A Review of Current Methods for Analysis of Mycotoxins in Herbal Medicines

Abstract
:1. Introduction
2. Sampling, Extraction and Cleanup
2.1. Sampling
2.2. Extraction Procedure
2.1.1. Extraction Solution
2.2.2. Extraction Method
2.3. Cleanup
2.3.1. SPE
Conventional SPE
Special SPE
Home-Made Cartridge
New Absorbents
2.3.2. IAC
2.3.3. Aptamer-Affinity Column (AAC)
2.3.4. Molecularly Imprinted Polymers (MIPs)
2.3.5. QuEChERS
2.3.6. One-Step Extraction
3. Analytical Techniques of Mycotoxins
3.1. Chromatographic Techniques for Detecting/Quantifying Mycotoxins
3.1.1. TLC Method
3.1.2. LC Technique
3.1.3. GC Technique
3.2. Rapid Screening Technologies for Mycotoxin Analysis
3.2.1. Enzyme-Linked Immunosorbent Assay (ELISA)
3.2.2. Lateral Flow Immunoassay (LFIA)
3.2.3. Aptamer-Based Lateral Flow Assay
3.2.4. Cytometric bead array (CBA)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization (WHO). WHO Traditional Medicine Strategy 2002–2005; WHO: Geneva, Switzerland, 2002. [Google Scholar]
- Zhu, Y.-P.; Woerdenbag, H.J. Traditional Chinese herbal medicine. Pharm. World Sci. 1995, 17, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Ramawat, K.G.; Goyal, S. The Indian herbal drugs scenario in global perspectives. In Bioactive Molecules and Medicinal Plants; Ramawat, K.G., Merillon, J.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 325–347. [Google Scholar]
- Baldé, N.M.; Youla, A.; Baldé, M.D.; Kaké, A.; Diallo, M.M.; Baldé, M.A.; Maugendre, D. Herbal medicine and treatment of diabetes in Africa: An example from Guinea. Diabetes Metab. 2006, 32, 171–175. [Google Scholar] [CrossRef]
- Wu, C.H.; Wang, C.C.; Kennedy, J. Changes in herb and dietary supplement use in the U.S. adult population: A comparison of the 2002 and 2007 National Health Interview Surveys. Clin. Ther. 2011, 33, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Natural Health Products Directorate-Health Canada. Natural Health Product Tracking Survey-2010 Final Report; Ipsos-Reid: Toronto, ON, Canada, 2011. [Google Scholar]
- World Health Organization (WHO). WHO Traditional Medicine Strategy: 2014–2023; WHO: Geneva, Switzerland, 2013. [Google Scholar]
- Abeywickrama, K.; Bean, G.A. Toxigenic Aspergillus flavus and aflatoxins in Sri Lankan medicinal plant material. Mycopathologia 1991, 113, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Halt, M. Moulds and mycotoxins in herb tea and medicinal plants. Eur. J. Epidemiol. 1998, 14, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Logrieco, A.; Moretti, A.; Solfrizzo, M. Alternaria toxins and plant diseases: An overview of origin, occurrence and risks. World Mycotoxin J. 2009, 2, 129–140. [Google Scholar] [CrossRef]
- Abramson, D.; Mills, J.T.; Marquardt, R.R.; Frohlich, A.A. Mycotoxins in fungal contaminated samples of animal feed from western Canada, 1982–1994. Can. J. Vet. Res. 1997, 61, 49–52. [Google Scholar] [PubMed]
- Diana Di Mavungu, J.; Monbaliu, S.; Scippo, M.L.; Maghuin-Rogister, G.; Schneider, Y.J.; Larondelle, Y.; Callebaut, A.; Robbens, J.; Van Peteghem, C.; De Saeger, S. LC-MS/MS multi-analyte method for mycotoxin determination in food supplements. Food Addit. Contam. Part A 2009, 26, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Ren, Y.P.; Zhu, J.F.; Cai, Z.X.; Chen, Y.; Luan, L.J.; Wu, Y.J. Multianalysis of 35 mycotoxins in traditional Chinese medicines by ultra-high-performance liquid chromatography-tandem mass spectrometry coupled with accelerated solvent extraction. J. Agric. Food Chem. 2012, 60, 8233–8247. [Google Scholar] [CrossRef] [PubMed]
- Zain, M.E. Impact of mycotoxins on humans and animals. J. Saudi Chem. Soc. 2011, 15, 129–144. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 1993; Volume 56. [Google Scholar]
- Ashiq, S.; Hussain, M.; Ahmad, B. Natural occurrence of mycotoxins in medicinal plants: A review. Fungal Genet. Biol. 2014, 66, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Domijan, A.M.; Peraica, M. 7.07—Carcinogenic Mycotoxins. In Comprehensive Toxicology, 3rd ed.; McQueen, C.A., Ed.; Elsevier: Oxford, UK, 2018; pp. 154–167. [Google Scholar]
- Brera, C.; De Santis, B.; Debegnach, F.; Miraglia, M. Chapter 12 Mycotoxins. In Comprehensive Analytical Chemistry; Picó, Y., Ed.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 51, pp. 363–427. [Google Scholar]
- Pitt, J.I. Mycotoxins: Deoxynivalenol and Other Trichothecenes A. In Encyclopedia of Food Safety; Academic Press: Waltham, MA, USA, 2014; pp. 295–298. [Google Scholar]
- Ostry, V. Alternaria mycotoxins: An overview of chemical characterization, producers, toxicity, analysis and occurrence in foodstuffs. World Mycotoxin J. 2008, 1, 175–188. [Google Scholar] [CrossRef]
- Schade, J.E.; King, A. Analysis of the major Alternaria toxins. J. Food Prot. 1984, 47, 978–995. [Google Scholar] [CrossRef]
- Frank, H.K. Citrinin. Zeitschrift für Ernährungswissenschaft 1992, 31, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Septien, I.; Cutuli, M.T.; Garcia, M.E.; Suarez, G.; Blanco, J.L. Solubility and stability of sterigmatocystin in different organic solvents. Toxicon 1993, 31, 1337–1340. [Google Scholar] [CrossRef]
- McCrone, W.C. Crystallographic Data. 87. Gliotoxin. Anal. Chem. 1954, 26, 1662–1663. [Google Scholar] [CrossRef]
- Cole, R.J.; Kirksey, J.W.; Moore, J.H.; Blankenship, B.R.; Diener, U.L.; Davis, N.D. Tremorgenic Toxin from Penicillium verruculosum. Appl. Microbiol. 1972, 24, 248–250. [Google Scholar] [PubMed]
- Eriksen, G.S.; Moldes-Anaya, A.; Fæste, C.K. Penitrem A and analogues: Toxicokinetics, toxicodynamics including mechanism of action and clinical significance. World Mycotoxin J. 2013, 6, 263–272. [Google Scholar] [CrossRef]
- Ciegler, A.; Detroy, R.W.; Lillehoj, E.B. Patulin, penicillic acid, and other carcinogenic lactones. In Microbial Toxins; Ciegler, A., Ed.; Academic: New York, NY, USA, 1971; Volume 6, p. 416. [Google Scholar]
- Silverton, J.V.; Akiyama, T.; Kabuto, C.; Sekita, S.; Yoshihira, K.; Natori, S. X-ray analysis of chaetoglobosin A, an indol-3-yl-13cytochalasan from Chaetomium globosum. Tetrahedron Lett. 1976, 17, 1349–1350. [Google Scholar] [CrossRef]
- Sekita, S.; Yoshihira, K.; Natori, S.; Kuwano, H. Structures of chaetoglobosin A and B, cytotoxic metabolites of chaetomium globosum. Tetrahedron Lett. 1973, 14, 2109–2112. [Google Scholar] [CrossRef]
- Frisvad, J.C.; Smedsgaard, J.; Larsen, T.O.; Samson, R.A. Mycotoxins, drugs and other extrolites produced by species in Penicillium subgenus Penicillium. Stud. Mycol. 2004, 49, 201–241. [Google Scholar]
- Steinrauf, L.K. Beauvericin and the other enniatins. Metal Ions Biol. Syst. 1985, 19, 139–171. [Google Scholar]
- Hu, L.; Rychlik, M. Occurrence of enniatins and beauvericin in 60 Chinese medicinal herbs. Food Addit. Contam. Part A 2014, 31, 1240–1245. [Google Scholar] [CrossRef] [PubMed]
- Trucksess, M.W.; Scott, P.M. Mycotoxins in botanicals and dried fruits: A review. Food Addit. Contam. Part A 2008, 25, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Kabak, B.; Dobson, A.D. Mycotoxins in spices and herbs: An update. Crit. Rev. Food Sci. Nutr. 2017, 57, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Do, K.H.; An, T.J.; Oh, S.K.; Moon, Y. Nation-based occurrence and endogenous biological reduction of mycotoxins in medicinal herbs and spices. Toxins 2015, 7, 4111–4130. [Google Scholar] [CrossRef] [PubMed]
- Azzoune, N.; Mokrane, S.; Riba, A.; Bouras, N.; Verheecke, C.; Sabaou, N.; Mathieu, F. Contamination of common spices by aflatoxigenic fungi and aflatoxin B1 in Algeria. Qual. Assur. Saf. Crops Foods 2015, 8, 137–144. [Google Scholar] [CrossRef]
- Keter, L.; Too, R.; Mwikwabe, N.; Mutai, C.; Orwa, J.; Mwamburi, L.; Ndwigah, S.; Bii, C.; Korir, R. Risk of fungi associated with aflatoxin and fumonisin in medicinal herbal products in the Kenyan market. Sci. World J. 2017, 2017, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jalili, M. Natural occurrence of aflatoxins contamination in commercial spices in Iran. Iran. J. Health Saf. Environ. 2016, 3, 513–517. [Google Scholar]
- Nejad, A.S.M.; Ghannad, M.S.; Kamkar, A. Determination of aflatoxin B1 levels in Iranian and Indian spices by ELISA method. Toxin Rev. 2014, 33, 151–154. [Google Scholar] [CrossRef]
- Veprikova, Z.; Zachariasova, M.; Dzuman, Z.; Zachariasova, A.; Fenclova, M.; Slavikova, P.; Vaclavikova, M.; Mastovska, K.; Hengst, D.; Hajslova, J. Mycotoxins in plant-based dietary supplements: Hidden health risk for consumers. J. Agric. Food Chem. 2015, 63, 6633–6643. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.S.J.; Lau, J.S.H.; El-Nezami, H. Chapter 10—Herbal medicine: Toxicity and recent trends in assessing their potential toxic effects. In Advances in Botanical Research; Shyur, L.-F., Lau, A.S.Y., Eds.; Academic Press: Cambridge, MA, USA, 2012; Volume 62, pp. 365–384. [Google Scholar]
- Food and Agriculture Organization of the United Nations. Worldwide Regulations for Mycotoxins in Food and Feed in 2003; Food and Agriculture Organization of the United Nations: Rome, Italy, 2004. [Google Scholar]
- European Union. Commission Regulation (EC) No 1881/2006 Setting maximum levels for certain contaminants in foodstuffs. Off. J. Eur. Uion 2006, L 364, 5–24. [Google Scholar]
- European Pharmacopoeia Commission. Determination of aflatoxin B1 in herbal drugs. In European Pharmacopoeia 9th Edition 2.8.18; Council of Europe: Strasbourg, France, 2016; Volume 1, p. 289. [Google Scholar]
- British Pharmacopoeia Commission. British Pharmacopoeia 2013 Appendix XI S. Determination of Mycotoxins in Herbal Drugs; Stationery Office: London, UK, 2012. [Google Scholar]
- World Health Organization (WHO). WHO Guidelines for Assessing Quality of Herbal Medicines With Reference to Contaminants and Residues; WHO: Geneva, Switzerland, 2007. [Google Scholar]
- United States Pharmacopeial Convention. USP 38-NF 33 Chapter 561: Articles of Botanical Origin; United States Pharmacopeial Convention: Rockville, MD, USA, 2014. [Google Scholar]
- United States Pharmacopeial Convention. USP Herbal Medicines Compendium; United States Pharmacopeial Convention: Rockville, MD, USA, 2017. [Google Scholar]
- Government of Canada, Natural and Non-prescription Health Products Directorate. Health Canada, Guidance Documents-Legislation and Guidelines-Natural Health Products, Quality of Natural Health Products Guide; Government of Canada, Natural and Non-prescription Health Products Directorate: Ottawa, ON, Canda, 2015; pp. 19–20.
- Chinese Pharmacopoeia Commission. Chinese Pharmacopoeia 2015 Edition volume IV; Chinese Medicine Science and Technology Press: Beijing, China, 2015. [Google Scholar]
- Korean Food & Drug. Korean Pharmacopoeia 10th Edition (English Version), General Tests, Processes and Apparatu; Korean Food & Drug: Chungcheongbuk-do, Korea, 2012; pp. 1673–1675.
- Japanese Pharmacopoeia Commentary Editorial Committee. The Japanese Pharmacopoeia 17th Edition (English Version), Analytical Methods for Aflatoxins in Crude Drug and Crude Drug Preparations; Japanese Pharmacopoeia Commentary Editorial Committee: Tokyo, Japan, 2016; pp. 2513–2515.
- United States Department of Agriculture Foreign Agricultural Service. Vietnam: Technical Regulations on Mycotoxins and Heavy Metals MRLs in Food; GAIN Report Number VM 3070; United States Department of Agriculture Foreign Agricultural Service: Washington, DC, USA, 2013; pp. 2–4.
- Kosalec, I.; Cvek, J.; Tomic, S. Contaminants of medicinal herbs and herbal products. Arh. Hig. Rada Toksikol. 2009, 60, 485–501. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.; Marín, S.; Sanchis, V.; Ramos, A.J. Mycotoxin in medicinal/aromatic herbs—A review. Bol. Latinoam. Caribe Plant. Med. Aromat. 2013, 12, 119–142. [Google Scholar]
- Dubey, N.K.; Mishra, P.K.; Kedia, A.; Prakash, B. Fungal and mycotoxin contamination of herbal raw materials and prospects of higher plant products as plant-based preservatives during post-harvest processing. In Microbial Diversity and Biotechnology in Food Security; Kharwar, R.N., Upadhyay, R.S., Dubey, N.K., Raghuwanshi, R., Eds.; Springer India: New Delhi, India, 2014; pp. 495–504. [Google Scholar]
- Rai, M.K.; Bonde, S.R.; Ingle, A.P.; Gade, A.K. Mycotoxin: rapid detection, differentiation and safety. J. Pharm. Educ. Res. 2012, 3, 22–34. [Google Scholar]
- Feng, X.; Kong, W.J.; Yang, M.H.; Ouyang, Z. Latest advancement for detection methods of mycotoxins in Traditional Chinese Medicine. World Sci. Tech. 2012, 14, 1944–1952. [Google Scholar]
- Whitaker, T.B. Standardisation of mycotoxin sampling procedures: An urgent necessity. Food Control 2003, 14, 233–237. [Google Scholar] [CrossRef]
- European Union. Commission Regulation (EC) No 401/2006 of 23 February 2006 laying down the methods of sampling and analysis for the official control of the levels of mycotoxins in foodstuffs. Off. J. Eur. Uion 2006, L70, 12–34. [Google Scholar]
- European Union. Commission Regulation (EU) No 178/2010 of 2 March 2010 amending Regulation (EC) No 401/2006 as regards groundnuts (peanuts), other oilseeds, tree nuts, apricot kernels, liquorice and vegetable oil. Off. J. Eur. Uion 2010, L52, 32–43. [Google Scholar]
- European Union. Commission Regulation (EU) No 519/2014 of 16 May 2014 amending Regulation (EC) No 401/2006 as regards methods of sampling of large lots, spices and food supplements, performance criteria for T-2, HT-2 toxin and citrinin and screening methods of analysis. Off. J. Eur. Uion 2014, L147, 29–43. [Google Scholar]
- European Union. Commission Directive 2002/27/EC of 13 March 2002 amending Directive laying down the sampling methods and the methods of analysis for the official control of the levels for certain contaminants in foodstuffs. Off. J. Eur. Commun. 2002, L75, 44–45. [Google Scholar]
- World Health Organization (WHO). Quality Control Methods for Herbal Materials; WHO: Geneva, Switzerland, 2011. [Google Scholar]
- Müller, P.; Basedow, T. Aflatoxin contamination of pods of Indian Cassia senna L. (Caesalpinaceae) before harvest, during drying and in storage: Reasons and possible methods of reduction. J. Stored Prod. Res. 2007, 43, 323–329. [Google Scholar] [CrossRef]
- Lee, D.; Lyu, J.; Lee, K.-G. Analysis of aflatoxins in herbal medicine and health functional foods. Food Control 2015, 48, 33–36. [Google Scholar] [CrossRef]
- Romagnoli, B.; Menna, V.; Gruppioni, N.; Bergamini, C. Aflatoxins in spices, aromatic herbs, herb-teas and medicinal plants marketed in Italy. Food Control 2007, 18, 697–701. [Google Scholar] [CrossRef]
- Braga, S.M.L.F.M.; de Medeiros, F.D.; de Jesus Oliveira, E.; Macedo, R.O. Development and validation of a method for the quantitative determination of aflatoxin contaminants in Maytenus ilicifolia by HPLC with fluorescence detection. Phytochem. Anal. 2005, 16, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Ren, Y.P.; Liu, X.S.; Luan, L.J.; Wu, Y.J. A reliable isotope dilution method for simultaneous determination of fumonisins B1, B2 and B3 in traditional Chinese medicines by ultra-high-performance liquid chromatography-tandem mass spectrometry. J. Sep. Sci. 2010, 33, 2723–2733. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Ren, Y.P.; Zhou, H.L.; Luan, L.J.; Cai, Z.X.; Wu, Y.J. A rapid method for simultaneous determination of zearalenone, alpha-zearalenol, beta-zearalenol, zearalanone, alpha-zearalanol and beta-zearalanol in traditional Chinese medicines by ultra-high-performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. B 2011, 879, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.-T.; Zhang, X.-F.; Yang, M.-H.; Ou-Yang, Z.; Liu, H.-B. Simultaneous determination of deoxynivalenol and nivalenol in Traditional Chinese Medicine by SPE and LC. Chromatographia 2010, 72, 551–555. [Google Scholar] [CrossRef]
- Wu, C.L.; Kuo, Y.H.; Lee, C.L.; Hsu, Y.W.; Pan, T.M. Synchronous high-performance liquid chromatography with a photodiode array detector and mass spectrometry for the determination of citrinin, monascin, ankaflavin, and the lactone and acid forms of monacolin K in red mold rice. J. AOAC Int. 2011, 62, 179–190. [Google Scholar]
- Prado, G.; Altoé, A.F.; Gomes, T.C.; Leal, A.S.; Morais, V.A.; Oliveira, M.S.; Ferreira, M.B.; Gomes, M.B.; Paschoal, F.N.; Von, S.S.R. Occurrence of aflatoxin B1 in natural products. Braz. J. Microbiol. 2012, 43, 1428–1436. [Google Scholar] [CrossRef] [PubMed]
- Monbaliu, S.; Wu, A.B.; Zhang, D.B.; Van Peteghem, C.; De Saeger, S. Multimycotoxin UPLC-MS/MS for tea, herbal infusions and the derived drinkable products. J. Agric. Food Chem. 2010, 58, 12664–12671. [Google Scholar] [CrossRef] [PubMed]
- Implvo, F.; Mendes, E.; Mbpp, O. Quantification of aflatoxins B1, B2, G1, and G2 in pepper by HPLC/fluorescence. J. Liq. Chromatogr. Relat. Technol. 2004, 27, 325–334. [Google Scholar]
- Fazekas, B.; Tar, A.; Kovács, M. Aflatoxin and ochratoxin A content of spices in Hungary. Food Addit. Contam. 2005, 22, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Z.; Wang, Z.; Gao, W.W.; Chen, J.; Yang, M.H.; Kuang, Y.; Huang, L.F.; Chen, S.L. Simultaneous determination of aflatoxin B1 and ochratoxin A in licorice roots and fritillary bulbs by solid-phase extraction coupled with high-performance liquid chromatography-tandem mass spectrometry. Food Chem. 2013, 138, 1048–1054. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.F.; Cheng, L.; Ji, S.; Wang, K. Simultaneous determination of seventeen mycotoxins residues in Puerariae lobatae radix by liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2014, 98, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.P.; Zhang, D.; Tan, L.H.; Yu, B.; Cao, W.G. Analysis of aflatoxins in traditional Chinese medicines: Classification of analytical method on the basis of matrix variations. Sci. Rep. 2016, 6, 30822. [Google Scholar] [CrossRef] [PubMed]
- Trucksess, M.W.; Weaver, C.M.; Oles, C.J.; Rump, L.V.; White, K.D.; Betz, J.M.; Rader, J.I. Use of multitoxin immunoaffinity columns for determination of aflatoxins and ochratoxin A in ginseng and ginger. J. AOAC Int. 2007, 90, 1042–1049. [Google Scholar] [PubMed]
- Whitaker, T.B.; Trucksess, M.W.; Weaver, C.M.; Slate, A. Sampling and analytical variability associated with the determination of aflatoxins and ochratoxin A in bulk lots of powdered ginger marketed in 1-lb bags. Anal. Bioanal. Chem. 2009, 395, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Weaver, C.M.; Trucksess, M.W. Determination of aflatoxins in botanical roots by a modification of AOAC Official Method 991.31: Single-laboratory validation. J. AOAC Int. 2010, 93, 184–189. [Google Scholar] [PubMed]
- Wei, R.W.; Yang, X.L.; Qiu, F.; Yang, M.H.; Qin, J.P. Simultaneous determination of aflatoxin B1, B2, G1, G2 and ochratoxin A in Glycyrrhiza uralensis by HPLC-FLD after immunoaffinity column with online post-column photochemical derivatization. China J. Chin. Mater. Med. 2011, 36, 2342–2346. [Google Scholar]
- Liu, S.Y.; Qiu, F.; Yang, M.H. Determination of aflatoxins in nelumbinis semen by immunoaffinity column clean-up and HPLC-FLD with on-line post-column photochemical derivatization and LC-MS/MS confirmation. China J. Chin. Mater. Med. 2012, 37, 305–309. [Google Scholar]
- Hao, A.; Zhao, L.; Liu, Y.; Wang, G.; Jin, H.; Bi, X.; Men, Q. HPLC determination of aflatoxin residues in traditional Chinese medicine Yinpian with post column photochemical derivation and fluorescence detection. Chin. J. Pharm. Anal. 2012, 32, 2203–2207. [Google Scholar]
- Wen, J.; Kong, W.J.; Wang, J.; Yang, M.H. Simultaneous determination of four aflatoxins and ochratoxin A in ginger and related products by HPLC with fluorescence detection after immunoaffinity column clean-up and postcolumn photochemical derivatization. J. Sep. Sci. 2013, 36, 3709–3716. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.J.; Liu, S.Y.; Qiu, F.; Xiao, X.H.; Yang, M.H. Simultaneous multi-mycotoxin determination in nutmeg by ultrasound-assisted solid-liquid extraction and immunoaffinity column clean-up coupled with liquid chromatography and on-line post-column photochemical derivatization-fluorescence detection. Analyst 2013, 138, 2729–2739. [Google Scholar] [CrossRef] [PubMed]
- Hao, A.Y.; Zhao, L.Y.; Liu, Y.H.; Wang, G.; Jin, H.Y.; Bi, X.L.; Men, Q.M. False positive research on HPLC determining aflatoxin residues in Chinese herbal pieces. Chin. J. Pharm. Anal. 2013, 33, 458–464. [Google Scholar]
- Kong, W.J.; Li, J.Y.; Qiu, F.; Wei, J.H.; Xiao, X.H.; Zheng, Y.; Yang, M.H. Development of a sensitive and reliable high performance liquid chromatography method with fluorescence detection for high-throughput analysis of multi-class mycotoxins in Coix seed. Anal. Chim. Acta 2013, 799, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.L.; Zhou, S.J.; Kong, W.J.; Ma, X.C.; Yang, M.H.; Wan, L.; Yang, S.H. Simultaneous determination of aflatoxins B1, B2, G1, G2 in Fructus Bruceae by high-performance liquid chromatography with online postcolumn photochemical derivatization. J. Sep. Sci. 2014, 37, 2771–2778. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Yang, H.L.; Leng, J.; Zheng, W.B. Determination of aflatoxin (AF) G2, G1, B2 and B1 in 72 batches of Shujin Huoxue pills by HPLC with post-column photochemical derivation. Chin. J. Pharm. Anal. 2014, 34, 437–441. [Google Scholar]
- Su, J.-H.; Zhang, C.; Zhong, S.-S.; Wang, M.-Z. Quantitative analysis of aflatoxin in Sterculiae Lychnophorae Semen by HPLC-FLD after immunoaffinity column with post-column photochemical derivatization. Chin. J. Exp. Trad. Med. Formulae 2014, 20, 75–78. [Google Scholar]
- Ali, N.; Hashim, N.H.; Shuib, N.S. Natural occurrence of aflatoxins and ochratoxin A in processed spices marketed in Malaysia. Food Addit. Contam. Part A 2015, 32, 518–532. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dou, X.W.; Kong, W.J.; Liu, C.M.; Han, X.; Yang, M.H. Assessment of critical points and development of a practical strategy to extend the applicable scope of immunoaffinity column cleanup for aflatoxin detection in medicinal herbs. J. Chromatogr. A 2017, 1483, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.L.; Martins, H.M.; Bernardo, F. Aflatoxins in spices marked in Portugal. Food Addit. Contam. 2001, 18, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Chen, J.M. HPLC analysis of alfatoxins in medicinal herb extracts by immunoaffinity column cleanup and post-column bromination. China J. Chin. Mater. Med. 2005, 30, 182–184. [Google Scholar]
- Zhang, X.H.; Liu, H.L.; Chen, J.M. Immunoaffinity column cleanup with liquid chromatography using post-column bromination for aflatoxins in medicinal herbs and plant extracts. J. Chromatogr. Sci. 2005, 43, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Ledzion, E.; Rybińska, K.; Postupolski, J.; Kurpińska-Jaworska, J.; Szczesna, M. Studies and safety evaluation of aflatoxins in herbal plants. Rocz. Panstw. Zakl. Hig. 2011, 62, 377–381. [Google Scholar] [PubMed]
- Yang, M.-H.; Chen, J.-M.; Zhang, X.-H. Immunoaffinity column clean-up and liquid chromatography with post-column derivatization for analysis of aflatoxins in Traditional Chinese Medicine. Chromatographia 2005, 62, 499–504. [Google Scholar] [CrossRef]
- Ip, S.-P.; Che, C.-T. Determination of aflatoxins in Chinese medicinal herbs by high-performance liquid chromatography using immunoaffinity column cleanup Improvement of recovery. J. Chromatogr. A 2006, 1135, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Katerere, D.R.; Stockenström, S.; Thembo, K.M.; Rheeder, J.P.; Shephard, G.S.; Vismer, H.F. A preliminary survey of mycological and fumonisin and aflatoxin contamination of African traditional herbal medicines sold in South Africa. Hum. Exp. Toxicol. 2008, 27, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Sha, D.X.; Uwe, G.; Sun, L.; Zhang, M.; Hou, F.; Kang, Q.; Wang, W. Comparison of two post-column derivatization systems for HPLC determination of aflatoxins in Citri Reticulatae Pericarpium. Chin. J. Pharm. Anal. 2013, 33, 1367–1371. [Google Scholar]
- Ran, C.; Chen, D.; Ma, H.; Jiang, Y. Graphene oxide adsorbent based dispersive solid phase extraction coupled with multi-pretreatment clean-up for analysis of trace aflatoxins in traditional proprietary Chinese medicines. J. Chromatogr. B 2017, 1044–1045, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Tassaneeyakul, W.; Razzazi-Fazeli, E.; Porasuphatana, S.; Bohm, J. Contamination of aflatoxins in herbal medicinal products in Thailand. Mycopathologia 2004, 158, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Catalán, J.; Piqué, E.; Falcó, G.; Borrego, N.; Rodamilans, M.; Llobet, J.M. Determination of aflatoxins in medicinal herbs by HPLC. An efficient method for routine analysis. Phytochem. Anal. 2005, 16, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Heperkan, D.; Güler, F.K.; Oktay, H.I. Mycoflora and natural occurrence of aflatoxin, cyclopiazonic acid, fumonisin and ochratoxin A in dried figs. Food Addit. Contam. Part A 2012, 29, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Hashim, N.H.; Saad, B.; Safan, K.; Nakajima, M.; Yoshizawa, T. Evaluation of a method to determine the natural occurrence of aflatoxins in commercial traditional herbal medicines from Malaysia and Indonesia. Food Chem. Toxicol. 2005, 43, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- D’Ovidio, K.; Trucksess, M.; Weaver, C.; Horn, E.; Mcintosh, M.; Bean, G. Aflatoxins in ginseng roots. Food Addit. Contam. 2006, 23, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.-H.; Lee, C.-H.; Jang, M.-R.; Son, Y.-W.; Lee, S.-M.; Choi, I.-S.; Kim, S.-H.; Kim, D.-B. Aflatoxins contamination in spices and processed spice products commercialized in Korea. Food Chem. 2008, 107, 1283–1288. [Google Scholar] [CrossRef]
- Zhao, S.R.; Xu, Y.X.; Xu, F.; Gao, W. High sensitive method for the detection of aflatoxins in extract from Chinese medicinal herb. J. Cap. Med. Univ. 2011, 32, 379–383. [Google Scholar]
- Li, W.G.; Xu, K.L.; Xiao, R.; Yin, G.F.; Liu, W.W. Development of an HPLC-based method for the detection of aflatoxins in Pu-erh tea. Int. J. Food Prop. 2015, 18, 842–848. [Google Scholar] [CrossRef]
- Garcia, J.I.G.; Moreno, M.C.; Callejas, F.R.; Velasco, S.R. Detection of aflatoxins, mutagens and carcinogens in black, white and green peppers (Piper Nigrum L.). J. Microb. Biochem. Technol. 2017, 09. [Google Scholar] [CrossRef]
- Wen, J.; Kong, W.J.; Hu, Y.C.; Wang, J.; Yang, M.H. Multi-mycotoxins analysis in ginger and related products by UHPLC-FLR detection and LC-MS/MS confirmation. Food Control 2014, 43, 82–87. [Google Scholar] [CrossRef]
- Yang, L.; Wang, L.; Pan, J.; Xiang, L.; Yang, M.; Logrieco, A.F. Determination of ochratoxin A in traditional Chinese medicinal plants by HPLC-FLD. Food Addit. Contam. Part A 2010, 27, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.L.; Zhou, S.J.; Kong, W.J.; Yang, M.H.; Wan, L.; Yang, S.H. Molecularly imprinted polymer-based solid phase clean-up for analysis of ochratoxin A in ginger and LC-MS/MS confirmation. Food Control 2013, 33, 337–343. [Google Scholar] [CrossRef]
- Yang, X.H.; Kong, W.J.; Hu, Y.C.; Yang, M.H.; Huang, L.Q.; Zhao, M.; Ouyang, Z. Aptamer-affinity column clean-up coupled with ultra high performance liquid chromatography and fluorescence detection for the rapid determination of ochratoxin A in ginger powder. J. Sep. Sci. 2014, 37, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, Y.-C.; Yang, M.-H.; Ou-Yang, Z. Natural occurrence of citrinin in widely consumed traditional Chinese food red yeast rice, medicinal plants and their related products. Food Chem. 2012, 132, 1040–1045. [Google Scholar] [CrossRef]
- Mornar, A.; Sertic, M.; Nigovic, B. Development of a rapid LC/DAD/FLD/MSn method for the simultaneous determination of monacolins and citrinin in red fermented rice products. J. Agric. Food Chem. 2013, 61, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, W.; Logrieco, A.F.; Yang, M.; Ou-yang, Z.; Wang, X.; Guo, Q. Determination of zearalenone in traditional Chinese medicinal plants and related products by HPLC-FLD. Food Addit. Contam. Part A 2011, 28, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Zhao, R.H.; Chen, B.; Yang, M.H. Determination of zearalenone in barley by high-performance liquid chromatography coupled with evaporative light scattering detection and natural occurrence of zearalenone in functional food. Food Chem. 2011, 126, 1508–1511. [Google Scholar] [CrossRef]
- Liau, B.C.; Jong, T.T.; Lee, M.R.; Chang, C.M. Supercritical fluid extraction and quantification of aflatoxins in Zizyphi Fructus by liquid chromatography/atmospheric pressure chemical ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Jin, H.Y.; Sun, L.; Ma, S.C.; Lin, R.C. Determination of aflatoxins in medicinal herbs by high-performance liquid chromatography-tandem mass spectrometry. Phytochem. Anal. 2012, 23, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Qiu, F.; Kong, W.J.; Wei, J.H.; Xiao, X.H.; Yang, M.H. Development and validation of an accurate and rapid LC-ESI-MS/MS method for the simultaneous quantification of aflatoxin B1, B2, G1 and G2 in lotus seeds. Food Control 2013, 29, 156–161. [Google Scholar] [CrossRef]
- Siddique, N.A.; Mujeeb, M.; Ahmad, S.; Panda, B.P.; Makhmoor, M. Determination of aflatoxins in medicinal plants by high-performance liquid chromatography-tandem mass spectrometry. J. Pharm. Pharmaceut. Sci. 2013, 16, 321–330. [Google Scholar] [CrossRef]
- Zheng, R.S.; Xu, H.; Wang, W.L.; Zhan, R.T.; Chen, W.W. Determination of aflatoxin B1, B2, G1, G2 in armeniacae semen amarum by high-performance liquid chromatography-tandem mass spectrometry. China J. Chin. Mater. Med. 2013, 38, 3534–3538. [Google Scholar]
- Li, X.-P.; Zhao, Y.-G.; Chen, X.-H.; Pan, S.-D.; Jin, M.-C. Simultaneous determination of four aflatoxins in walnut kernel using dispersive solid-phase extraction combined with ultra fast liquid chromatography-tandem mass spectrometry. Chin. J. Health Lab. Technol. 2014, 24, 2647–2650. [Google Scholar]
- Wei, R.W.; Qiu, F.; Kong, W.J.; Wei, J.H.; Yang, M.H.; Luo, Z.L.; Qin, J.P.; Ma, X.J. Co-occurrence of aflatoxin B1, B2, G1, G2 and ochrotoxin A in Glycyrrhiza uralensis analyzed by HPLC-MS/MS. Food Control 2013, 32, 216–221. [Google Scholar] [CrossRef]
- Zheng, R.S.; Xu, H.; Wang, W.L.; Zhan, R.T.; Chen, W.W. Simultaneous determination of aflatoxin B1, B2, G1, G2, ochratoxin A, and sterigmatocystin in traditional Chinese medicines by LC-MS-MS. Anal. Bioanal. Chem. 2014, 406, 3031–3039. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Zheng, Y.; Luan, L.; Cai, Z.; Ren, Y.; Wu, Y. An ultra-high-performance liquid chromatography-tandem mass spectrometry method for simultaneous determination of aflatoxins B1, B2, G1, G2, M1 and M2 in traditional Chinese medicines. Anal. Chim. Acta 2010, 664, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Chun-Yan, B.; Zhu, B.; Jiang, S.J. Simultaneous determination of 5 mycotoxins in Chinese patent medicines by high performance liquid chromatography-tandem mass spectrometry. Chin. J. Health Lab. Technol. 2014, 24, 2654–2656. [Google Scholar]
- Yang, X.H.; Hu, Y.C.; Kong, W.J.; Chu, X.F.; Yang, M.H.; Zhao, M.; Ouyang, Z. Ultra-fast liquid chromatography with tandem mass spectrometry determination of ochratoxin A in traditional Chinese medicines based on vortex-assisted solid-liquid microextraction and aptamer-affinity column clean-up. J. Sep. Sci. 2014, 37, 3052–3059. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-M.; Zhang, S.; Mao, D.; Chen, J.; Ji, S. Determination of patulin in Chinese herbs by UHPLC- triple quadrupole MS with dispersive solid phase extraction. Chin. J. Health Lab. Technol. 2014, 24, 337–340. [Google Scholar]
- Waskiewicz, A.; Beszterda, M.; Bocianowski, J.; Golinski, P. Natural occurrence of fumonisins and ochratoxin A in some herbs and spices commercialized in Poland analyzed by UPLC-MS/MS method. Food Microbiol. 2013, 36, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.H.; Qiu, F.; Yang, M.H.; Zhen, O.Y. Simultaneous determination of zearalenone and α-zearalenol in traditional Chinese medicines by high performance liquid chromatography-tandem mass spectrometry. Chin. J. Health Lab. Technol. 2011, 21, 1322–1327. [Google Scholar]
- Dong, M.F.; Si, W.S.; Wang, W.M.; Bai, B.; Nie, D.X.; Song, W.G.; Zhao, Z.H.; Guo, Y.R.; Han, Z. Determination of type A trichothecenes in coix seed by magnetic solid-phase extraction based on magnetic multi-walled carbon nanotubes coupled with ultra-high performance liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2016, 408, 6823–6831. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qiu, F.; Yang, M.H.; Ouyang, Z. LC-MS/MS determination of citrinin in traditional Chinese medicines. Chin. J. Pharm. Anal. 2011, 31, 1726–1730. [Google Scholar]
- Arroyo-Manzanares, N.; García-Campaña, A.M.; Gámiz-Gracia, L. Multiclass mycotoxin analysis in Silybum marianum by ultra high performance liquid chromatography–tandem mass spectrometry using a procedure based on QuEChERS and dispersive liquid–liquid microextraction. J. Chromatogr. A 2013, 1282, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, C.J.; Li, J.; Luan, L.J.; Liu, X.S.; Wu, Y.J. Determination of 10 mycotoxin contaminants in Panax notoginseng by ultra performance liquid chromatography-tandem mass spectrometry. Acta Pharm. Sin. 2015, 50, 81–85. [Google Scholar]
- Liu, H.M.; Kong, W.J.; Liu, C.M.; Liu, Q.T.; Hu, Y.C.; Yang, M.H. Rapid analysis and identification of multi-class mycotoxins in Morinda officinalis by UFLC-ESI-MS/MS. RSC Adv. 2015, 5, 63561–63571. [Google Scholar] [CrossRef]
- Liu, Q.T.; Kong, W.J.; Guo, W.Y.; Yang, M.H. Multi-class mycotoxins analysis in Angelica sinensis by ultra fast liquid chromatography coupled with tandem mass spectrometry. J. Chromatogr. B 2015, 988, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.; Kong, W.J.; Li, Y.J.; Liu, H.M.; Liu, Q.T.; Dou, X.W.; Ou-yang, Z.; Yang, M.H. High-throughput determination of multi-mycotoxins in Chinese yam and related products by ultra fast liquid chromatography coupled with tandem mass spectrometry after one-step extraction. J. Chromatogr. B 2016, 1022, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Meng, W.; Sun, W.; Li, D.; Yu, Z.; Tong, L.; Zhao, Y. Simultaneous qualitative and quantitative analysis of 21 mycotoxins in Radix Paeoniae Alba by ultra-high performance liquid chromatography quadrupole linear ion trap mass spectrometry and QuEChERS for sample preparation. J. Chromatogr. B 2016, 1031, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.M.; Luo, J.Y.; Kong, W.J.; Liu, Q.; Hu, Y.C.; Yang, M.H. UFLC-ESI-MS/MS analysis of multiple mycotoxins in medicinal and edible Areca catechu. Chemosphere 2016, 150, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kong, W.J.; Yang, M.H. Simultaneous determination of 11 mycotoxins in malt by isotope internal standard-UPLC-MS/MS. Acta Pharm. Sin. 2016, 51, 110–115. [Google Scholar]
- Reinholds, I.; Pugajeva, I.; Bavrins, K.; Kuckovska, G.; Bartkevics, V. Mycotoxins, pesticides and toxic metals in commercial spices and herbs. Food Additi. Contam. Part B 2017, 10, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.S.; Kong, W.J.; Wang, S.; Wei, J.H.; Yang, M.H. Simultaneous analysis of multiple mycotoxins in Alpinia oxyphylla by UPLC-MS/MS. World Mycotoxin J. 2017, 10, 41–51. [Google Scholar] [CrossRef]
- Jiang, K.; Huang, P.; Luan, L.; Fan, K.; Guo, W.; Zhao, Z.; Wu, Y.; Han, Z. Iron (II, III) oxide/multi-walled carbon nanotube composite as solid-phase extraction sorbent followed by ultra-high performance liquid chromatography tandem mass spectrometry for simultaneous determination of zearalenone and type A trichothecenes in Salviae miltiorrhizae Radix et Rhizoma (Danshen). J. Chromatogr. A 2017, 1482, 1–10. [Google Scholar] [PubMed]
- Zhang, S.; Lu, J.; Wang, S.; Mao, D.; Miao, S.; Ji, S. Multi-mycotoxins analysis in Pheretima using ultra-high-performance liquid chromatography tandem mass spectrometry based on a modified QuEChERS method. J. Chromatogr. B 2016, 1035, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Pereira, V.L.; Fernandes, J.O.; Cunha, S.C. Mycotoxins in cereals and related foodstuffs: A review on occurrence and recent methods of analysis. Trends Food Sci. Technol. 2014, 36, 96–136. [Google Scholar] [CrossRef]
- Masinde, L.A.; Sheng, W.; Xu, X.; Zhang, Y.; Yuan, M.; Kennedy, I.R.; Wang, S. Colloidal gold based immunochromatographic strip for the simple and sensitive determination of aflatoxin B1 and B2 in corn and rice. Microchim. Acta 2013, 180, 921–928. [Google Scholar] [CrossRef]
- Dzuman, Z.; Zachariasova, M.; Lacina, O.; Veprikova, Z.; Slavikova, P.; Hajslova, J. A rugged high-throughput analytical approach for the determination and quantification of multiple mycotoxins in complex feed matrices. Talanta 2014, 121, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Fung, K.Y.; Lau, Y.T.; Ng, K.M.; Lau, D.T.W. Relationship between maceration and extraction yield in the production of Chinese herbal medicine. Food Bioprod. Process. 2016, 98, 236–243. [Google Scholar] [CrossRef]
- Richter, B.E.; Jones, B.A.; Ezzell, J.L.; Porter, N.L.; Avdalovic, N.; Pohl, C.; Chem, A. Accelerated solvent extraction: A technique for sample preparation. Anal. Chem. 1996, 68, 1033–1039. [Google Scholar] [CrossRef]
- Meneely, J.P.; Ricci, F.; van Egmond, H.P.; Elliott, C.T. Current methods of analysis for the determination of trichothecene mycotoxins in food. TrAC-Trend. Anal. Chem. 2011, 30, 192–203. [Google Scholar] [CrossRef]
- Wang, X.; Liu, B.; Lu, Q.; Qu, Q. Graphene-based materials: Fabrication and application for adsorption in analytical chemistry. J. Chromatogr. A 2014, 1362, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Si, W.; Jiang, K.; Nie, D.; Wu, Y.; Zhao, Z.; De Saeger, S.; Han, Z. Multi-walled carbon nanotubes as solid-phase extraction sorbents for simultaneous determination of type A trichothecenes in maize, wheat and rice by ultra-high performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2015, 1423, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Hamula, C.; Guthrie, J.; Zhang, H.; Li, X.; Le, X. Selection and analytical applications of aptamers. TrAC-Trend. Anal. Chem. 2006, 25, 681–691. [Google Scholar] [CrossRef]
- McKeague, M.; Bradley, C.R.; De Girolamo, A.; Visconti, A.; Miller, J.D.; DeRosa, M.C. Screening and initial binding assessment of fumonisin B1 aptamers. Int. J. Mol. Sci. 2010, 11, 4864–4881. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Long, Y.; Pan, J.; Li, K.; Liu, F. Application of molecularly imprinted polymers to solid-phase extraction of analytes from real samples. J. Biochem. Biophys. Methods 2007, 70, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Haupt, K.; Mosbach, K. Molecularly imprinted polymers and their use in biomimetic sensors. Chem. Rev. 2000, 100, 2495–2504. [Google Scholar] [CrossRef] [PubMed]
- Appell, M.; Kendra, D.F.; Kim, E.K.; Maragos, C.M. Synthesis and evaluation of molecularly imprinted polymers as sorbents of moniliformin. Food Addit. Contam. 2007, 24, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Anastassiades, M.; Lehotay, S.J.; Stajnbaher, D.; Schenck, F.J. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J. AOAC Int. 2003, 86, 412–431. [Google Scholar] [PubMed]
- González-Curbelo, M.Á.; Socas-Rodríguez, B.; Herrera-Herrera, A.V.; González-Sálamo, J.; Hernández-Borges, J.; Rodríguez-Delgado, M.Á. Evolution and applications of the QuEChERS method. TrAC-Trend. Anal. Chem. 2015, 71, 169–185. [Google Scholar] [CrossRef]
- Sargeant, K.; O’Kelly, J.; Carnaghan, R.B.A.; Allcroft, R. The assay of a toxic principle in certain groundnut meals. Vet. Rec. 1961, 73, 1219–1222. [Google Scholar]
- Honma, Y.; Naito, S.; Earnshaw, A.; Nagashima, H.; Goto, T. Progress in the accuracy of mycotoxin analysis in the last quarter century. Mycotoxins 2004, 54, 33–38. [Google Scholar] [CrossRef]
- Rizzo, I.; Vedoya, G.; Maurutto, S.; Haidukowski, M.; Varsavsky, E. Assessment of toxigenic fungi on Argentinean medicinal herbs. Microbiol. Res. 2004, 159, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Bugno, A.; Almodovar, A.A.B.; Pereira, T.C.; Pinto, T.J.A.; Sabino, M. Occurrence of toxigenic fungi in herbal drugs. Braz. J. Microbiol. 2006, 37, 47–51. [Google Scholar] [CrossRef]
- Singh, P.; Srivastava, B.; Kumar, A.; Dubey, N.K. Fungal contamination of raw materials of some herbal drugs and recommendation of Cinnamomum camphora oil as herbal fungitoxicant. Microb. Ecol. 2008, 56, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Gautam, A.; Sharma, S.; Bhadauria, R. Detection of toxigenic fungi and mycotoxins in medicinally important powdered herbal drugs. Internet J. Microbiol. 2010, 7, 2. [Google Scholar]
- Gautam, A.K.; Bhadauria, R. Mycoflora and mycotoxins in some important stored crude and powdered herbal drugs. Biol. Forum 2009, 1, 1–7. [Google Scholar]
- Aquino, S.; Gonçalez, E.; Rossi, M.H.; Nogueira, J.H.; Reis, T.A.; Corrêa, B. Evaluation of fungal burden and aflatoxin presence in packed medicinal plants treated by gamma radiation. J. Food Prot. 2010, 73, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Delgado, S.; Núñez, F.; Sánchez, B.; Bermúdez, E.; Rodríguez, J.M. Toxigenic microorganisms in medicinal plants used for ritual protection of infants. Food Res. Int. 2011, 44, 304–309. [Google Scholar] [CrossRef]
- Ahmad, B.; Ashiq, S.; Hussain, A.; Bashir, S.; Hussain, M. Evaluation of mycotoxins, mycobiota, and toxigenic fungi in selected medicinal plants of Khyber Pakhtunkhwa, Pakistan. Fungal Biol. 2014, 118, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Khati, P. Mycoflora and aflatoxin assessment of crude herbal drugs during storage in Haridwar, Uttarakhand, India. Indian Phytopathol. 2014, 67, 407–411. [Google Scholar]
- Ezekwesiliofili, J.O.; Onyemelukwe, N.F.; Agwaga, P.; Orji, I. The bioload and aflatoxin content of herbal medicines from selected states in Nigeria. Afr. J. Tradit. Complement. Altern. Med. 2014, 11, 143–147. [Google Scholar] [CrossRef]
- Aiko, V.; Mehta, A. Prevalence of toxigenic fungi in common medicinal herbs and spices in India. 3 Biotech 2016, 6, 159. [Google Scholar] [CrossRef] [PubMed]
- Rajeshwari, P.; Raveesha, K. Mycological analysis and aflatoxin B1 contaminant estimation of herbal drug raw materials. Afr. J. Tradit. Complement. Altern. Med. 2016, 13, 123–131. [Google Scholar] [PubMed]
- Yue, Y.-T.; Zhang, X.-F.; Pan, J.; Ou-Yang, Z.; Wu, J.; Yang, M.-H. Determination of deoxynivalenol in medicinal herbs and related products by GC-ECD and confirmation by GC-MS. Chromatographia 2010, 71, 533–538. [Google Scholar] [CrossRef]
- Antignac, J.-P.; de Wasch, K.; Monteau, F.; De Brabander, H.; Andre, F.; Le Bizec, B. The ion suppression phenomenon in liquid chromatography–mass spectrometry and its consequences in the field of residue analysis. Anal. Chim. Acta 2005, 529, 129–136. [Google Scholar] [CrossRef]
- Rychlik, M.; Asam, S. Stable isotope dilution assays in mycotoxin analysis. Anal. Bioanal. Chem. 2008, 390, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Krska, R.; Baumgartner, S.; Josephs, R. The state-of-the-art in the analysis of type-A and -B trichothecene mycotoxins in cereals. Fresen. J. Anal. Chem. 2001, 371, 285–299. [Google Scholar] [CrossRef]
- Li, P.; Zhang, Z.; Hu, X.; Zhang, Q. Advanced hyphenated chromatographic-mass spectrometry in mycotoxin determination: Current status and prospects. Mass Spectrom. Rev. 2013, 32, 420–452. [Google Scholar] [CrossRef] [PubMed]
- Lattanzio, V.M.T.; Pascale, M.; Visconti, A. Current analytical methods for trichothecene mycotoxins in cereals. TrAC-Trend. Anal. Chem. 2009, 28, 758–768. [Google Scholar] [CrossRef]
- Yue, Y.-T.; Zhang, X.-F.; Ou-Yang, Z.; Gao, W.-W.; Wu, J.; Yang, M.-H. Determination of T-2 toxin in Traditional Chinese Herbal Medicines by GC-ECD. Chromatographia 2009, 70, 1495–1499. [Google Scholar] [CrossRef]
- Radová, Z.; Holadová, K.; Hajšlová, J. Comparison of two clean-up principles for determination of trichothecenes in grain extract. J. Chromatogr. A 1998, 829, 259–267. [Google Scholar] [CrossRef]
- Kotal, F.; Holadová, K.; Hajšlová, J.; Poustka, J.; Radová, Z. Determination of trichothecenes in cereals. J. Chromatogr. A 1999, 830, 219–225. [Google Scholar] [CrossRef]
- Kong, W.J.; Zhang, X.F.; Shen, H.H.; Ou-Yang, Z.; Yang, M.H. Validation of a gas chromatography-electron capture detection of T-2 and HT-2 toxins in Chinese herbal medicines and related products after immunoaffinity column clean-up and pre-column derivatization. Food Chem. 2012, 132, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Lequin, R.M. Enzyme immunoassay (EIA)/enzyme-linked immunosorbent assay (ELISA). Clin. Chem. 2005, 51, 2415–2418. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, A.; Jinap, S.; Soleimany, F. Qualitative and quantitative analysis of mycotoxins. Comp. Rev. Food Sci. Food Saf. 2009, 8, 202–251. [Google Scholar] [CrossRef]
- Ok, H.E.; Kim, H.J.; Shim, W.B.; Lee, H.; Bae, D.-H.; Chung, D.-H.; Chun, H.S. Natural occurrence of aflatoxin B1 in marketed foods and risk estimates of dietary exposure in Koreans. J. Food Protect. 2007, 70, 2824–2828. [Google Scholar] [CrossRef]
- Chen, J.M.; Zhang, X.H.; Yang, M.H.; Jin, Y. A survey of determination of aflatoxin in Chinese materia medicia. Chin. Trad. Herbal Drugs 2006, 37, 463–466. [Google Scholar]
- Thirumala-Devi, K.; Mayo, M.A.; Reddy, G.; Emmanuel, K.E.; Larondelle, Y.; Reddy, D.V.R. Occurrence of ochratoxin A in black pepper, coriander, ginger and turmeric in India. Food Addit. Contam. 2001, 18, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.F.; Dou, X.W.; Kong, W.J.; Yang, M.H.; Zhao, C.; Zhao, M.; Ouyang, Z. Contamination level of aflatoxin B1 in lotus seeds rapid screening by indirect competitive ELISA method. China J. Chin. Mater. Med. 2015, 40, 704–709. [Google Scholar]
- Santos, L.; Marín, S.; Sanchis, V.; Ramos, A.J. Screening of mycotoxin multicontamination in medicinal and aromatic herbs sampled in Spain. J. Sci. Food Agric. 2009, 89, 1802–1807. [Google Scholar] [CrossRef]
- Tosun, H.; Arslan, R. Determination of aflatoxin B1 levels in organic spices and herbs. Sci. World J. 2013, 2013, 874093. [Google Scholar] [CrossRef] [PubMed]
- Jeswal, P.; Kumar, D. Mycobiota and natural incidence of aflatoxins, ochratoxin A, and citrinin in Indian spices confirmed by LC-MS/MS. Int. J. Microbiol. 2015, 2015, 242486. [Google Scholar] [CrossRef] [PubMed]
- Tonti, S.; Mandrioli, M.; Nipoti, P.; Pisi, A.; Toschi, T.G.; Prodi, A. Detection of fumonisins in fresh and dehydrated commercial garlic. J. Agric. Food Chem. 2017, 65, 7000–7005. [Google Scholar] [CrossRef] [PubMed]
- Colak, H.; Bingol, E.B.; Hampikyan, H.; Nazli, B. Determination of aflatoxin contamination in red-scaled, red and black pepper by ELISA and HPLC. J. Food Drug Anal. 2006, 14, 292–296. [Google Scholar]
- Gray, S.L.; Lackey, B.R.; Tate, P.L.; Riley, M.B.; Camper, N.D. Mycotoxins in root extracts of American and Asian ginseng bind estrogen receptors alpha and beta. Exp. Biol. Med. 2004, 229, 560–568. [Google Scholar] [CrossRef]
- Shim, W.B.; Kim, K.; Ofori, J.A.; Chung, Y.C.; Chung, D.H. Occurrence of aflatoxins in herbal medicine distributed in South Korea. J. Food Protect. 2012, 75, 1991–1999. [Google Scholar] [CrossRef] [PubMed]
- Kononenko, G.P.; Burkin, A.A. Distribution of mycotoxins and usnic acid in the thalli of epigeous lichens. Biol. Bull. Russ. Acad. Sci. 2015, 42, 213–219. [Google Scholar] [CrossRef]
- Saleh, A.; Abdullahi, I.O.; Olonitola, O.S. Determination of aflatoxin contamination in cassava flour sold in selected markets in Zaria, Kaduna state; Nigeria. Bayer. J. Pure Appl. Sci. 2016, 9, 145–147. [Google Scholar] [CrossRef]
- Dungkokkruad, P.; Jutirak, J.; Wongkamsom, N.; Kongsee, P.; Saichi, M.; Meesaad, A. The ability of Thai herbal household plant crude extracts (Alpinia Galangal) in growth inhibition of mold Aspergillus Flavus and destruction of aflatoxin B1. Eur. J. Sustain. Dev. 2017, 6, 210–216. [Google Scholar]
- Campbell, R.L.; Wagner, D.B.; O’Connell, J.P. Solid Phase Assay with Visual Readout. U.S. Patent 4,703,017, 1987. [Google Scholar]
- Sun, X.L.; Zhao, X.L.; Tang, J.; Zhou, J.; Chu, F.S. Preparation of gold-labeled antibody probe and its use in immunochromatography assay for detection of aflatoxin B1. Int. J. Food Microbiol. 2005, 99, 185–194. [Google Scholar]
- Anfossi, L.; Baggiani, C.; Giovannoli, C.; D’Arco, G.; Giraudi, G. Lateral-flow immunoassays for mycotoxins and phycotoxins: A review. Anal. Bioanal. Chem. 2013, 405, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, P.W.; Zhang, Q.; Li, R.; Zhang, W.; Zhang, Z.W.; Ding, X.X.; Tang, X.Q. Multi-component immunochromatographic assay for simultaneous detection of aflatoxin B1, ochratoxin A and zearalenone in agro-food. Biosens. Bioelectron. 2013, 49, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xie, Y.J.; Kong, W.J.; Yang, M.H.; Wang, J. Determination of aflatoxin B1 in lotus seeds by colloidal gold test strip based on lateral flow immune chromatography technique. Cent. South Pharm. 2015, 23, 779–780. [Google Scholar]
- Xie, Y.-J.; Yang, Y.; Kong, W.-J.; Yang, S.-H.; Yang, M.-H. Application of nanoparticle probe-based lateral flow immunochromatographic assay in mycotoxins detection. Chin. J. Anal. Chem. 2015, 43, 618–628. [Google Scholar] [CrossRef]
- Wang, L.B.; Ma, W.W.; Chen, W.; Liu, L.Q.; Ma, W.; Zhu, Y.Y.; Xu, L.G.; Kuang, H.; Xu, C.L. An aptamer-based chromatographic strip assay for sensitive toxin semi-quantitative detection. Biosens. Bioelectron. 2011, 26, 3059–3062. [Google Scholar] [CrossRef] [PubMed]
- Shim, W.B.; Kim, M.J.; Mun, H.; Kim, M.G. An aptamer-based dipstick assay for the rapid and simple detection of aflatoxin B1. Biosens. Bioelectron. 2014, 62, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.L.; Kong, W.J.; Dou, X.W.; Zhao, M.; Ouyang, Z.; Yang, M.H. An aptamer based lateral flow strip for on-site rapid detection of ochratoxin A in Astragalus membranaceus. J. Chromatogr. B 2016, 1022, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Aqai, P.; Peters, J.; Gerssen, A.; Haasnoot, W.; Nielen, M.W. Immunomagnetic microbeads for screening with flow cytometry and identification with nano-liquid chromatography mass spectrometry of ochratoxins in wheat and cereal. Anal. Bioanal. Chem. 2011, 400, 3085–3096. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, H.; Guo, L.; Zheng, Y.; Guo, Y. Microsphere-based flow cytometric immunoassay for the determination of citrinin in red yeast rice. Food Chem. 2012, 134, 2540–2545. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.Y.; Zhang, Y.; Su, L.; Chang, J.; Wang, H.J. Application of upconversion luminescent-magnetic microbeads with weak background noise and facile separation in ochratoxin A detection. J. Nanopart. Res. 2017, 19, 60. [Google Scholar] [CrossRef]
- Czeh, A.; Mandy, F.; Feher-Toth, S.; Torok, L.; Mike, Z.; Koszegi, B.; Lustyik, G. A flow cytometry based competitive fluorescent microsphere immunoassay (CFIA) system for detecting up to six mycotoxins. J. Immunol. Methods 2012, 384, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.; Bienenmann-Ploum, M.; de Rijk, T.; Haasnoot, W. Development of a multiplex flow cytometric microsphere immunoassay for mycotoxins and evaluation of its application in feed. Mycotoxin Res. 2011, 27, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.; Thomas, D.; Boers, E.; de Rijk, T.; Berthiller, F.; Haasnoot, W.; Nielen, M.W. Colour-encoded paramagnetic microbead-based direct inhibition triplex flow cytometric immunoassay for ochratoxin A, fumonisins and zearalenone in cereals and cereal-based feed. Anal. Bioanal. Chem. 2013, 405, 7783–7794. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.-B.; Liu, Q.-T.; Dou, X.-W.; Yang, M.-H.; Kong, W.-J.; Wan, L. Rapid detection of ochratoxin A in malt by cytometric bead array based on indirect competition principle. Chin. J. Anal. Chem. 2016, 44, 625–632. [Google Scholar] [CrossRef]
- Kong, W.J.; Xiao, C.B.; Ying, G.Y.; Liu, X.F.; Zhao, X.H.; Wang, R.L.; Wan, L.; Yang, M.H. Magnetic microspheres-based cytometric bead array assay for highly sensitive detection of ochratoxin A. Biosens. Bioelectron. 2017, 94, 420–428. [Google Scholar] [CrossRef] [PubMed]
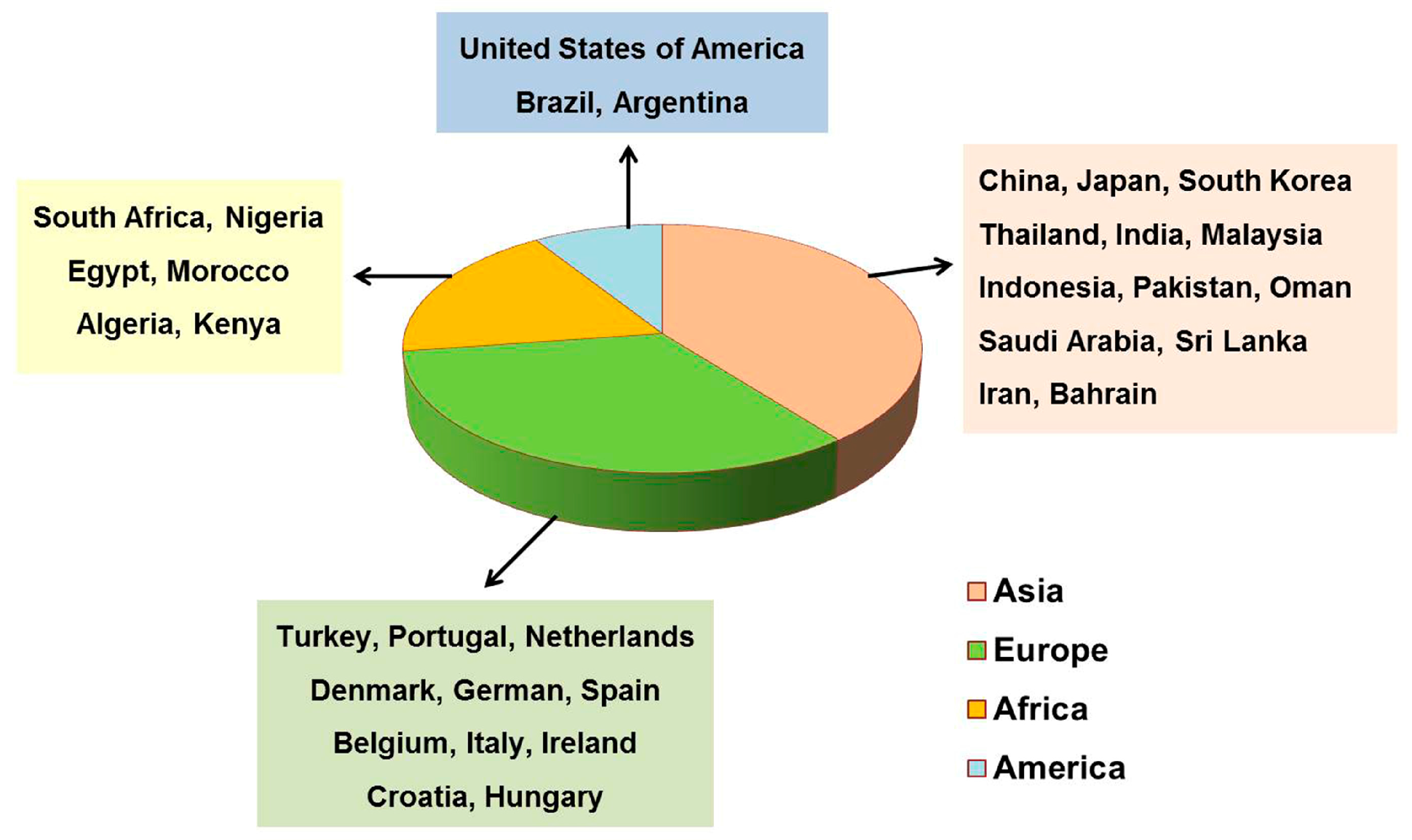
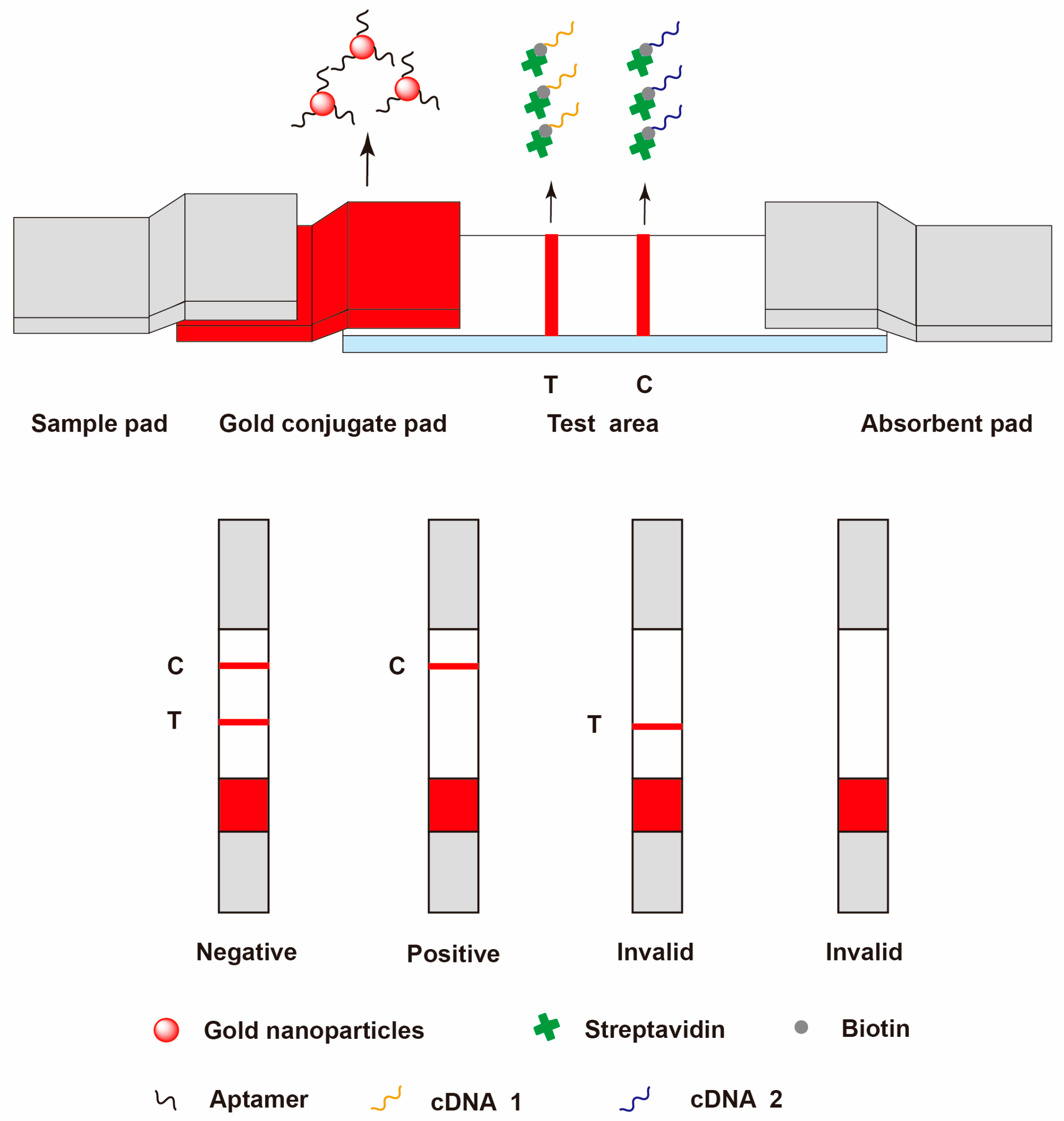


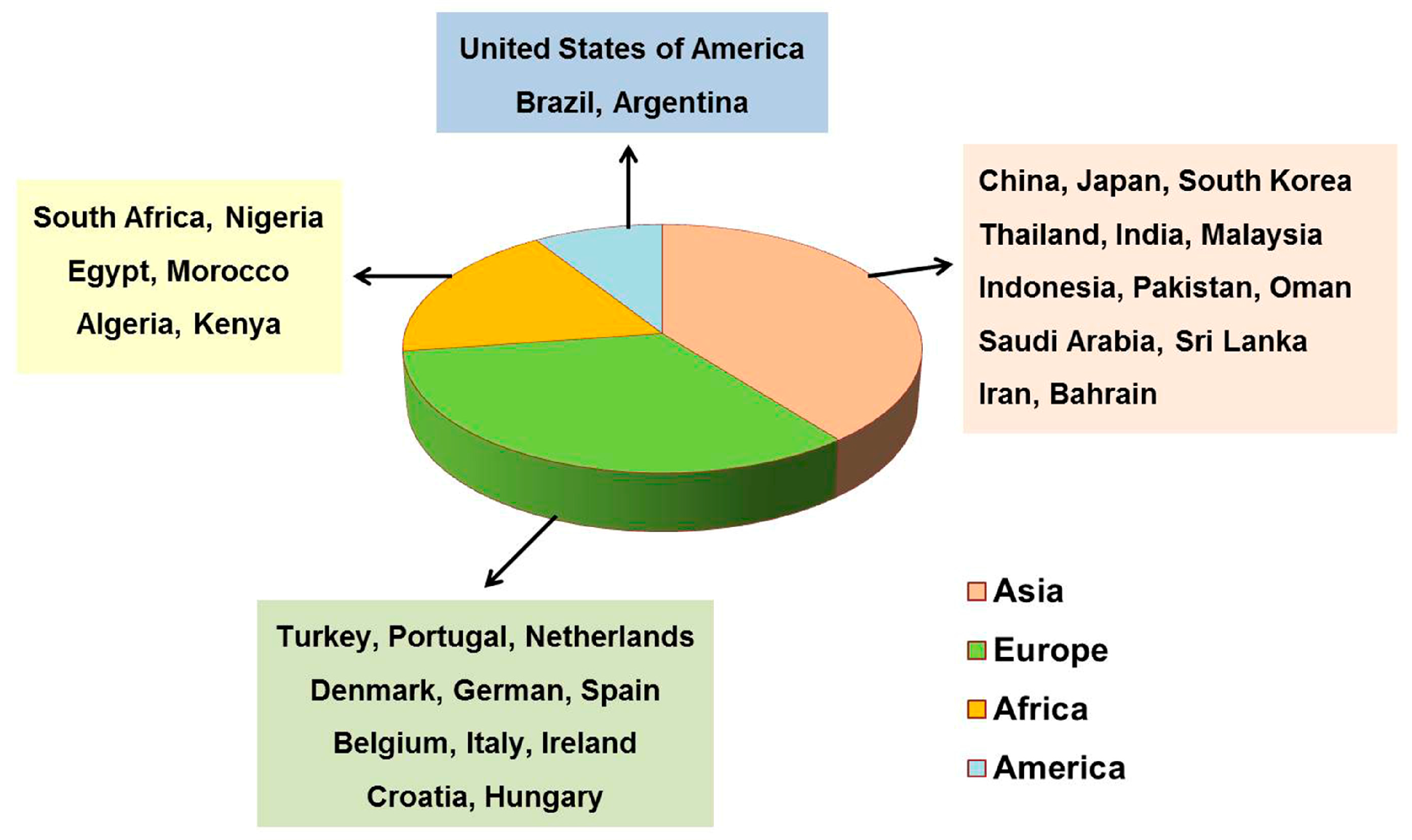{kind=link}
{kind=link}
{kind=link}
| Type of Mycotoxin | Source and Solubility | References | |
|---|---|---|---|
| Source | Solubility | ||
| Aflatoxin (AFB1, AFB2, AFG1, AFG2, AFM1) | Main source: Aspergillus Solubility: soluble in moderately polar organic solvents (e.g., chloroform, methanol, dimethysulfoxide), scarcely soluble in water (10–30 mg/mL) and insoluble in non-polar organic solvents | [16,17,18] | [18] |
| Ochratoxins (OTA, OTB) | Main source: Aspergillus and Penicillium Solubility: OTA: moderately soluble in polar organic solvents (e.g., chloroform, methanol) and dissolves in dilute aqueous sodium bicarbonate | [16,17,18] | [18] |
| Trichothecenes (Type A trichothecenes: (T-2, HT-2, NEO, DAS), Type B trichothecenes (DON, NIV, DOM-1, Fusarenone-X)) | Main source: Fusarium, Myrothecium, Stachybotrys, Trichoderma, Cephalosporium, Trichothecium and Verticimonosporium Solubility: Type A trichothecenes: highly soluble in ethyl acetate, acetone, chloroform, dichloromethane and diethyl ether; Type B trichothecenes: soluble in methanol, acetonitrile and ethanol | [18,19] | [18] |
| Zearalenones (ZEN, α-ZOL, β-ZOL, ZAN) | Main source: Fusarium Solubility: ZEN: soluble in water, slightly soluble in hexane and progressively more soluble in benzene, acetonitrile, dichloromethane, methanol, ethanol and acetone | [16,18] | [18] |
| Fumonisins (FB1, FB2, FB3) | Main source: Fusarium Solubility: soluble in water, acetonitrile–water or methanol, and insoluble in chloroform and hexane | [16,17,18] | [18] |
| Alternaria toxins (AOH, AME, TEA, TEN) | Main source: Alternaria Solubility: AME: insoluble in aqueous NaHCO3 or water, slightly soluble in ether, sparingly soluble in benzene AOH: insoluble in hexane, light petroleum, benzene, aqueous NaHCO3 and water, more soluble than AME in ethanol, methanol, acetone TEA: slightly soluble in water TEN: slightly soluble in benzene | [10,20] | [21] |
| Patulin | Main source: Penicillium Solubility: soluble in water, methanol, ethanol, acetone and ethyl or amyl acetate and less soluble in diethyl ether and benzene | [18] | [18] |
| Citrinin | Main source: Aspergillus, Penicillium and related species Solubility: practically insoluble in water, soluble in ethanol, dioxane, dilute alkali, acetone, benzene, and chloroform | [18] | [22] |
| Cyclopiazonic acid | Main source: Penicillium and other fungi species including Aspergillus Solubility: soluble in chloroform and dimethyl sulfoxide | [18] | [18] |
| Sterigmatocystin | Main source: Aspergillus Solubility: highly soluble in pyridine | [18] | [23] |
| Gliotoxin | Main source: a wide variety of widespread moulds Solubility: soluble in pyridine, dioxane, dimethylformamide, acetic acid, and chloroform, slightly soluble in benzene, acetone, carbonate trachloride, and ethyl alcohol | [18] | [24] |
| Tremorgenic mycotoxins (Penitrem A, Verruculogen) | Main source: a wide spectrum of fungi belonging to the genera Penicillium, Aspergillus, Claviceps and Acremonium Solubility: Penitrem A: soluble in acetone, methanol and dimethyl sulfoxide. Verruculogen: soluble in benzene, ethyl acetate, and acetone, slightly soluble in ethanol, and very soluble in chloroform | [18] | [25,26] |
| Penicillic acid | Main source: several species of Aspergillus and Penicillium Solubility: moderately soluble (2%) in cold water and in cold benzene, highly soluble in hot water, alcohol, ether, and chloroform, and insoluble in pentane-hexane | [18] | [27] |
| Chaetoglobosin A | Main source: Chaetomium globosum and some species of Penicillium Solubility: soluble in acetone, methanol | [28,29,30] | [28,29] |
| Beauvericin and other enniatins (BEA, ENN A, ENN A1, ENN B, ENN B1) | Main source: Fusarium Solubility: having low solubility in water | [31] | [31,32] |
| Moniliformin | Main source: Fusarium Solubility: soluble in water and polar solvents | [18] | [18] |
| Country/Region | Product (Group) | AFB1 μg kg−1 | Total AFs μg kg−1 | OTA μg kg−1 | Reference |
|---|---|---|---|---|---|
| Europe a | Herbal drugs | 2 | 4 | [44] | |
| United States | Some types of raw medicinal herb materials, as well as their powder and/or dry extract | 5 | 20 | [47,48] | |
| China | A total of nineteen different types of TCMs | 5 | 10 | [50] | |
| Britain | Herbal drugs | 2 | 4 | [45] | |
| Korea | Armeniacae Semen, Arecae Semen, Cassiae Semen, Crotonis Semen, Curcumae Radix, Dolichoris Semen, Glycyrrhizae Radix et Rhizoma, Nelumbinis Semen, Myristicae Semen, Persicae Semen, Pinelliae Tuber, Polygalae Radix, Carthami Flos, Thujae Semen, Trichosanthis Semen, Zizyphi Semen | 10 | 15 | [51] | |
| Indonesia | Coconut, spices and traditional drug medicines/herbs | 20 | [42] | ||
| Canada | Products containing ginseng or any substance derived from this source, Evening Primrose Oil, sugar cane, sugar beets, cottonseed | 5 | 20 | [49] | |
| Japan | Crude drug and preparations containing crude drugs as main ingredient (crude drug preparations) | 10 | [52] | ||
| Vietnam | Nutmeg | 5 | 10 | 30 | [53] |
| Ginger and turmeric | |||||
| Black and white pepper | |||||
| Licorice root used for herbal tea | 20 | ||||
| Licorice extract for beverage or to mix | 80 | ||||
| Germany | Any materials used in manufacture of medicinal products (including medicinal herbal products) | 2 | 4 | [46] | |
| Argentina | Herbs, herbal materials and herbal preparations used for herbal tea infusions | 5 | 20 | [46] | |
| Europe b | Nutmeg | 5 | 10 | 15 | [43] |
| Ginger | |||||
| Turmeric | |||||
| White and black pepper | |||||
| Dried figs | 6 | 10 | |||
| Liquorice root, ingredient for herbal infusion | 20 | ||||
| Liquorice extract, for use in food in particular beverages and confectionary | 80 |
| Mycotoxin | Detection | Sample | Extraction Solution | Extraction Method | Cleanup | LOD | LOQ | Reference |
|---|---|---|---|---|---|---|---|---|
| AFs | HPLC-FLD post-column Photochemical derivatization | Ginseng, ginger | Methanol-10 mM PBS containing 1% Tween 20 (80:20, v/v) | Shaking | IAC | 0.1 ng g−1 for AFB1 | 1 ng g−1 for AFB1 | [80] |
| AFs | HPLC-FLD post-column Photochemical derivatization | Ginger | Methanol-0.5% NaHCO3 solution (70:30, v/v) | Shaking | IAC | [81] | ||
| AFs | HPLC-FLD post-column Photochemical derivatization | Ginseng, ginger, kava kava, black cohosh, echinacea, valerian | Acetonitrile–water (84:16, v/v) | Shaking | IAC | [82] | ||
| AFs | HPLC-FLD post-column Photochemical derivatization | Glycyrrhiza uralensis | Methanol-water (80:20, v/v) | Sonicating | IAC | 0.015–0.06 μg kg−1 | 0.05–0.2 μg kg−1 | [83] |
| AFs | HPLC-FLD post-column Photochemical derivatization | Nelumbinis semen | Methanol-water (80:20, v/v) | Homogenizing | IAC | 0.03–0.10 μg kg−1 | 0.06–0.25 μg kg−1 | [84] |
| AFs | HPLC-FLD post-column Photochemical derivatization | Traditional Chinese medicine Yinpian | Methanol-water (70:30, v/v) | Sonicating | IAC | 0.12–0.44 pg | 0.31–1.09 pg | [85] |
| AFs | HPLC-FLD post-column Photochemical derivatization | Ginger and related products | Methanol-water (80:20, v/v) | Sonicating | IAC | 0.03–0.2 μg kg−1 | 0.1–0.6 μg kg−1 | [86] |
| AFs | HPLC-FLD post-column Photochemical derivatization | Nutmeg | Methanol-water (80:20, v/v) | Sonicating | IAC | 0.02–0.06 μg kg−1 | 0.06–0.2 μg kg−1 | [87] |
| AFs | HPLC-FLD post-column Photochemical derivatization | Chinese herbal pieces | Methanol-water (70:30, v/v) | Sonicating | IAC | [88] | ||
| AFs | HPLC-FLD post-column Photochemical derivatization | Coix seed | Methanol-water (80:20, v/v) | Sonicating | IAC | 0.01–0.11 μg kg−1 | 0.04–0.32 μg kg−1 | [89] |
| AFs | HPLC-FLD post-column Photochemical derivatization | Fructus Bruceae | Methanol-water (80:20, v/v) | Sonicating | IAC | 0.02–0.08 ng mL−1 | 0.05–0.20 ng mL−1 | [90] |
| AFs | HPLC-FLD post-column Photochemical derivatization | Shujin Huoxue pills | Methanol-water (70:30, v/v) | Sonicating | IAC | 0.26–1.04 pg | [91] | |
| AFs | HPLC-FLD post-column Photochemical derivatization | Sterculiae Lychnophorae | Methanol-water (70:30, v/v) | Sonicating | IAC | 0.0144–0.0528 μg L−1 | 0.0288–0.1056 μg L−1 | [92] |
| AFs | HPLC-FLD post-column Photochemical derivatization | Spices | Methanol-water (70:30, v/v) or Methanol-water (80:20, v/v) | Shaking | IAC | 0.01 ng g−1 for each AF | [93] | |
| AFs | HPLC-FLD post-column Photochemical derivatization | Red pepper, black pepper, turmeric and cinnamon | Methanol-water (80:20, v/v) | Homogenizing | IAC | 0.02–0.08 ng g−1 | [38] | |
| AFs | HPLC-FLD post-column Photochemical derivatization | Six kinds of medicinal herbs | Methanol-water (70:30, v/v) | Homogenizing | IAC | 0.04–0.2 μg kg−1 | 0.25–1.0 μg kg−1 | [94] |
| AFs | HPLC-FLD post-column bromination derivatization | Twelve kinds of spices | Methanol-water (80:20, v/v) | IAC | 1 μg kg−1 | [95] | ||
| AFs | HPLC-FLD post-column bromination derivatization | Thirty seven TCMs | Methanol-water (70:30, v/v) | Sonicating | IAC | 0.06–0.20 μg kg−1 | [96] | |
| AFs | HPLC-FLD post-column bromination derivatization | Thirty three species of medicinal herbs and 11 kinds of patent medicines | Methanol-water (70:30, v/v) | Sonicating | IAC | [97] | ||
| AFs | HPLC-FLD post-column bromination derivatization | Herbal plants | Methanol-water | IAC | 0.03–0.3 μg kg−1 | 0.05–0.7 μg kg−1 | [98] | |
| AFs | HPLC-FLD post-column iodine derivatization | Nighteen TCMs | Methanol-water (70:30, v/v) | Sonicating | IAC | 0.22–0.75 μg kg−1 | [99] | |
| AFs | HPLC-FLD post-column iodine derivatization | Bulbus Fritillariae Thunbergii, Fructus Schisandrae Chinensis, Fructus Crataegi, Fructus Mume | Methanol-water (70:30, v/v) | Sonicating | IAC | 0.06 μg kg−1 | 0.3 μg kg−1 | [100] |
| AFs | HPLC-FLD post-column iodine derivatization | Sixteen plant species | Methanol-water (80:20, v/v) | Homogenizing | IAC | 0.5 μg kg−1 for AFB1 | [101] | |
| AFs | HPLC-FLD post-column iodine derivatization | Citri Reticulatae Pericarpium | Methanol-water (70:30, v/v) | Shaking | IAC | 0.19–0.24 μg kg−1 | [102] | |
| AFs | HPLC-FLD post-column iodine derivatization | Proprietary Chinese medicines | Methanol | Sonicating | GO-based dSPE | 0.020–0.041 ng mL−1 | 0.061–0.125 ng mL−1 | [103] |
| AFs | HPLC-FLD post-column derivatization with electrochemically generated bromine | Twenty-eight samples of herbal medicinal products | Methanol-water (80:20, v/v) | Homogenizing | IAC | 0.04 ng g−1 | [104] | |
| AFs | HPLC-FLD post-column derivatization with electrochemically generated bromine | Five kinds of medicinal herbs | Methanol-water (70:30, v/v) | Sonicating | IAC | 0.05–0.1 ng g−1 | [105] | |
| AFs | HPLC-FLD post-column derivatization with electrochemically generated bromine | One hundred and three samples of different kinds of spices and herbs | Methanol-water (80:20, v/v) | Shaking | IAC | 0.2–0.5 μg kg−1 | 0.6–1.5 μg kg−1 | [67] |
| AFs | HPLC-FLD post-column derivatization with electrochemically generated bromine | Dried figs | Methanol-water (80:20, v/v) | IAC | 0.1 ng g−1 | [106] | ||
| AFs | HPLC-FLD post-column derivatization with electrochemically generated bromine | Citri Reticulatae Pericarpium | Methanol-water (70:30, v/v) | Shaking | IAC | 0.10–0.18 μg kg−1 | [102] | |
| AFs | HPLC-FLD post-column derivatization with electrochemically generated bromine | One hundred and eighty five functional food and 56 herbal medicines | Methanol-water (70:30, v/v) | Shaking | IAC | 0.07–0.32 ng g−1 | 0.21–0.96 ng g−1 | [66] |
| AFs | HPLC-FLD pre-column derivatization with TFA | White pepper | Chloroform-water (100:10, v/v) | Silica cartridge and C18 cartridge | 0.006–0.009 μg L−1 | [75] | ||
| AFs | HPLC-FLD pre-column derivatization with TFA | Ninety one spice samples | Methanol-water (80:20, v/v) | Shaking | IAC | 0.1–0.2 μg kg−1 | [76] | |
| AFs | HPLC-FLD pre-column derivatization with TFA | Twenty three commercial traditional herbal medicines | Methanol-water (70:30, v/v) | Shaking | IAC | 0.01 μg kg−1 | [107] | |
| AFs | HPLC-FLD pre-column derivatization with TFA | Ginseng roots | Methanol-water (80:20, v/v) | Shaking | IAC | 0.1 ng g−1 for AFB1 | [108] | |
| AFs | HPLC-FLD pre-column derivatization with TFA | Eighty eight spices and processed spice products | Methanol-water (70:30, v/v) | Shaking | IAC | 0.01–0.15 μg kg−1 | 0.03–0.45 μg kg−1 | [109] |
| AFs | HPLC-FLD pre-column derivatization with TFA | Eight kinds of medicinal herbs | Methanol-water (80:20, v/v) | Blending | IAC | 0.02–0.09 ppb | [110] | |
| AFs | HPLC-FLD pre-column derivatization with TFA | Pu-erh tea | Acetonitrile-water (84:16, v/v) | Shaking | SPE | [111] | ||
| AFs | HPLC-FLD pre-column derivatization with TFA | One hundred and eighty five functional food and 56 herbal medicines | Methanol-water (70:30, v/v) | Shaking | IAC | 0.32–2.28 ng g−1 | 0.95–6.83 ng g−1 | [66] |
| AFs | HPLC-FLD pre-column derivatization with TFA | Black, White and Green Peppers | Acetonitrile-water (60:40, v/v) | Blending | IAC | 0.01–0.5 ng mL−1 | 0.05–2.5 ng mL−1 | [112] |
| AFs | HPLC-FLD | Maytenus ilicifolia | Methanol-water (70:30, v/v) | Sonicating | IAC | 0.1–3.5 ng g−1 | [68] | |
| AFs | UPLC-FLD | Ginger and related products | Methanol-water (80:20, v/v) | Sonicating | IAC | 0.005–0.2 μg kg−1 | 0.0125–0.5 μg kg−1 | [113] |
| OTA | HPLC-FLD | Fifty-seven traditional Chinese medicinal plants | Methanol-water (80:20, v/v) | Sonicating | IAC | 0.3 μg kg−1 | 0.8 μg kg−1 | [114] |
| OTA | UPLC-FLD | Ginger | Acetonitrile–water (60:40, v/v) | Sonicating | MIP-SPE | 0.09 ng mL−1 | 0.30 ng mL−1 | [115] |
| OTA | UPLC-FLD | Ginger powder | Acetonitrile–water (60:40, v/v) | Sonicating | AAC | 0.5 μg kg−1 | 1.5 μg kg−1 | [116] |
| OTA | HPLC-FLD | Ginseng, Ginger | Methanol-1% NaHCO3 solution (70:30, v/v) | Shaking | IAC | 0.1 ng g−1 | 1 ng g−1 | [80] |
| OTA | HPLC-FLD | Ginger | Methanol-0.5% NaHCO3 solution (70:30, v/v) | Shaking | IAC | [81] | ||
| OTA | HPLC-FLD | Glycyrrhiza uralensis | Methanol-water (80:20, v/v) | Sonicating | IAC | 0.25 μg kg−1 | 0.75 μg kg−1 | [83] |
| OTA | HPLC-FLD | Ginger and related products | Methanol-water (80:20, v/v) | Sonicating | IAC | 0.3 μg kg−1 | 0.9 μg kg−1 | [86] |
| OTA | HPLC-FLD | Nutmeg | Methanol-water (80:20, v/v) | Sonicating | IAC | 0.25 μg kg−1 | 0.8 μg kg−1 | [87] |
| OTA | HPLC-FLD | Spices | Acetonitrile–water (60:40, v/v) | Shaking | IAC | 0.10 ng g−1 | [93] | |
| OTA | HPLC-FLD | Black pepper, white pepper and spice mixture samples | 1M phosphoric acid-chloroform (10:100, v/v) | Shaking | IAC | 0.2 μg kg−1 | [76] | |
| OTA | UPLC-FLD | Ginger and related products | Methanol-water (80:20, v/v) | Sonicating | IAC | 0.1 μg kg−1 | 0.3 μg kg−1 | [113] |
| CIT | HPLC-FLD | Red mold rice | Ethanol-water (75:25, v/v) | Shaking | [72] | |||
| CIT | HPLC-FLD | Red yeast rice, medicinal plants and their related products | Methanol-water (70:30, v/v) | Vortexing | IAC | 0.8 μg kg−1 | 2 μg kg−1 | [117] |
| CIT | HPLC-FLD | Red fermented rice | Methanol-water (80:20, v/v) | Sonicating | 0.0005 μg mL−1 | 0.001 μg mL−1 | [118] | |
| DON, NIV | HPLC-UV | Thirty samples of TCMs | Acetonitrile-water (80:20, v/v) | Homogenizing | SPE | 63 μg kg−1 for DON and 50.0 μg kg−1 for NIV | 125.0 μg kg−1 for DON and 100.0 μg kg−1 for NIV | [71] |
| ZEN | HPLC-FLD | One hundred and seven samples of Chinese medicinal herbs | Methanol-water (80:20, v/v) | Homogenizing | IAC | 9.5 μg kg−1 | [119] | |
| ZEN | HPLC-ELSD | Barley | Methanol | Blending | QuEChERS | 1.56 ng g−1 | [120] | |
| ZEN, α-ZOL, β-ZOL | HPLC-FLD | Coix seed | Methanol-water (80:20, v/v) | Sonicating | IAC | 11.7–50.2 μg kg−1 | 29.3–125.5 μg kg−1 | [89] |
| FB1, FB2, FB3 | HPLC-FLD pre-column derivatization with o-phthaldialdehyde | Sixteen plant species | Methanol | Homogenizing | SPE | 5 μg kg−1 for FB1 | [101] |
| Mycotoxin | Sample | Extraction Solution | Extraction Method | Cleanup | LOD | LOQ | Reference |
|---|---|---|---|---|---|---|---|
| AFs | Zizyphi Fructus | SFE | Without purification | 0.17–0.32 ng g−1 | 0.56–1.05 ng g−1 | [121] | |
| AFs | One hundred and seventy four samples from 50 medicinal herb species | Methanol-water (70:30, v/v) | Sonicating | IAC | 0.135–0.883 μg kg−1 | [122] | |
| AFs | Lotus seeds | Methanol-water (80:20, v/v) | Blending | IAC | 0.003–0.007 μg kg−1 | 0.010–0.020 μg kg−1 | [123] |
| AFs | Mucuna pruriens, Delphinium denudatum, and Portulaca oleraceae | Methanol-water (70:30, v/v) | Sonicating | IAC | 0.28–1.10 μg kg−1 | 0.79–3.34 μg kg−1 | [124] |
| AFs | Armeniacae Semen Amarum | Acetonitrile-water (84:16, v/v) | Vortexing | Without purification | 5.200–6.300 ng L−1 | 10.40–12.60 ng L−1 | [125] |
| AFs | Walnut kernel | Methanol-water (70:30, v/v) | Sonicating | Self-made amino -function nanometre Fe3O4 magnetic polymer SPE | 0.004–0.013 μg kg−1 | 0.012–0.042 μg kg−1 | [126] |
| AFs | Twenty two TCMs matrix types | Methanol-water (70:30, v/v), Methanol-water (75:25, v/v), Methanol-water (85:15, v/v) | Sonicating, shaking, and homogenizing | C18-SPE | 0.008–0.022 μg kg−1 | 0.011–0.029 μg kg−1 | [79] |
| AFs, OTA | Glycyrrhiza uralensis | Methanol-water (80:20, v/v) | Sonicating | IAC | 0.003–0.007 μg kg−1 | 0.010–0.020 μg kg−1 | [127] |
| AFB1, OTA | Licorice roots, fritillary bulbs | Methanol-water (85:15, v/v) | Sonicating | C18-SPE | 0.012 μg kg−1 for AFB1, 0.024 μg kg−1 for OTA | 0.035 μg kg−1 for AFB1, 0.095 μg kg−1 for OTA | [77] |
| AFs, OTA, ST | Two-hundred and forty-four samples of 25 types of widely used TCMs | Acetonitrile–water (84:16, v/v) | Soaking and shaking | 0.1–25.0 ng L−1 | [128] | ||
| AFB1, AFB2, AFG1, AFG2, AFM1, AFM2 | Thirty TCMs | Acetonitrile–water (84:16, v/v) | Homogenizing | Home-made mixed cartridge | 0.07–0.26 μg kg−1 | 0.10–0.73 μg kg−1 | [129] |
| AFs, PAT | Chinese patent medicines | Acetonitrile–water (84:16, v/v) | Vortexing | Mycosep 228 Aflapat mutifutional column | 0.1–1 μg kg−1 | [130] | |
| OTA | Five types of TCMs | Acetonitrile–water (60:40, v/v) | Soaking | AAC | 0.5–0.8 μg kg−1 | 1.5–2.5 μg kg−1 | [131] |
| PAT | Fructus crataegi, fructus mume, pericarpium citri reticulatae, fructus aurantii | Pectinase enzymolysis and acetonitrile-water (60:20, v/v) extraction | Blending | dSPE and Mycrosep228AlaPat | 0.3–0.5 μg kg−1 | [132] | |
| OTA, PAT | Seventy nine samples of various spices and herbs | Methanol-water (3:1, v/v) for FBs, acetonitrile–water (60:40, v/v) for OTA | Homogenizing for FBs, soaking for OTA | SAX cartridge for FBs, and IAC for OTA | 0.1 ng g−1 for OTA, 0.5–1.0 ng g−1 for FBs | [133] | |
| ZEN, α-ZOL | Twenty five TCMs | Methanol-water (80:20, v/v) | Shaking | IAC | 0.6 μg kg−1 | 1.2 μg kg−1 | [134] |
| ZEN, α-ZOL, β-ZOL, ZAN, α-ZAL, β-ZAL | Thirty-three commercially available dried TCMs | Acetonitrile–water (60:40, v/v) | Soaking and Homogenizing | Home-made cleanup cartridge | 0.06–0.79 ng mL−1 | 0.13–0.99 ng mL−1 | [70] |
| T2, HT-2, NEO, and DAS | Coix seed | Acetonitrile–water (84:16, v/v) | Sonicating | Magnetic SPE | 0.3–1.5 μg kg−1 | [135] | |
| FB1, FB2 and FB3 | Four types of dried TCMs | Acetonitrile–water (50:50, v/v) | Soaking and homogenizing | MultiSep 211 Fum columns | 0.05–0.10 ng mL−1 | 0.08–0.16 ng mL−1 | [69] |
| CIT | Twenty seven TCMs | Methanol-water (70:30, v/v) | Shaking | IAC | 1.0 μg kg−1 | 2.5 μg kg−1 | [136] |
| ENNs and BEA | Sixty types of dried Chinese medicinal herbs | Methanol | Shaking | Without purification | 0.8–1.2 μg kg−1 | 2.5–3.7 μg kg−1 | [32] |
| 23 mycotoxins | Botanical food supplements | Ethyl acetate-formic acid (95:5, v/v) | Shaking | Oasis HLBTM SPE cartridges | 0.3–30 ng g−1 | 1–100 ng g−1 | [12] |
| 22 mycotoxins | Raw tea and herbal infusion materials | Ethyl acetate-formic acid (99:1, v/v) | Shaking | NH2-SPE and C18-SPE column | 2.1–122 μg kg−1 | 4.1–243 μg kg−1 | [74] |
| 35 mycotoxins | Four types of dried TCMs | Acetonitrile–water (84:16, v/v) | ASE | Homemade Cleanup Cartridges | 0.01–1.56 μg kg−1 | 0.11–1.86 μg kg−1 | [13] |
| 15 mycotoxins | Milk thistle samples (seeds and extract) | 30 mM NaH2PO4 buffer pH 7.1 and 5% formic acid in acetonitrile | Vortexing | QuEChERS | 0.45–459 μg kg−1 | 1.5–1530 μg kg−1 | [137] |
| 17 mycotoxins | Puerariae lobatae radix | Acetonitrile–water (90:10, v/v) | Sonicating | PuriToxSR TC-M160 MultiPurification Column | 0.00203–1.06 μg kg−1 | 0.0488–4.97 μg kg−1 | [78] |
| 10 mycotoxins | Panax notoginseng | Acetonitrile | Sonicating | HLB multifunction cleanup column | 0.043–2.9 μg kg−1 | 0.15–8.6 μg kg−1 | [138] |
| 11 mycotoxins | Morinda officinalis | Methanol-water (80:20, v/v) containing 0.1% formic acid | Vortexing | Without purification | 0.02–4.00 ng mL−1 | 0.06–10 ng mL−1 | [139] |
| 8 mycotoxins | Angelica sinensis | PBS and 5% formic acid in acetonitrile | vortexing | QuEChERS | 0.005–0.125 μg kg−1 | 0.0625–0.25 μg kg−1 | [140] |
| 8 mycotoxins | Chinese yam and related products | Methanol-water-formic acid (79:20:1, v/v/v) | Sonicating | Without purification | 0.02–0.15 ng mL−1 | 0.06–0.50 ng mL−1 | [141] |
| 21 mycotoxins | Radix Paeoniae Alba | PBS and 5% formic acid in acetonitrile | Vortexing | Modified QuEChERS | 0.03–5.36 μg kg−1 | 0.20–22.50 μg kg−1 | [142] |
| 11 mycotoxins | Areca catechu | Methanol-water (80:20, v/v) | Soaking and vortexing | Without purification | 0.1–20 μg kg−1 | 0.25–50 μg kg−1 | [143] |
| 11 mycotoxins | Malt | Acetonitrile-water-acetic acid (80:19:1, v/v/v) | Sonicating | Without purification | 0.01–5.85 ng mL−1 | 0.03–17.5 ng mL−1 | [144] |
| 11 mycotoxins | Three types of ground herbs | Acetonitrile-water (50:50, v/v) | Shaking | A buffered QuEChERS SPE | 0.5–4.0 μg kg−1 | 1.5–12 μg kg−1 | [145] |
| 11 mycotoxins | Alpinia oxyphylla | Acetonitrile -water-acetic acid (79:20:1, v/v/v). | Sonicating | Without purification | 0.03–6.00 μg kg−1 | 0.10–20.0 μg kg−1 | [146] |
| ZEN and type A trichothecenes | Salviae Miltiorrhizae Radix et Rhizoma | Acetonitrile-water (84:16, v/v) | Soaking and sonicating | Fe3O4/MWCNT | 0.45–1.80 μg kg−1 | 1.20–4.80 μg kg−1 | [147] |
| Sample | Mycotoxin | Reference |
|---|---|---|
| A total of 152 samples, belonging to 56 species of medicinal herbs | AFB1, AFB2, AFG1, AFG2, ZEN, T-2, NEO, DON | [167] |
| Ninety-one samples of medicinal herbs, composed by 65 different plant species | AFB1, AFB2, AFG1, AFG2, OTA | [168] |
| A total of 30 raw materials comprising five samples of each medicinal | AFB1 | [169] |
| A total of 68 powdered samples | AFB1, AFB2, AFG1, AFG2, CIT, ST | [170] |
| A total of 25 sun dried freshly stored fruit samples of and 25 powdered of Emblica officinalis, Terminalia bellirica, Terminalia chebula | AFB1, AFB2, AFG1, AFG2 | [171] |
| Eighty samples consisting of 20 each of four medicinal plants | AFB1, AFB2, AFG1, AFG2 | [172] |
| Two random samples of two different plant materials | AFB1, AFG1, CIT, Griseofulvin, OTA, ST | [173] |
| Thirty different samples of medicinal plants | AFB1, AFB2, AFG1, AFG2, OTA | [174] |
| Ten sun dried one year stored crude drug samples | AFB1, AFB2, AFG1, AFG2 | [175] |
| A total of 210 samples randomly bought from traditional medical practitioners | AFB1, AFB2, AFG1, AFG2 | [176] |
| A total of 63 samples which includes 38 different types of commonly used herbs, herbal products, spices, and food materials | AFB1, AFB2, AFG1, AFG2 | [177] |
| Eighteen samples of 6 different types | AFB1 | [178] |
| Sample | Mycotoxin | LOD | Reference |
|---|---|---|---|
| Red scaled, red and black pepper | AFs and AFB1 | 0.25 μg kg−1 for AFs, 1.0 μg kg−1 for AFB1 | [199] |
| Black pepper, coriander, ginger and turmeric | OTA | [193] | |
| P. ginseng, P. quinquefolius | ZEN | [200] | |
| Eighty-four medicinal and/or aromatic herb samples | OTA, FBs, AFs, ZEN, T-2, DON, CIT | 0.025 μg kg−1 for OTA, 83 μg kg−1 for FBs, 1.4 μg kg−1 for AFs, 0.14 μg kg−1 ZEN, 0.28 μg kg−1 for T-2, 14.80 μg kg−1 for DON and 16.5 μg kg−1 for CIT | [195] |
| A total of 700 herbal medicine samples (70 types and10 samples of each type) | AFB1 | 0.05 ng mL−1 | [201] |
| Ninety three organic spice and 37 organic herb samples | AFB1 | [196] | |
| A total of 36 samples of spices | AFB1 | [39] | |
| Red chilli, black pepper, turmeric, coriander, cumin, fennel, caraway, fenugreek, and dry ginger | AFs, OTA, CIT | 4 ng g−1 for AFs, 2 ng g−1 for OTA, and 15 ng g−1 for CTN | [197] |
| Lichens | AOL, AFB1, DON, DAS, ZEN, Mycophenolic acid (MPA), OTA, PR toxin (PR), ST, T2, FB1, Cyclopiazonic acid (CPA), CIT, Emodin (EMO), EA, Roridin A (ROA) | 2 (AFB1, T2, EA), 4 (ST), 8 (OTA, ROA), 20 (MPA, CIT, AOL, ZEN), 40 (DON, EMO), 50 (FB1) and 100 (DAS, CPA, PR) ng g−1 | [202] |
| Lotus seeds | AFB1 | 0.128 μg L−1 | [194] |
| Cassava flour | AFs | [203] | |
| Garlic | FB1, FB2 | 0.17 ppm | [198] |
| Ginger, galangal, garlic, elephant garlic | AFB1 | [204] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Dou, X.-W.; Zhang, C.; Logrieco, A.F.; Yang, M.-H. A Review of Current Methods for Analysis of Mycotoxins in Herbal Medicines. Toxins 2018, 10, 65. https://doi.org/10.3390/toxins10020065
Zhang L, Dou X-W, Zhang C, Logrieco AF, Yang M-H. A Review of Current Methods for Analysis of Mycotoxins in Herbal Medicines. Toxins. 2018; 10(2):65. https://doi.org/10.3390/toxins10020065
Chicago/Turabian StyleZhang, Lei, Xiao-Wen Dou, Cheng Zhang, Antonio F. Logrieco, and Mei-Hua Yang. 2018. "A Review of Current Methods for Analysis of Mycotoxins in Herbal Medicines" Toxins 10, no. 2: 65. https://doi.org/10.3390/toxins10020065