, C.Y.; Vaisar, T.; Shimokado, K.; Kargi, A.; Chen, M.H.; Wang, S.; McDonald, T.O.; O’Brien, K.D.; Heinecke, J.W.; et al. Serum amyloid A3 does not contribute to circulating SAA levels. J. Lipid Res. 2009, 50, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Den Hartigh, L.J.; Wang, S.R.; Goodspeed, L.; Ding, Y.L.; Averill, M.; Subramanian, S.; Wietecha, T.; O’Brien, K.D.; Chait, A. Deletion of serum amyloid A3 improves high fat high sucrose diet-induced adipose tissue inflammation and hyperlipidemia in female mice. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Faty, A.; Ferre, P.; Commans, S. The acute phase protein serum amyloid A induces lipolysis and inflammation in human adipocytes through distinct pathways. PLoS ONE 2012, 7, e34031. [Google Scholar] [CrossRef] [PubMed]
- Luche, E.; Cousin, B.; Garidou, L.; Serino, M.; Waget, A.; Barreau, C.; André, M.; Valet, P.; Courtney, M.; Casteilla, L.; et al. Metabolic endotoxemia directly increases the proliferation of adipocyte precursors at the onset of metabolic diseases through a cd14-dependent mechanism. Mol. Metab. 2013, 2, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Badolato, R.; Wang, J.M.; Murphy, W.J.; Lloyd, A.R.; Michiel, D.F.; Bausserman, L.L.; Kelvin, D.J.; Oppenheim, J.J. Serum amyloid-A is a chemoattractant—Induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J. Exp. Med. 1994, 180, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Sandri, S.; Hatanaka, E.; Franco, A.G.; Pedrosa, A.M.C.; Monteiro, H.P.; Campa, A. Serum amyloid A induces ccl20 secretion in mononuclear cells through mapk (p38 and ERK1/2) signaling pathways. Immunol. Lett. 2008, 121, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Svatikova, A.; Wolk, R.; Shamsuzzaman, A.S.; Kara, T.; Olson, E.J.; Somers, V.K. Serum amyloid A in obstructive sleep apnea. Circulation 2003, 108, 1451–1454. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, E.M.; Sandri, S.; Knebel, F.H.; Iglesias Contesini, C.G.; Campa, A.; Filippin-Monteiro, F.B. Hypoxia increases serum amyloid A3 (SAA3) in differentiated 3t3-l1 adipocytes. Inflammation 2013, 36, 1107–1110. [Google Scholar] [CrossRef] [PubMed]
- Broussard, J.L.; Chapotot, F.; Abraham, V.; Day, A.; Delebecque, F.; Whitmore, H.R.; Tasali, E. Sleep restriction increases free fatty acids in healthy men. Diabetologia 2015, 58, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.D.; Kip, K.E.; Marroquin, O.C.; Ridker, P.M.; Kelsey, S.F.; Shaw, L.J.; Pepine, C.J.; Sharaf, B.; Merz, C.N.B.; Sopko, G.; et al. Serum amyloid A as a predictor of coronary artery disease and cardiovascular outcome in women—The national heart, lung, and blood institute-sponsored women’s ischemia syndrome evaluation (wise). Circulation 2004, 109, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.C.; Jayne, C.; Thompson, J.; Wilson, P.G.; Yoder, M.H.; Webb, N.; Tannock, L.R. A brief elevation of serum amyloid a is sufficient to increase atherosclerosis. J. Lipid Res. 2015, 56, 286–293. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Oliveira, E.M.; Visniauskas, B.; Tufik, S.; Andersen, M.L.; Chagas, J.R.; Campa, A. Serum Amyloid A Production Is Triggered by Sleep Deprivation in Mice and Humans: Is That the Link between Sleep Loss and Associated Comorbidities? Nutrients 2017, 9, 311. https://doi.org/10.3390/nu9030311
De Oliveira EM, Visniauskas B, Tufik S, Andersen ML, Chagas JR, Campa A. Serum Amyloid A Production Is Triggered by Sleep Deprivation in Mice and Humans: Is That the Link between Sleep Loss and Associated Comorbidities? Nutrients. 2017; 9(3):311. https://doi.org/10.3390/nu9030311
Chicago/Turabian StyleDe Oliveira, Edson M., Bruna Visniauskas, Sergio Tufik, Monica L. Andersen, Jair R. Chagas, and Ana Campa. 2017. "Serum Amyloid A Production Is Triggered by Sleep Deprivation in Mice and Humans: Is That the Link between Sleep Loss and Associated Comorbidities?" Nutrients 9, no. 3: 311. https://doi.org/10.3390/nu9030311