Connective Tissue Growth Factor Is Related to All-cause Mortality in Hemodialysis Patients and Is Lowered by On-line Hemodiafiltration: Results from the Convective Transport Study

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Patient and Dialysis Characteristics
2.2. Determinants of Plasma CTGF
2.3. CTGF and All-cause Mortality
2.4. CTGF and Fatal- and Non-fatal Cardiovascular Events and Infectious Mortality
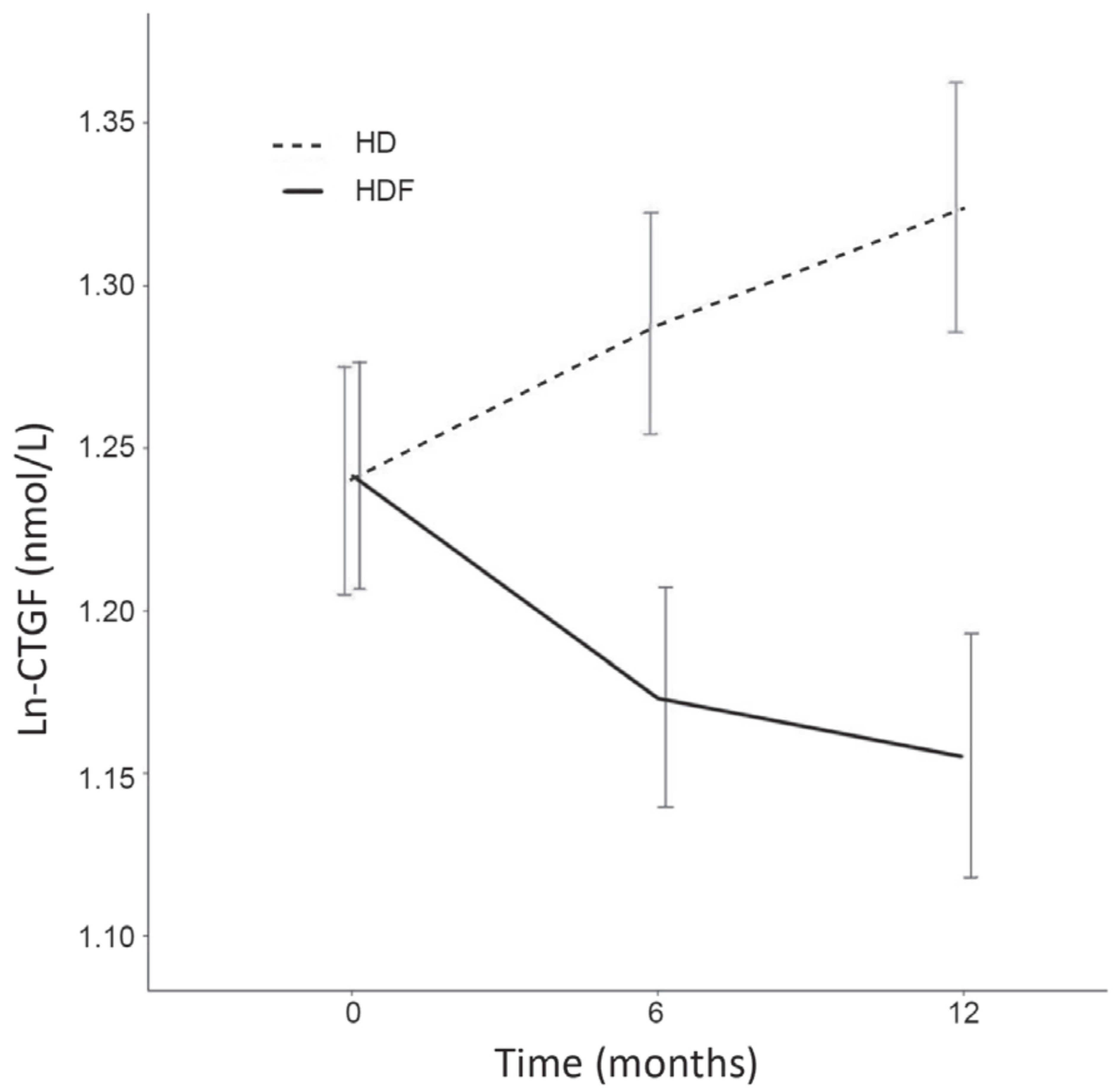
2.5. CTGF Change over Time, HDF versus HD
3. Discussion
3.1. Determinants of Plasma CTGF Concentrations
3.2. CTGF and All-cause Mortality
3.3. CTGF Change over Time, HDF versus HD
4. Methods
4.1. General Methods
4.2. Dialysis Procedures
4.3. Data Collection
4.4. Laboratory Measurements
4.5. Data Analysis
4.5.1. Determinants of Plasma CTGF
4.5.2. CTGF and All-cause Mortality, Cardiovascular Events, Cardiovascular Mortality and Infectious Mortality
4.5.3. CTGF Change over Time, HDF versus HD
Author Contributions
Funding
Acknowledgments
Local Investigators
Conflicts of Interest
References
- Ramazani, Y.; Knops, N.; Elmonem, M.A.; Nguyen, T.Q.; Arcolino, F.O.; van den Heuvel, L.; Levtchenko, E.; Kuypers, D.; Goldschmeding, R. Connective tissue growth factor (CTGF) from basics to clinics. Matrix Biol. J. Int. Soc. Matrix Biol. 2018, 68–69, 44–66. [Google Scholar] [CrossRef]
- Dendooven, A.; Gerritsen, K.G.; Nguyen, T.Q.; Kok, R.J.; Goldschmeding, R. Connective tissue growth factor (CTGF/CCN2) ELISA: A novel tool for monitoring fibrosis. Biomark. Biochem. Indic. Expo. Response Susceptibility Chem. 2011, 16, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Daniels, A.; van Bilsen, M.; Goldschmeding, R.; van der Vusse, G.J.; van Nieuwenhoven, F.A. Connective tissue growth factor and cardiac fibrosis. Acta Physiol. (Oxf. Engl.) 2009, 195, 321–338. [Google Scholar] [CrossRef] [PubMed]
- Ruperez, M.; Lorenzo, O.; Blanco-Colio, L.M.; Esteban, V.; Egido, J.; Ruiz-Ortega, M. Connective tissue growth factor is a mediator of angiotensin II-induced fibrosis. Circulation 2003, 108, 1499–1505. [Google Scholar] [CrossRef]
- Ruster, C.; Wolf, G. Angiotensin II as a morphogenic cytokine stimulating renal fibrogenesis. J. Am. Soc. Nephrol. JASN 2011, 22, 1189–1199. [Google Scholar] [CrossRef]
- Gerritsen, K.G.; Abrahams, A.C.; Peters, H.P.; Nguyen, T.Q.; Koeners, M.P.; den Hoedt, C.H.; Dendooven, A.; van den Dorpel, M.A.; Blankestijn, P.J.; Wetzels, J.F.; et al. Effect of GFR on plasma N-terminal connective tissue growth factor (CTGF) concentrations. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2012, 59, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Gerritsen, K.G.; Peters, H.P.; Nguyen, T.Q.; Koeners, M.P.; Wetzels, J.F.; Joles, J.A.; Christensen, E.I.; Verroust, P.J.; Li, D.; Oliver, N.; et al. Renal proximal tubular dysfunction is a major determinant of urinary connective tissue growth factor excretion. Am. J. Physiol. Ren. Physiol. 2010, 298, F1457–F1464. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, H.; Oka, T.; Kusachi, S.; Nakanishi, T.; Takeda, K.; Nakahama, M.; Doi, M.; Murakami, T.; Ninomiya, Y.; Takigawa, M.; et al. Increased expression of connective tissue growth factor in the infarct zone of experimentally induced myocardial infarction in rats. J. Mol. Cell. Cardiol. 1998, 30, 2411–2422. [Google Scholar] [CrossRef]
- Koitabashi, N.; Arai, M.; Kogure, S.; Niwano, K.; Watanabe, A.; Aoki, Y.; Maeno, T.; Nishida, T.; Kubota, S.; Takigawa, M.; et al. Increased connective tissue growth factor relative to brain natriuretic peptide as a determinant of myocardial fibrosis. Hypertension (Dallas Tex. 1979) 2007, 49, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Koitabashi, N.; Arai, M.; Niwano, K.; Watanabe, A.; Endoh, M.; Suguta, M.; Yokoyama, T.; Tada, H.; Toyama, T.; Adachi, H.; et al. Plasma connective tissue growth factor is a novel potential biomarker of cardiac dysfunction in patients with chronic heart failure. Eur. J. Heart Fail. 2008, 10, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Rickard, A.J.; Morgan, J.; Chrissobolis, S.; Miller, A.A.; Sobey, C.G.; Young, M.J. Endothelial cell mineralocorticoid receptors regulate deoxycorticosterone/salt-mediated cardiac remodeling and vascular reactivity but not blood pressure. Hypertension (Dallas Tex. 1979) 2014, 63, 1033–1040. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Oie, E.; Vinge, L.E.; Yndestad, A.; Oystein Andersen, G.; Andersson, Y.; Attramadal, T.; Attramadal, H. Connective tissue growth factor—A novel mediator of angiotensin II-stimulated cardiac fibroblast activation in heart failure in rats. J. Mol. Cell. Cardiol. 2004, 36, 393–404. [Google Scholar] [CrossRef]
- Dorn, L.E.; Petrosino, J.M.; Wright, P.; Accornero, F. CTGF/CCN2 is an autocrine regulator of cardiac fibrosis. J. Mol. Cell. Cardiol. 2018, 121, 205–211. [Google Scholar] [CrossRef]
- Cicha, I.; Yilmaz, A.; Klein, M.; Raithel, D.; Brigstock, D.R.; Daniel, W.G.; Goppelt-Struebe, M.; Garlichs, C.D. Connective tissue growth factor is overexpressed in complicated atherosclerotic plaques and induces mononuclear cell chemotaxis in vitro. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1008–1013. [Google Scholar] [CrossRef] [PubMed]
- Leeuwis, J.W.; Nguyen, T.Q.; Theunissen, M.G.; Peeters, W.; Goldschmeding, R.; Pasterkamp, G.; Vink, A. Connective tissue growth factor is associated with a stable atherosclerotic plaque phenotype and is involved in plaque stabilization after stroke. Stroke 2010, 41, 2979–2981. [Google Scholar] [CrossRef] [PubMed]
- Ponticos, M. Connective tissue growth factor (CCN2) in blood vessels. Vasc. Pharmacol. 2013, 58, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Gerritsen, K.G.; Falke, L.L.; van Vuuren, S.H.; Leeuwis, J.W.; Broekhuizen, R.; Nguyen, T.Q.; de Borst, G.J.; Nathoe, H.M.; Verhaar, M.C.; Kok, R.J.; et al. Plasma CTGF is independently related to an increased risk of cardiovascular events and mortality in patients with atherosclerotic disease: The SMART study. Growth Factors (Chur Switz.) 2016, 34, 149–158. [Google Scholar] [CrossRef]
- Jaffa, A.A.; Usinger, W.R.; McHenry, M.B.; Jaffa, M.A.; Lipstiz, S.R.; Lackland, D.; Lopes-Virella, M.; Luttrell, L.M.; Wilson, P.W. Connective tissue growth factor and susceptibility to renal and vascular disease risk in type 1 diabetes. J. Clin. Endocrinol. Metab. 2008, 93, 1893–1900. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.Q.; Tarnow, L.; Jorsal, A.; Oliver, N.; Roestenberg, P.; Ito, Y.; Parving, H.H.; Rossing, P.; van Nieuwenhoven, F.A.; Goldschmeding, R. Plasma connective tissue growth factor is an independent predictor of end-stage renal disease and mortality in type 1 diabetic nephropathy. Diabetes Care 2008, 31, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K.; Rocco, M.V.; Yan, G.; Leypoldt, J.K.; Levin, N.W.; Greene, T.; Agodoa, L.; Bailey, J.; Beck, G.J.; Clark, W.; et al. Serum beta-2 microglobulin levels predict mortality in dialysis patients: Results of the HEMO study. J. Am. Soc. Nephrol. JASN 2006, 17, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.Y.; Lai, K.N. The importance of residual renal function in dialysis patients. Kidney Int. 2006, 69, 1726–1732. [Google Scholar] [CrossRef] [Green Version]
- London, G.M. Cardiovascular calcifications in uremic patients: Clinical impact on cardiovascular function. J. Am. Soc. Nephrol. JASN 2003, 14, S305–S309. [Google Scholar] [CrossRef]
- Pecoits-Filho, R.; Barany, P.; Lindholm, B.; Heimburger, O.; Stenvinkel, P. Interleukin-6 is an independent predictor of mortality in patients starting dialysis treatment. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2002, 17, 1684–1688. [Google Scholar] [CrossRef] [Green Version]
- Roestenberg, P.; van Nieuwenhoven, F.A.; Wieten, L.; Boer, P.; Diekman, T.; Tiller, A.M.; Wiersinga, W.M.; Oliver, N.; Usinger, W.; Weitz, S.; et al. Connective tissue growth factor is increased in plasma of type 1 diabetic patients with nephropathy. Diabetes Care 2004, 27, 1164–1170. [Google Scholar] [CrossRef]
- Ito, Y.; Aten, J.; Nguyen, T.Q.; Joles, J.A.; Matsuo, S.; Weening, J.J.; Goldschmeding, R. Involvement of connective tissue growth factor in human and experimental hypertensive nephrosclerosis. Nephron. Exp. Nephrol. 2011, 117, e9–e20. [Google Scholar] [CrossRef]
- Cozzolino, M.; Biondi, M.L.; Banfi, E.; Riser, B.L.; Mehmeti, F.; Cusi, D.; Gallieni, M. CCN2 (CTGF) gene polymorphism is a novel prognostic risk factor for cardiovascular outcomes in hemodialysis patients. Blood Purif. 2010, 30, 272–276. [Google Scholar] [CrossRef]
- Yao, Y.; Li, B.; Fu, C.; Teng, G.; Ma, G.; Liu, N. Anti-connective tissue growth factor detects and reduces plaque inflammation in early-stage carotid atherosclerotic lesions. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 2385–2394. [Google Scholar] [CrossRef]
- Sanchez-Lopez, E.; Rayego, S.; Rodrigues-Diez, R.; Rodriguez, J.S.; Rodrigues-Diez, R.; Rodriguez-Vita, J.; Carvajal, G.; Aroeira, L.S.; Selgas, R.; Mezzano, S.A.; et al. CTGF promotes inflammatory cell infiltration of the renal interstitium by activating NF-kappaB. J. Am. Soc. Nephrol. JASN 2009, 20, 1513–1526. [Google Scholar] [CrossRef]
- Arizono, K.; Nomura, K.; Motoyama, T.; Matsushita, Y.; Matsuoka, K.; Miyazu, R.; Takeshita, H.; Fukui, H. Use of ultrapure dialysate in reduction of chronic inflammation during hemodialysis. Blood Purif. 2004, 22 Suppl. 2, 26–29. [Google Scholar] [CrossRef]
- Furuya, R.; Kumagai, H.; Takahashi, M.; Sano, K.; Hishida, A. Ultrapure dialysate reduces plasma levels of beta2-microglobulin and pentosidine in hemodialysis patients. Blood Purif. 2005, 23, 311–316. [Google Scholar] [CrossRef]
- Weng, L.; Wang, W.; Su, X.; Huang, Y.; Su, L.; Liu, M.; Sun, Y.; Yang, B.; Zhou, H. The Effect of cAMP-PKA Activation on TGF-beta1-Induced Profibrotic Signaling. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 36, 1911–1927. [Google Scholar] [CrossRef] [PubMed]
- Happe, H.; van der Wal, A.M.; Leonhard, W.N.; Kunnen, S.J.; Breuning, M.H.; de Heer, E.; Peters, D.J. Altered Hippo signalling in polycystic kidney disease. J. Pathol. 2011, 224, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Attanasio, M.; Uhlenhaut, N.H.; Sousa, V.H.; O’Toole, J.F.; Otto, E.; Anlag, K.; Klugmann, C.; Treier, A.C.; Helou, J.; Sayer, J.A.; et al. Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat. Genet. 2007, 39, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Hassane, S.; Leonhard, W.N.; van der Wal, A.; Hawinkels, L.J.; Lantinga-van Leeuwen, I.S.; ten Dijke, P.; Breuning, M.H.; de Heer, E.; Peters, D.J. Elevated TGFbeta-Smad signalling in experimental Pkd1 models and human patients with polycystic kidney disease. J. Pathol. 2010, 222, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Norman, J. Fibrosis and progression of autosomal dominant polycystic kidney disease (ADPKD). Biochim. Et Biophys. Acta 2011, 1812, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Gauer, S.; Holzmann, Y.; Kranzlin, B.; Hoffmann, S.C.; Gretz, N.; Hauser, I.A.; Goppelt-Struebe, M.; Geiger, H.; Obermuller, N. CTGF Is Expressed During Cystic Remodeling in the PKD/Mhm (cy/+) Rat Model for Autosomal-Dominant Polycystic Kidney Disease (ADPKD). J. Histochem. Cytochem. Off. J. Histochem. Soc. 2017, 65, 743–755. [Google Scholar] [CrossRef]
- Ito, Y.; Aten, J.; Bende, R.J.; Oemar, B.S.; Rabelink, T.J.; Weening, J.J.; Goldschmeding, R. Expression of connective tissue growth factor in human renal fibrosis. Kidney Int. 1998, 53, 853–861. [Google Scholar] [CrossRef] [Green Version]
- Leypoldt, J.K.; Cheung, A.K.; Deeter, R.B. Rebound kinetics of beta2-microglobulin after hemodialysis. Kidney Int. 1999, 56, 1571–1577. [Google Scholar] [CrossRef]
- Grooteman, M.P.; van den Dorpel, M.A.; Bots, M.L.; Penne, E.L.; van der Weerd, N.C.; Mazairac, A.H.; den Hoedt, C.H.; van der Tweel, I.; Levesque, R.; Nube, M.J.; et al. Effect of online hemodiafiltration on all-cause mortality and cardiovascular outcomes. J. Am. Soc. Nephrol. JASN 2012, 23, 1087–1096. [Google Scholar] [CrossRef]
- Ward, R.A.; Greene, T.; Hartmann, B.; Samtleben, W. Resistance to intercompartmental mass transfer limits beta2-microglobulin removal by post-dilution hemodiafiltration. Kidney Int. 2006, 69, 1431–1437. [Google Scholar] [CrossRef]
- Grotendorst, G.R.; Duncan, M.R. Individual domains of connective tissue growth factor regulate fibroblast proliferation and myofibroblast differentiation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 729–738. [Google Scholar] [CrossRef]
- Penne, E.L.; Blankestijn, P.J.; Bots, M.L.; van den Dorpel, M.A.; Grooteman, M.P.; Nube, M.J.; van der Tweel, I.; Ter Wee, P.M. Effect of increased convective clearance by on-line hemodiafiltration on all cause and cardiovascular mortality in chronic hemodialysis patients—The Dutch CONvective TRAnsport STudy (CONTRAST): Rationale and design of a randomised controlled trial [ISRCTN38365125]. Curr. Control. Trials Cardiovasc. Med. 2005, 6, 8. [Google Scholar] [CrossRef]
- Association for the Advancement of Medical Instrumentation. Quality of dialysis fluid for hemodialysis fluid and related therapies ANSI/AAMI 11663:2014; Association for the Advancement of Medical Instrumentation: Arlington, VA, USA, 2014. [Google Scholar]
- Daugirdas, J.T. Second generation logarithmic estimates of single-pool variable volume Kt/V: An analysis of error. J. Am. Soc. Nephrol. JASN 1993, 4, 1205–1213. [Google Scholar]
- Fouque, D.; Vennegoor, M.; ter Wee, P.; Wanner, C.; Basci, A.; Canaud, B.; Haage, P.; Konner, K.; Kooman, J.; Martin-Malo, A.; et al. EBPG guideline on nutrition. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2007, 22 (Suppl. 2), ii45–ii87. [Google Scholar] [CrossRef]


{kind=link}
| Characteristic | On-Line HDF (n = 199) | Low Flux HD (n = 205) |
|---|---|---|
| Age (years) | 64 ± 14.3 | 63 ± 13.6 |
| Male sex—n (%) | 121 (61) | 130 (63) |
| Region | ||
| Netherlands—n (%) | 193 (96) | 197 (96) |
| Norway—n (%) | 7 (4) | 8 (4) |
| History of cardiovascular disease—n (%) | 90 (45) | 87 (42) |
| Diabetes mellitus—n (%) | 46 (23) | 39 (19) |
| Dialysis vintage (years) | 1.7 (0.9 to 3.1) | 2.0 (0.9 to 3.7) |
| Underlying renal disease—n (%) | ||
| Glomerulonephritis | 10 (5%) | 11 (6%) |
| Tubulointerstitial nephritis | 17 (9%) | 14 (7%) |
| Polycystic kidney disease | 10 (5%) | 10 (5%) |
| Other congenital/hereditary diseases | 2 (1%) | 0 |
| Renal vascular disease | 37(19%) | 47 (24%) |
| Diabetic nephropathy | 22 (11%) | 22 (11%) |
| Multisystem disease | 8 (4%) | 7 (4%) |
| Other | 19 (10%) | 22 (11%) |
| Unknown | 74 (37%) | 72 (36%) |
| Systolic blood pressure (mmHg) ^ | 147 ± 22 | 149 ± 21 |
| Diastolic blood pressure (mmHg) ^ | 75 ± 12 | 76 ± 12 |
| Pulse pressure, pre-dialysis (mmHg) | 69 ± 18 | 68 ± 16 |
| Body mass index, after dialysis (kg/m2) | 24.7 ± 4.9 | 25.3 ± 4.7 |
| Vascular access– n (%) | ||
| Arteriovenous fistula | 162 (81) | 176 (86) |
| Graft | 31 (16) | 22 (11) |
| Central catheter | 5 (3) | 5 (2) |
| Duration of a dialysis session (hours) | 4.0 (3.5 to 4.0) | 4.0 (3.5 to 4.0) |
| Number of treatments/week | 2.90 ± 0.30 | 2.95 ± 0.23 |
| Blood flow (mL/min) | 300 (300 to 325) | 300 (299 to 326) |
| Dialysis single pool Kt/Vurea | 1.35 (1.23 to 1.52) | 1.33 (1.23 to 1.47) |
| Residual kidney function—n (%) * | 119 (60) | 111 (54) |
| Estimated glomerular filtration rate (mL/min/1.73m2) ^ | 2.8 (1.3 to 5.1) | 2.5 (1.1 to 5.1) |
| Hemoglobin (mmol/L) ^ | 7.4 ± 0.8 | 7.3±0.7 |
| Phosphorus (mmol/L) ^ | 1.68 ± 0.56 | 1.65 ± 0.47 |
| Beta-2-microglobulin (mg/L) ^ | 28.5 ± 11.5 | 31.7 ± 11.9 |
| Albumin (g/L) ^ | 35.8 ± 4.8 | 36.0 ± 4.8 |
| Creatinine (μmol/L) ^ | 837 ± 243 | 896 ± 255 |
| Connective Tissue Growth Factor (nmol/L) ^ | 3.5 (2.7 to 4.3) | 3.5 (2.8 to 4.4) |
| Ln-CTGF (nmol/L) ^ | 1.24 ± 0.39 | 1.26 ± 0.39 |
| Hs-CRP (mg/L) ^ | 4.06 (1.43 to 10.43) | 3.88 (1.31 to 10.28) |
| IL-6 (pg/mL) ^ | 2.11 (1.26 to 4.08) | 1.98 (1.18 to 3.42) |
| Prescribed medication—n (%) | ||
| RAAS blocker | 108 (54) | 107 (52) |
| Beta blocker | 101 (51) | 126 (61) |
| Calcium antagonist | 63 (32) | 72 (35) |
| Statin | 104 (52) | 97 (47) |
| Determinants | β | p | Change in CTGF (%) (95% CI) * |
|---|---|---|---|
| Previous CVD | 0.099 | 0.006 | 10.4 (2.8 to 18.5) |
| Dialysis vintage | 0.031 | <0.001 | 3.1 (1.7 to 4.5) |
| Pulse pressure | 0.002 | 0.059 | 0.2 (0 to 0.4) |
| Ln-IL-6 | 0.075 | <0.001 | 7.8 (4.1 to 11.7) |
| β2-microglobulin | 0.003 | 0.045 | 0.3 (0 to 0.7) |
| eGFR | –0.033 | <0.001 | −3.3 (−4.4 to −2.1) |
| RAAS-blocker use | –0.068 | 0.052 | −6.6 (−12.8 to 0.1) |
| UF | 0.034 | 0.091 | 3.5 (−0.6 to 7.7) |
| Underlying renal disease | |||
| Glomerulonephritis | 0.050 | 0.484 | 5.1 (−8.7 to 20.8) |
| Tubulointerstitial nephritis | 0.222 | 0.002 | 24.9 (8.5 to 43.6) |
| Polycystic kidney disease | 0.403 | <0.001 | 49.6 (28.1 to 74.5) |
| Renal vascular disease | 0.115 | 0.041 | 12.2 (5.0 to 25.4) |
| Diabetic nephropathy | 0.103 | 0.120 | 10.8 (−23.7 to 26.1) |
| Multisystem disease | 0.078 | 0.387 | 8.1 (−9.4 to 29.2) |
| Other | 0.030 | 0.682 | 3.0 (−10.8 to 18.9) |
| Unknown | reference |
| Outcome | Model | N | #Events | Hazard Ratio (95% CI) | P |
|---|---|---|---|---|---|
| All-cause mortality | I II III | 404 384 382 | 158 151 149 | 1.10 (1.01–1.19) 1.09 (1.00–1.19) 1.09 (0.99–1.19) | 0.02 0.05 0.07 |
| Fatal and non-fatal cardiovascular events | I IV | 404 362 | 131 121 | 1.06 (0.96–1.16) 1.03 (0.94–1.14) | 0.24 0.51 |
| Cardiovascular mortality | I V | 404 394 | 50 46 | 0.99 (0.84–1.18) 1.01 (0.85–1.19) | 0.93 0.96 |
| Infectious mortality | I VI | 404 391 | 40 39 | 1.14 (0.99–1.31) 1.06 (0.92–1.23) | 0.08 0.42 |
| Outcome | Model | Hazard Ratio (95% CI) | |||
|---|---|---|---|---|---|
| Quartile 1 | Quartile 2 | Quartile 3 | Quartile 4 | ||
| All-cause mortality | I II | 1.00 (reference) 1.00 (reference) | 1.17 (0.73–1.87) 1.15 (0.69–1.91) | 1.23 (0.78–1.96) 1.09 (0.63–1.88) | 1.60 (1.03–2.50) 1.71 (1.02–2.88) |
| Fatal and non-fatal cardiovascular events | I III | 1.00 (reference) 1.00 (reference) | 1.35 (0.80–2.26) 1.42 (0.81–2.50) | 1.85 (1.12–3.04) 1.91 (1.08–3.38) | 1.41 (0.83–2.38) 1.63 (0.86–3.08) |
| Ln-CTGF | Model | Change in Plasma CTGF Concentration (Δ) per 6 Months | |||||
|---|---|---|---|---|---|---|---|
| HD (N = 205) | HDF (N = 199) | HDF versus HD | |||||
| Δ | p | Δ | p | Δ | p | ||
| 0–6 Months | |||||||
| Ln-CTGF (nmol/L) (mean (95% CI)) | I II | 0.047 (0.013 to 0.081) 0.031 (−0.0055 to 0.062) | 0.007 0.10 | −0.050 (−0.086 to −0.015) −0.058 (−0.096 to −0.019) | 0.006 0.003 | −0.097 (−0.147 to −0.048) −0.088 (−0.133 to −0.036) | <0.001 0.001 |
| CTGF (%) * | I II | +4.9 +3.1 | −4.8 −5.4 | −8.9 −8.1 | |||
| 6–12 Months | |||||||
| Ln-CTGF (nmol/L) (mean (95% CI)) | I II | 0.039 (−0.002 to 0.080) 0.042 (−0.001 to 0.085) | 0.06 0.06 | −0.028 (−0.070 to 0.014) −0.034 (−0.077 to 0.011) | 0.19 0.13 | −0.067 (−0.123 to -0.009) −0.075 (−0.136 to -0.014) | 0.02 0.02 |
| CTGF (%) * | I II | +4.1 +4.3 | −2.7 −3.2 | −6.3 −7.0 | |||
| Mean Δ Ln-CTGF * (nmol/L/6 Months) (95% CI) | p | |
|---|---|---|
| Convection Tertile 1 | −0.038 (−0.070 to −0.003) | 0.03 |
| CTGF (%) ** | −3.6 | - |
| Convection Tertile 2 | −0.044 (−0.075 to -0.011) | 0.009 |
| CTGF (%) ** | −4.2 | - |
| Convection Tertile 3 | −0.046 (−0.075 to −0.015) | 0.004 |
| CTGF (%) ** | −4.4 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
den Hoedt, C.H.; van Gelder, M.K.; Grooteman, M.P.; Nubé, M.J.; Blankestijn, P.J.; Goldschmeding, R.; Kok, R.J.; Bots, M.L.; van den Dorpel, M.A.; Gerritsen, K.G.F. Connective Tissue Growth Factor Is Related to All-cause Mortality in Hemodialysis Patients and Is Lowered by On-line Hemodiafiltration: Results from the Convective Transport Study. Toxins 2019, 11, 268. https://doi.org/10.3390/toxins11050268
den Hoedt CH, van Gelder MK, Grooteman MP, Nubé MJ, Blankestijn PJ, Goldschmeding R, Kok RJ, Bots ML, van den Dorpel MA, Gerritsen KGF. Connective Tissue Growth Factor Is Related to All-cause Mortality in Hemodialysis Patients and Is Lowered by On-line Hemodiafiltration: Results from the Convective Transport Study. Toxins. 2019; 11(5):268. https://doi.org/10.3390/toxins11050268
Chicago/Turabian Styleden Hoedt, Claire H., Maaike K. van Gelder, Muriel P. Grooteman, Menso J. Nubé, Peter J. Blankestijn, Roel Goldschmeding, Robbert Jan Kok, Michiel L. Bots, Marinus A. van den Dorpel, and Karin G. F. Gerritsen. 2019. "Connective Tissue Growth Factor Is Related to All-cause Mortality in Hemodialysis Patients and Is Lowered by On-line Hemodiafiltration: Results from the Convective Transport Study" Toxins 11, no. 5: 268. https://doi.org/10.3390/toxins11050268