Botulinum Toxin Type A—A Modulator of Spinal Neuron–Glia Interactions under Neuropathic Pain Conditions

Abstract
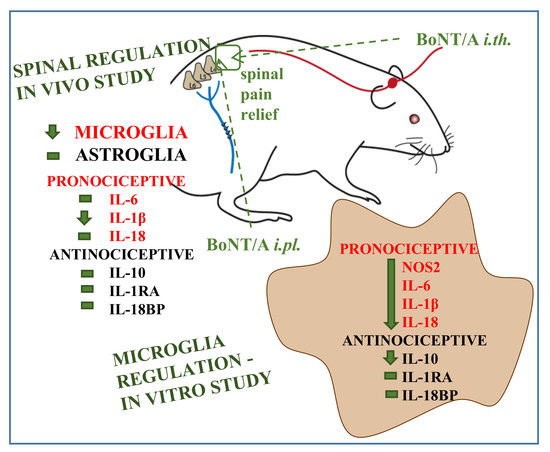
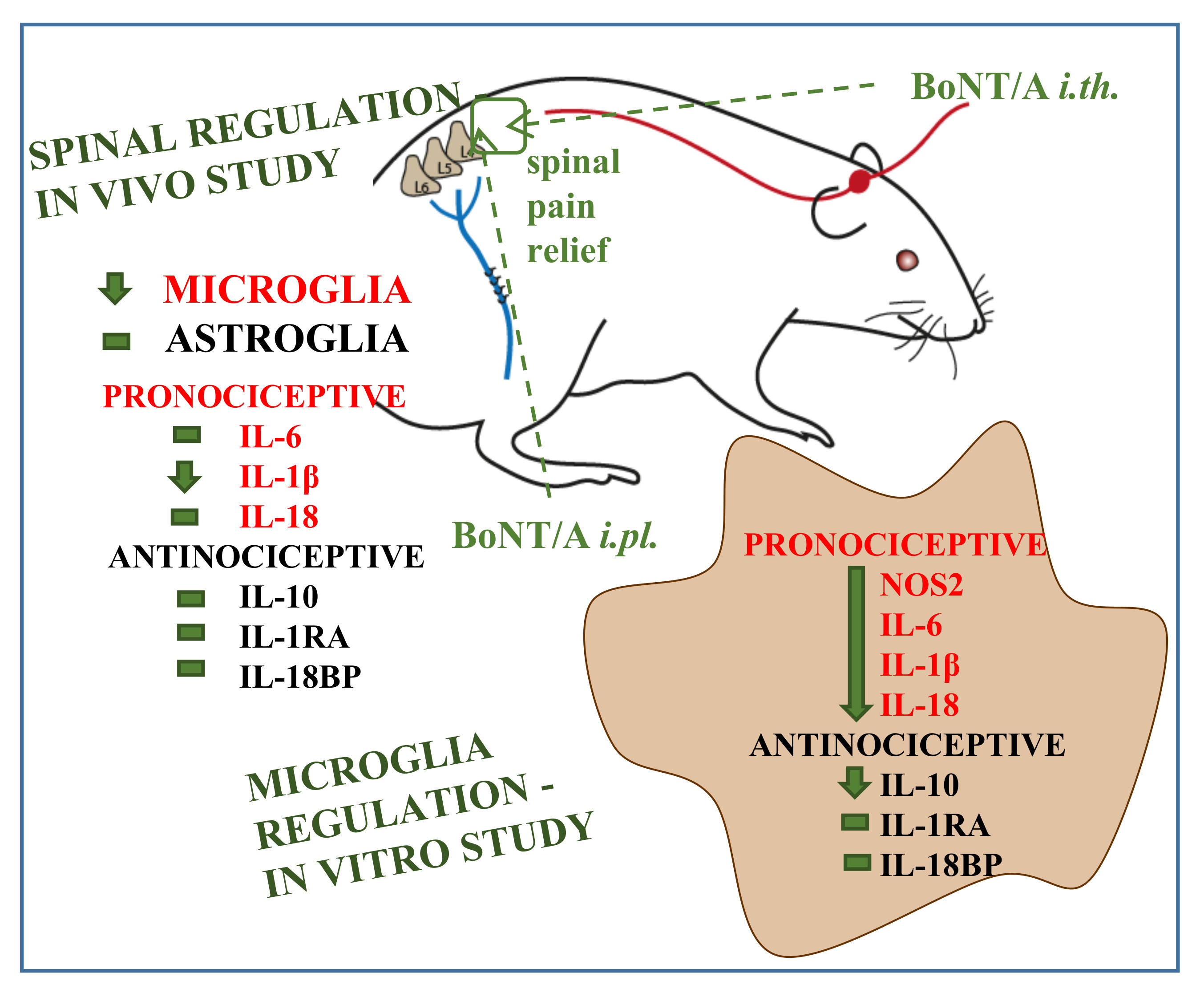
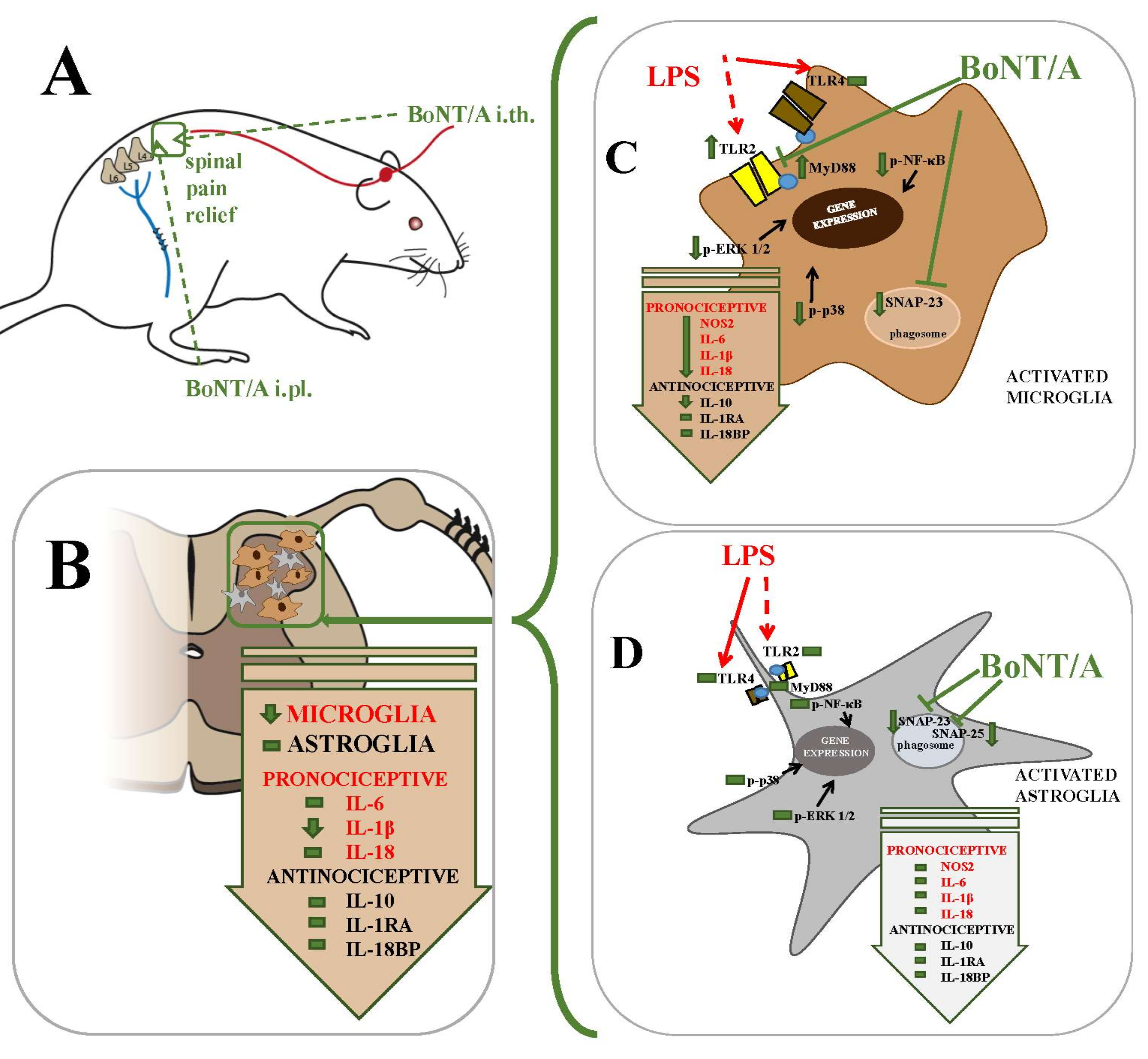
:
{kind=link}
{kind=link}
1. The Therapeutic Effect of Bont/A—Powerful Analgesic Agent against Neuropathic Pain
2. Mechanism-Based Evidence for the Analgesic Actions of Bont/A
3. Far Beyond the Neurons—The Role of Glial Cells in Bont/A-Induced Analgesia
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Austin, P.J.; Moalem-Taylor, G. The neuro-immune balance in neuropathic pain: Involvement of inflammatory immune cells, immune-like glial cells and cytokines. J. Neuroimmunol. 2010, 229, 26–50. [Google Scholar] [CrossRef] [PubMed]
- Mika, J.; Zychowska, M.; Popiolek-Barczyk, K.; Rojewska, E.; Przewlocka, B. Importance of glial activation in neuropathic pain. Eur. J. Pharmacol. 2013, 716, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Watkins, L.R.; Milligan, E.D.; Maier, S.F. Glial activation: A driving force for pathological pain. Trends Neurosci. 2001, 24, 450–455. [Google Scholar] [CrossRef]
- Watkins, L.R.; Maier, S.F. GLIA: A novel drug discovery target for clinical pain. Nat. Rev. Drug Discov. 2003, 2, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Gibson, W.; Wand, B.M.; O’Connell, N.E. Transcutaneous electrical nerve stimulation (TENS) for neuropathic pain in adults. Cochrane Database Syst. Rev. 2017, 2017, CD011976. [Google Scholar] [CrossRef] [PubMed]
- Hatch, M.N.; Cushing, T.R.; Carlson, G.D.; Chang, E.Y. Neuropathic pain and SCI: Identification and treatment strategies in the 21st century. J. Neurol. Sci. 2017, 384, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Çakici, N.; Fakkel, T.M.; van Neck, J.W.; Verhagen, A.P.; Coert, J.H. Systematic review of treatments for diabetic peripheral neuropathy. Diabet. Med. 2016, 33, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Tamburin, S.; Lacerenza, M.R.; Castelnuovo, G.; Agostini, M.; Paolucci, S.; Bartolo, M.; Bonazza, S.; Federico, A.; Formaglio, F.; Giusti, E.M.; et al. Italian Consensus Conference on Pain in Neurorehabilitation (ICCPN) Pharmacological and non-pharmacological strategies in the integrated treatment of pain in neurorehabilitation. Evidence and recommendations from the Italian Consensus Conference on Pain in Neurorehabilitation. Eur. J. Phys. Rehabil. Med. 2016, 52, 741–752. [Google Scholar] [PubMed]
- Boldt, I.; Eriks-Hoogland, I.; Brinkhof, M.W.G.; de Bie, R.; Joggi, D.; von Elm, E. Non-pharmacological interventions for chronic pain in people with spinal cord injury. Cochrane Database Syst. Rev. 2014, 11, CD009177. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.A. Clostridial toxins as therapeutic agents: Benefits of nature’s most toxic proteins. Annu. Rev. Microbiol. 1999, 53, 551–575. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Álvarez, F.; Hernando de la Bárcena, I.; Marzo-Sola, M.E. Botulinum toxin in trigeminal neuralgia. Med. Clin. (Barc) 2017, 148, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Alviar, M.J.M.; Hale, T.; Dungca, M. Pharmacologic interventions for treating phantom limb pain. Cochrane Database Syst. Rev. 2016, 10, CD006380. [Google Scholar] [CrossRef] [PubMed]
- Bruno, V.A.; Fox, S.H.; Mancini, D.; Miyasaki, J.M. Botulinum Toxin Use in Refractory Pain and Other Symptoms in Parkinsonism. Can. J. Neurol. Sci. 2016, 43, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.-J.; Song, T.-J.; Chu, M.K. Treatment Update of Chronic Migraine. Curr. Pain Headache Rep. 2017, 21, 26. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, M.L.; García-Moreno, H.; Arias, J.A.; Pareja, J.A. Botulinum neurotoxin type-A for the treatment of atypical odontalgia. Pain Med. 2016, 17, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Kleen, J.K.; Levin, M. Injection Therapy for Headache and Facial Pain. Oral Maxillofac. Surg. Clin. N. Am. 2016, 28, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Lunde, H.M.B.; Torkildsen, Ø.; Bø, L.; Bertelsen, A.K. Botulinum Toxin as Monotherapy in Symptomatic Trigeminal Neuralgia. Headache 2016, 56, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Song, H.; Dong, Y.; Ye, Y.; Li, J. Intra-articular injections of botulinum toxin a for refractory joint pain: A systematic review and meta-analysis. Clin. Rehabil. 2017, 31, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, C.; Molgó, J. Botulinal neurotoxins: Revival of an old killer. Curr. Opin. Pharmacol. 2005, 5, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Welch, M.J.; Purkiss, J.R.; Foster, K. A Sensitivity of embryonic rat dorsal root ganglia neurons to Clostridium botulinum neurotoxins. Toxicon 2000, 38, 245–258. [Google Scholar] [CrossRef]
- Durham, P.L.; Cady, R.; Cady, R.; Blumenfeld, A.J. Regulation of Calcitonin Gene-Related Peptide Secretion from Trigeminal Nerve Cells by Botulinum Toxin Type A: Implications for Migraine Therapy. Headache 2004, 44, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Krämer, H.H.; Angerer, C.; Erbguth, F.; Schmelz, M.; Birklein, F. Botulinum toxin A reduces neurogenic flare but has almost no effect on pain and hyperalgesia in human skin. J. Neurol. 2003, 250, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Burstein, R.; Zhang, X.C.; Levy, D.; Aoki, K.R.; Brin, M.F. Selective inhibition of meningeal nociceptors by botulinum neurotoxin type A: Therapeutic implications for migraine and other pains. Cephalalgia 2014, 34, 853–869. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, M.A.; Swope, D.M.; Grimes, D. Diffusion of botulinum toxins. Tremor Other Hyperkinet. Mov. 2012, 2, 319–322. [Google Scholar] [CrossRef]
- Klein, A.W. The therapeutic potential of botulinum toxin. Dermatol. Surg. 2004, 30, 452–455. [Google Scholar] [PubMed]
- Wheeler, A.H. Botulinum toxin A, adjunctive therapy for refractory headaches associated with pericranial muscle tension. Headache 1998, 38, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Binder, W.J.; Brin, M.F.; Blitzer, A.; Schoenrock, L.D.; Pogoda, J.M. Botulinum toxin type A (BOTOX) for treatment of migraine headaches: An open-label study. Otolaryngol. Head Neck Surg. 2000, 123, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A. Botulinum toxin type a for chronic migraine. Curr. Neurol. Neurosci. Rep. 2010, 10, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.Y.; Sheu, J.J.; Yu, J.M.; Chen, W.T.; Tseng, I.J.; Chang, H.H.; Hu, C.J. Botulinum toxin for diabetic neuropathic pain: A randomized double-blind crossover trial. Neurology 2009, 72, 1473–1478. [Google Scholar] [CrossRef] [PubMed]
- Ranoux, D.; Attal, N.; Morain, F.; Bouhassira, D. Botulinum toxin type A induces direct analgesic effects in chronic neuropathic pain. Ann. Neurol. 2008, 64, 274–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, Y.; Matsuka, Y.; Spigelman, I.; Ishihara, Y.; Yamamoto, Y.; Sonoyama, W.; Kuboki, T.; Oguma, K. Botulinum toxin type a (150 kDa) decreases exaggerated neurotransmitter release from trigeminal ganglion neurons and relieves neuropathy behaviors induced by infraorbital nerve constriction. Neuroscience 2009, 159, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Arbizu, R.A.; Rodriguez, L. Use of Clostridium botulinum toxin in gastrointestinal motility disorders in children. World J. Gastrointest. Endosc. 2015, 7, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Intiso, D.; Basciani, M.; Santamato, A.; Intiso, M.; Di Rienzo, F. Botulinum toxin type a for the treatment of neuropathic pain in neuro-rehabilitation. Toxins 2015, 7, 2454–2480. [Google Scholar] [CrossRef] [PubMed]
- Bach-Rojecky, L.; Lacković, Z. Antinociceptive effect of botulinum toxin type a in rat model of carrageenan and capsaicin induced pain. Croat. Med. J. 2005, 46, 201–208. [Google Scholar] [PubMed]
- Luvisetto, S.; Rossetto, O.; Montecucco, C.; Pavone, F. Toxicity of botulinum neurotoxins in central nervous system of mice. Toxicon 2003, 41, 475–481. [Google Scholar] [CrossRef]
- Luvisetto, S.; Marinelli, S.; Cobianchi, S.; Pavone, F. Anti-allodynic efficacy of botulinum neurotoxin A in a model of neuropathic pain. Neuroscience 2007, 145, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, S.; Luvisetto, S.; Cobianchi, S.; Makuch, W.; Obara, I.; Mezzaroma, E.; Caruso, M.; Straface, E.; Przewlocka, B.; Pavone, F. Botulinum neurotoxin type A counteracts neuropathic pain and facilitates functional recovery after peripheral nerve injury in animal models. Neuroscience 2010, 171, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Mika, J.; Rojewska, E.; Makuch, W.; Korostynski, M.; Luvisetto, S.; Marinelli, S.; Pavone, F.; Przewlocka, B. The effect of botulinum neurotoxin A on sciatic nerve injury-induced neuroimmunological changes in rat dorsal root ganglia and spinal cord. Neuroscience 2011, 175, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Zychowska, M.; Rojewska, E.; Makuch, W.; Luvisetto, S.; Pavone, F.; Marinelli, S.; Przewlocka, B.; Mika, J. Participation of pro- and anti-nociceptive interleukins in botulinum toxin A-induced analgesia in a rat model of neuropathic pain. Eur. J. Pharmacol. 2016, 791, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Vacca, V.; Marinelli, S.; Eleuteri, C.; Luvisetto, S.; Pavone, F. Botulinum neurotoxin A enhances the analgesic effects on inflammatory pain and antagonizes tolerance induced by morphine in mice. Brain Behav. Immun. 2012, 26, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Vacca, V.; Marinelli, S.; Luvisetto, S.; Pavone, F. Botulinum toxin A increases analgesic effects of morphine, counters development of morphine tolerance and modulates glia activation and μ opioid receptor expression in neuropathic mice. Brain Behav. Immun. 2013, 32, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Koga, K.; Chen, T.; Zhuo, M. Neuronal and microglial mechanisms for neuropathic pain in the spinal dorsal horn and anterior cingulate cortex. J. Neurochem. 2017, 141, 486–498. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, M.; Wu, G.; Wu, L.J. Neuronal and microglial mechanisms of neuropathic pain. Mol. Brain 2011, 4, 31. [Google Scholar] [CrossRef] [PubMed]
- Wasser, C.R.; Kavalali, E.T. Leaky synapses: Regulation of spontaneous neurotransmission in central synapses. Neuroscience 2009, 158, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.J.; Imlach, W.L.; Jiao, W.; Wolfram, V.; Wu, Y.; Grbic, M.; Cela, C.; Baines, R.A.; Nitabach, M.N.; McCabe, B.D. Miniature Neurotransmission Regulates Drosophila Synaptic Structural Maturation. Neuron 2014, 82, 618–634. [Google Scholar] [CrossRef] [PubMed]
- Molgó, J.; Siegel, L.S.; Tabti, N.; Thesleff, S. A study of synchronization of quantal transmitter release from mammalian motor endings by the use of botulinal toxins type A and D. J. Physiol. 1989, 411, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, O.; Seveso, M.; Caccin, P.; Schiavo, G.; Montecucco, C. Tetanus and botulinum neurotoxins: Turning bad guys into good by research. Toxicon 2001, 39, 27–41. [Google Scholar] [CrossRef]
- Katz, E.; Ferro, P.A.; Cherksey, B.D.; Sugimori, M.; Llinas, R.; Uchitel, O.D. Effects of Ca2+ channel blockers on transmitter release and presynaptic currents at the frog neuromuscular junction. J. Physiol. 1995, 486, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Südhof, T.C.; Rothman, J.E. Membrane fusion: Grappling with SNARE and SM proteins. Science 2009, 323, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Pantano, S.; Montecucco, C. The blockade of the neurotransmitter release apparatus by botulinum neurotoxins. Cell. Mol. Life Sci. 2014, 71, 793–811. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Khanijou, S.; Rubino, J.; Aoki, K.R. Subcutaneous administration of botulinum toxin a reduces formalin-induced pain. Pain 2004, 107, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Wang, J.; Lawrence, G.; Dolly, J.O. Synaptobrevin I mediates exocytosis of CGRP from sensory neurons and inhibition by botulinum toxins reflects their anti-nociceptive potential. J. Cell Sci. 2007, 120, 2864–2874. [Google Scholar] [CrossRef] [PubMed]
- Luvisetto, S.; Marinelli, S.; Lucchetti, F.; Marchi, F.; Cobianchi, S.; Rossetto, O.; Montecucco, C.; Pavone, F. Botulinum neurotoxins and formalin-induced pain: Central vs. peripheral effects in mice. Brain Res. 2006, 1082, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.C.; Yoshimura, N.; Huang, C.C.; Chiang, P.H.; Chancellor, M.B. Intravesical botulinum toxin a administration produces analgesia against acetic acid induced bladder pain responses in rats. J. Urol. 2004, 172 (4 Pt 1), 1529–1532. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, F.; Rossi, C.; Gianfranceschi, L.; Rossetto, O.; Caleo, M. Long-Distance Retrograde Effects of Botulinum Neurotoxin A. J. Neurosci. 2008, 28, 3689–3696. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.A.; Bigalke, H.; Aoki, K.R. Botulinum neurotoxin—From laboratory to bedside. Neurotox. Res. 2006, 9, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, B. Botulinum neurotoxins in the treatment of refractory pain. Nat. Clin. Pract. Neurol. 2008, 4, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Bach-Rojecky, L.; Relja, M.; Lacković, Z. Botulinum toxin type A in experimental neuropathic pain. J. Neural Transm. Vienna Austria 1996 2005, 112, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Habermann, E. 125I-labeled neurotoxin from clostridium botulinum A: Preparation, binding to synaptosomes and ascent to the spinal cord. Naunyn Schmiedebergs Arch. Pharmacol. 1974, 281, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, H.; Erdmann, G.; Wellhöner, H.H. 125I-Labelled botulinum a neurotoxin: Pharmacokinetics in cats after intramuscular injection. Naunyn. Schmiedebergs. Arch. Pharmacol. 1976, 292, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, S.; Vacca, V.; Ricordy, R.; Uggenti, C.; Tata, A.M.; Luvisetto, S.; Pavone, F. The Analgesic Effect on Neuropathic Pain of Retrogradely Transported botulinum Neurotoxin A Involves Schwann Cells and Astrocytes. PLoS ONE 2012, 7, e47977. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, A.; Popiolek-Barczyk, K.; Pavone, F.; Mika, J. Comparison of the Expression Changes after Botulinum Toxin Type A and Minocycline Administration in Lipopolysaccharide-Stimulated Rat Microglial and Astroglial Cultures. Front. Cell. Infect. Microbiol. 2017, 7, 147. [Google Scholar] [CrossRef] [PubMed]
- Hepp, R.; Perraut, M.; Chasserot-Golaz, S.; Galli, T.; Aunis, D.; Langley, K.; Grant, N.J. Cultured glial cells express the SNAP-25 analogue SNAP-23. Glia 1999, 27, 181–187. [Google Scholar] [CrossRef]
- Parpura, V.; Fang, Y.; Basarsky, T.; Jahn, R.; Haydon, P.G. Expression of synaptobrevin II, cellubrevin and syntaxin but not SNAP-25 in cultured astrocytes. FEBS Lett. 1995, 377, 489–492. [Google Scholar] [CrossRef] [PubMed]
- DeLeo, J.A.; Yezierski, R.P. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain 2001, 90, 1–6. [Google Scholar] [CrossRef]
- Popiolek-Barczyk, K.; Mika, J. Targeting the microglial signaling pathways: New insights in the modulation of neuropathic pain. Curr. Med. Chem. 2016, 23, 2908–2928. [Google Scholar] [CrossRef] [PubMed]
- Colburn, R.W.; DeLeo, J.A.; Rickman, A.J.; Yeager, M.P.; Kwon, P.; Hickey, W.F. Dissociation of microglial activation and neuropathic pain behaviors following peripheral nerve injury in the rat. J. Neuroimmunol. 1997, 79, 163–175. [Google Scholar] [CrossRef]
- Colburn, R.W.; Rickman, A.J.; Deleo, J.A. The effect of site and type of nerve injury on spinal glial activation and neuropathic pain behavior. Exp. Neurol. 1999, 157, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, M. Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science 1994, 263, 1768–1771. [Google Scholar] [CrossRef] [PubMed]
- Roh, D.H.; Yoon, S.Y.; Seo, H.S.; Kang, S.Y.; Han, H.J.; Beitz, A.J.; Lee, J.H. Intrathecal injection of carbenoxolone, a gap junction decoupler, attenuates the induction of below-level neuropathic pain after spinal cord injury in rats. Exp. Neurol. 2010, 224, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Zündorf, G.; Kahlert, S.; Reiser, G. Gap-junction blocker carbenoxolone differentially enhances NMDA-induced cell death in hippocampal neurons and astrocytes in co-culture. J. Neurochem. 2007, 102, 508–521. [Google Scholar] [CrossRef] [PubMed]
- Haber, M.; Murai, K.K. Reshaping neuron-glial communication at hippocampal synapses. Neuron Glia Biol. 2006, 2, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Panatier, A.; Vallée, J.; Haber, M.; Murai, K.K.; Lacaille, J.C.; Robitaille, R. Astrocytes are endogenous regulators of basal transmission at central synapses. Cell 2011, 146, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Oliet, S.H.R.; Panatier, A.; Piet, R.; Mothet, J.P.; Poulain, D.A.; Theodosis, D.T. Neuron-glia interactions in the rat supraoptic nucleus. Prog. Brain Res. 2008, 170, 109–117. [Google Scholar] [PubMed]
- Romero-Sandoval, A.; Chai, N.; Nutile-McMenemy, N.; DeLeo, J.A. A comparison of spinal Iba1 and GFAP expression in rodent models of acute and chronic pain. Brain Res. 2008, 1219, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Tanga, F.Y.; Raghavendra, V.; DeLeo, J.A. Quantitative real-time RT-PCR assessment of spinal microglial and astrocytic activation markers in a rat model of neuropathic pain. Neurochem. Int. 2004, 45, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Mika, J. Modulation of microglia can attenuate neuropathic pain symptoms and enhance morphine effectiveness. Pharmacol. Rep. 2008, 60, 297–307. [Google Scholar] [PubMed]
- Clark, A.K.; Gentry, C.; Bradbury, E.J.; McMahon, S.B.; Malcangio, M. Role of spinal microglia in rat models of peripheral nerve injury and inflammation. Eur. J. Pain 2007, 11, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Coyle, D.E. Partial peripheral nerve injury leads to activation of astroglia and microglia which parallels the development of allodynic behavior. Glia 1998, 23, 75–83. [Google Scholar] [CrossRef]
- Zychowska, M.; Rojewska, E.; Przewlocka, B.; Mika, J. Mechanisms and pharmacology of diabetic neuropathy—Experimental and clinical studies. Pharmacol. Rep. 2013, 65, 1601–1610. [Google Scholar] [CrossRef]
- Amin, A.R.; Attur, M.G.; Thakker, G.D.; Patel, P.D.; Vyas, P.R.; Patel, R.N.; Patel, I.R.; Abramson, S.B. A novel mechanism of action of tetracyclines: Effects on nitric oxide synthases. Proc. Natl. Acad. Sci. USA 1996, 93, 14014–14019. [Google Scholar] [CrossRef] [PubMed]
- Colovic, M.; Caccia, S. Liquid chromatographic determination of minocycline in brain-to-plasma distribution studies in the rat. J. Chromatogr. A 2003, 791, 337–343. [Google Scholar] [CrossRef]
- Mika, J.; Osikowicz, M.; Rojewska, E.; Korostynski, M.; Wawrzczak-Bargiela, A.; Przewlocki, R.; Przewlocka, B. Differential activation of spinal microglial and astroglial cells in a mouse model of peripheral neuropathic pain. Eur. J. Pharmacol. 2009, 623, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, R.; Ekstrøm, P.; Giercksky, K.E. Pentoxifylline improves survival and reduces tumor necrosis factor, interleukin-6, and endothelin-1 in fulminant intra-abdominal sepsis in rats. Shock 1995, 3, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Mika, J.; Osikowicz, M.; Makuch, W.; Przewlocka, B. Minocycline and pentoxifylline attenuate allodynia and hyperalgesia and potentiate the effects of morphine in rat and mouse models of neuropathic pain. Eur. J. Pharmacol. 2007, 560, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Sweitzer, S.M.; Schubert, P.; DeLeo, J.A. Propentofylline, a glial modulating agent, exhibits antiallodynic properties in a rat model of neuropathic pain. J. Pharmacol. Exp. Ther. 2001, 297, 1210–1217. [Google Scholar] [PubMed]
- Kim, Y.J.; Kim, J.-H.; Lee, K.-J.; Choi, M.-M.; Kim, Y.H.; Rhie, G.; Yoo, C.-K.; Cha, K.; Shin, N.-R. Botulinum Neurotoxin Type A Induces TLR2-Mediated Inflammatory Responses in Macrophages. PLoS ONE 2015, 10, e0120840. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.X.; Zhuang, Z.Y.; Woolf, C.J.; Ji, R.R. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J. Neurosci. 2003, 23, 4017–4022. [Google Scholar] [PubMed]
- Tsuda, M.; Mizokoshi, A.; Shigemoto-Mogami, Y.; Koizumi, S.; Inoue, K. Activation of p38 Mitogen-Activated Protein Kinase in Spinal Hyperactive Microglia Contributes to Pain Hypersensitivity Following Peripheral Nerve Injury. Glia 2004, 45, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Quirion, R. The ERK/MAPK pathway, as a target for the treatment of neuropathic pain. Expert Opin. Ther. Targets 2005, 9, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Rojewska, E.; Popiolek-Barczyk, K.; Jurga, A.M.; Makuch, W.; Przewlocka, B.; Mika, J. Involvement of pro- and antinociceptive factors in minocycline analgesia in rat neuropathic pain model. J. Neuroimmunol. 2014, 277, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Rojewska, E.; Piotrowska, A.; Makuch, W.; Przewlocka, B.; Mika, J. Pharmacological kynurenine 3-monooxygenase enzyme inhibition significantly reduces neuropathic pain in a rat model. Neuropharmacology 2016, 102, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Mika, J.; Popiolek-Barczyk, K.; Rojewska, E.; Makuch, W.; Starowicz, K.; Przewlocka, B. Delta-opioid receptor analgesia is independent of microglial activation in a rat model of neuropathic pain. PLoS ONE 2014, 9, e104420. [Google Scholar] [CrossRef] [PubMed]
- Popiolek-Barczyk, K.; Kolosowska, N.; Piotrowska, A.; Makuch, W.; Rojewska, E.; Jurga, A.M.; Pilat, D.; Mika, J. Parthenolide relieves pain and promotes M2 microglia/macrophage polarization in rat model of neuropathy. Neural Plast. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, A.; Kwiatkowski, K.; Rojewska, E.; Makuch, W.; Mika, J. Maraviroc reduces neuropathic pain through polarization of microglia and astroglia—Evidence from in vivo and in vitro studies. Neuropharmacology 2016, 108, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Bisby, M. a Increased activation of nuclear factor kappa B in rat lumbar dorsal root ganglion neurons following partial sciatic nerve injuries. Brain Res. 1998, 797, 243–254. [Google Scholar] [CrossRef]
- Meunier, A.; Latrémolière, A.; Dominguez, E.; Mauborgne, A.; Philippe, S.; Hamon, M.; Mallet, J.; Benoliel, J.J.; Pohl, M. Lentiviral-mediated targeted NF-κB blockade in dorsal spinal cord glia attenuates sciatic nerve injury-induced neuropathic pain in the rat. Mol. Ther. 2007, 15, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, K.; Obata, K.; Kondo, T.; Okamura, H.; Noguchi, K. Interleukin-18-Mediated Microglia/Astrocyte Interaction in the Spinal Cord Enhances Neuropathic Pain Processing after Nerve Injury. J. Neurosci. 2008, 28, 12775–12787. [Google Scholar] [CrossRef] [PubMed]
- Jurga, A.M.; Rojewska, E.; Piotrowska, A.; Makuch, W.; Pilat, D.; Przewlocka, B.; Mika, J. Blockade of toll-like receptors (TLR2, TLR4) attenuates pain and potentiates buprenorphine analgesia in a rat neuropathic pain model. Neural Plast. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, M.A.; Cho, I.-H.; Kim, M.S.; Lee, S.; Jo, E.-K.; Choi, S.-Y.; Park, K.; Kim, J.S.; Akira, S.; et al. A critical role of toll-like receptor 2 in nerve injury-induced spinal cord glial cell activation and pain hypersensitivity. J. Biol. Chem. 2007, 282, 14975–14983. [Google Scholar] [CrossRef] [PubMed]
- Lehnardt, S.; Massillon, L.; Follett, P.; Jensen, F.E.; Ratan, R.; Rosenberg, P.A.; Volpe, J.J.; Vartanian, T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 8514–8519. [Google Scholar] [CrossRef] [PubMed]
- Tanga, F.Y.; Nutile-McMenemy, N.; DeLeo, J.A. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc. Natl. Acad. Sci. USA 2005, 102, 5856–5861. [Google Scholar] [CrossRef] [PubMed]
- Borrello, S.; Nicolò, C.; Delogu, G.; Pandolfi, F.; Ria, F. TLR2: A crossroads between infections and autoimmunity? Int. J. Immunopathol. Pharmacol. 2011, 24, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A. A human homologue of the Drosophila toll protein signals activation of adaptive immunity. Nature 1997, 388, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Gao, Y.-J.; Ji, R.-R. Emerging role of Toll-like receptors in the control of pain and itch. Neurosci. Bull. 2012, 28, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K. Innate immune sensing of pathogens and danger signals by cell surface Toll-like receptors. Semin. Immunol. 2007, 19, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Nair-Gupta, P.; Baccarini, A.; Tung, N.; Seyffer, F.; Florey, O.; Huang, Y.; Banerjee, M.; Overholtzer, M.; Roche, P.A.; Tampé, R.; et al. TLR signals induce phagosomal MHC-I delivery from the endosomal recycling compartment to allow cross-presentation. Cell 2014, 158, 506–521. [Google Scholar] [CrossRef] [PubMed]
- Beauvillain, C.; Donnou, S.; Jarry, U.; Scotet, M.; Gascan, H.; Delneste, Y.; Guermonprez, P.; Jeannin, P.; Couez, D. Neonatal and adult microglia cross-present exogenous antigens. Glia 2008, 56, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Holm, T.H.; Draeby, D.; Owens, T. Microglia are required for astroglial toll-like receptor 4 response and for optimal TLR2 and TLR3 response. Glia 2012, 60, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Lee, H.; Berg, A.H.; Lisanti, M.P.; Shapiro, L.; Scherer, P.E. The lipopolysaccharide-activated Toll-like receptor (TLR)-4 induces synthesis of the closely related receptor TLR-2 in adipocytes. J. Biol. Chem. 2000, 275, 24255–24263. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojewska, E.; Piotrowska, A.; Popiolek-Barczyk, K.; Mika, J. Botulinum Toxin Type A—A Modulator of Spinal Neuron–Glia Interactions under Neuropathic Pain Conditions. Toxins 2018, 10, 145. https://doi.org/10.3390/toxins10040145
Rojewska E, Piotrowska A, Popiolek-Barczyk K, Mika J. Botulinum Toxin Type A—A Modulator of Spinal Neuron–Glia Interactions under Neuropathic Pain Conditions. Toxins. 2018; 10(4):145. https://doi.org/10.3390/toxins10040145
Chicago/Turabian StyleRojewska, Ewelina, Anna Piotrowska, Katarzyna Popiolek-Barczyk, and Joanna Mika. 2018. "Botulinum Toxin Type A—A Modulator of Spinal Neuron–Glia Interactions under Neuropathic Pain Conditions" Toxins 10, no. 4: 145. https://doi.org/10.3390/toxins10040145
APA StyleRojewska, E., Piotrowska, A., Popiolek-Barczyk, K., & Mika, J. (2018). Botulinum Toxin Type A—A Modulator of Spinal Neuron–Glia Interactions under Neuropathic Pain Conditions. Toxins, 10(4), 145. https://doi.org/10.3390/toxins10040145